
Synthesis and In Vitro Characterization of Glycopeptide Drug Candidates Related to PACAP1–23

 ,
, 
Abstract
:1. Introduction
1.1. Previous SAR Work and Evidence of PACAP1–23 Neuroprotection In Vitro
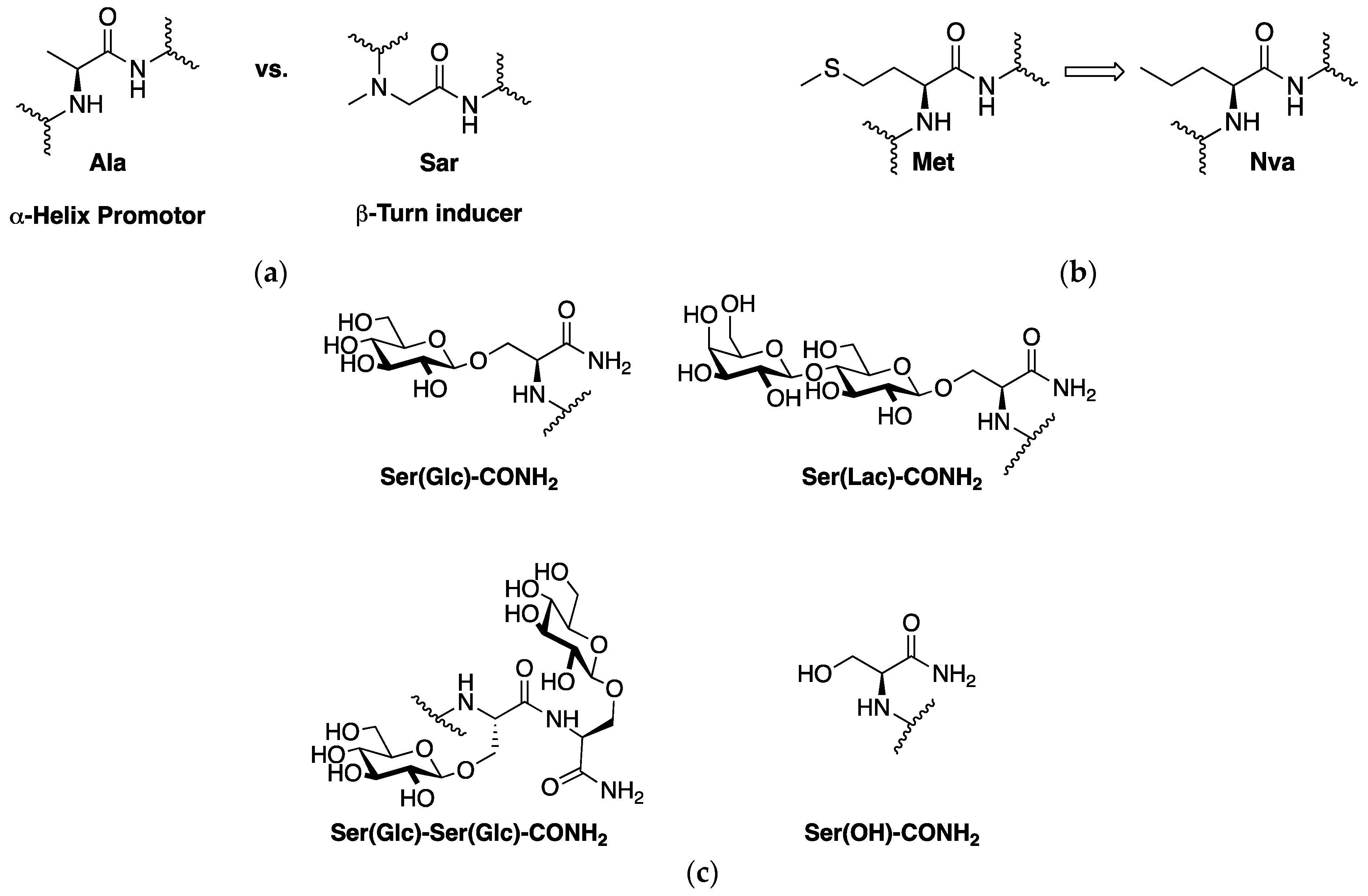
1.2. PACAP1-23 Glycopeptide Design Considerations
2. Results
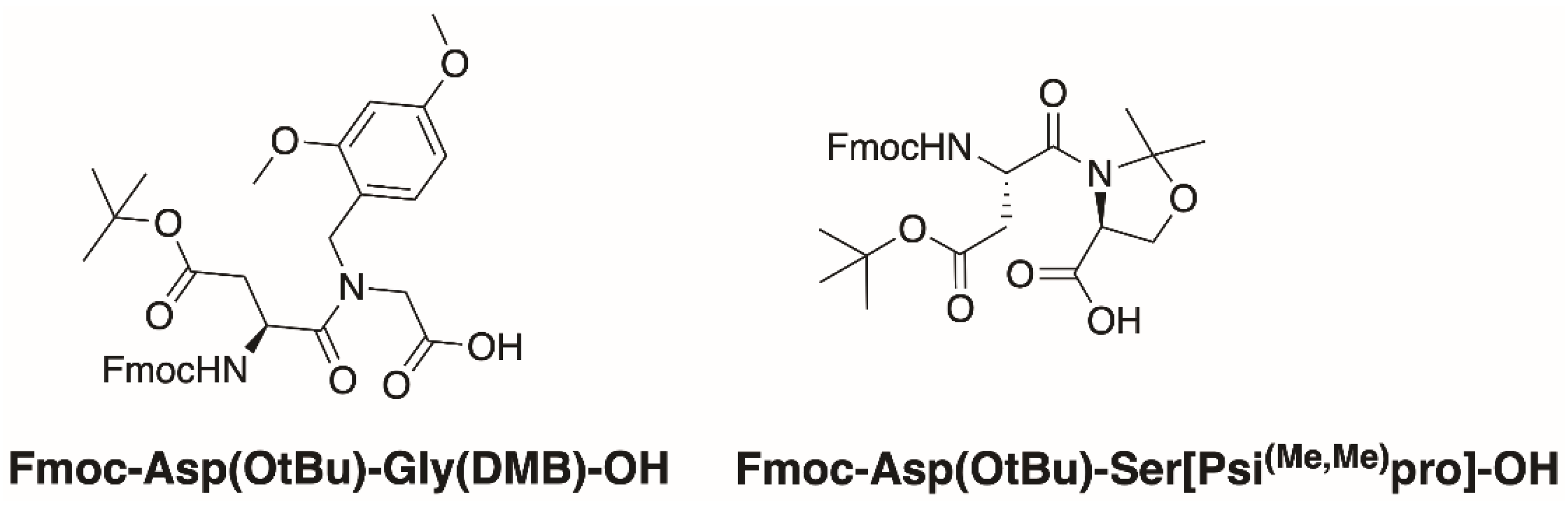
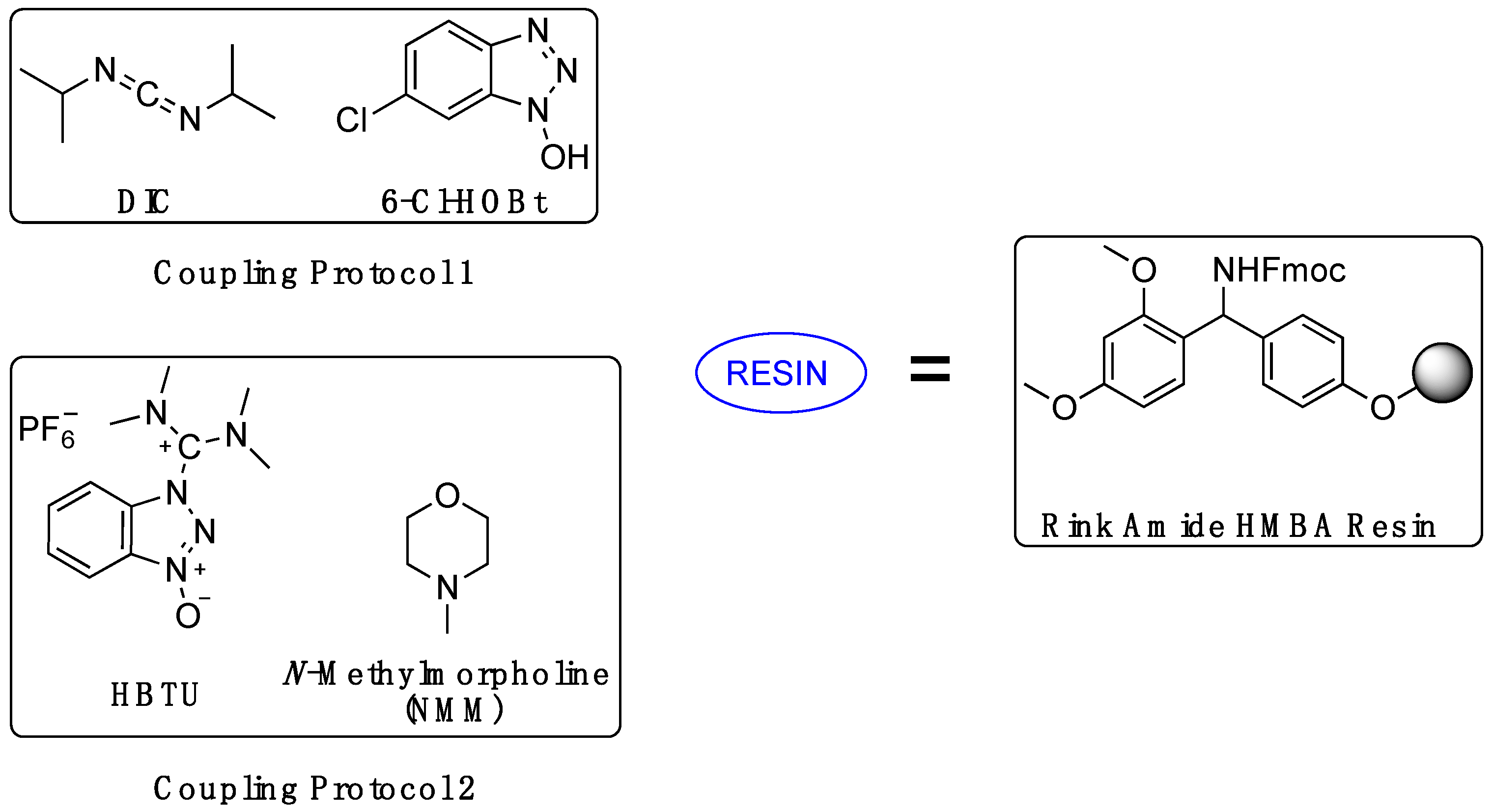
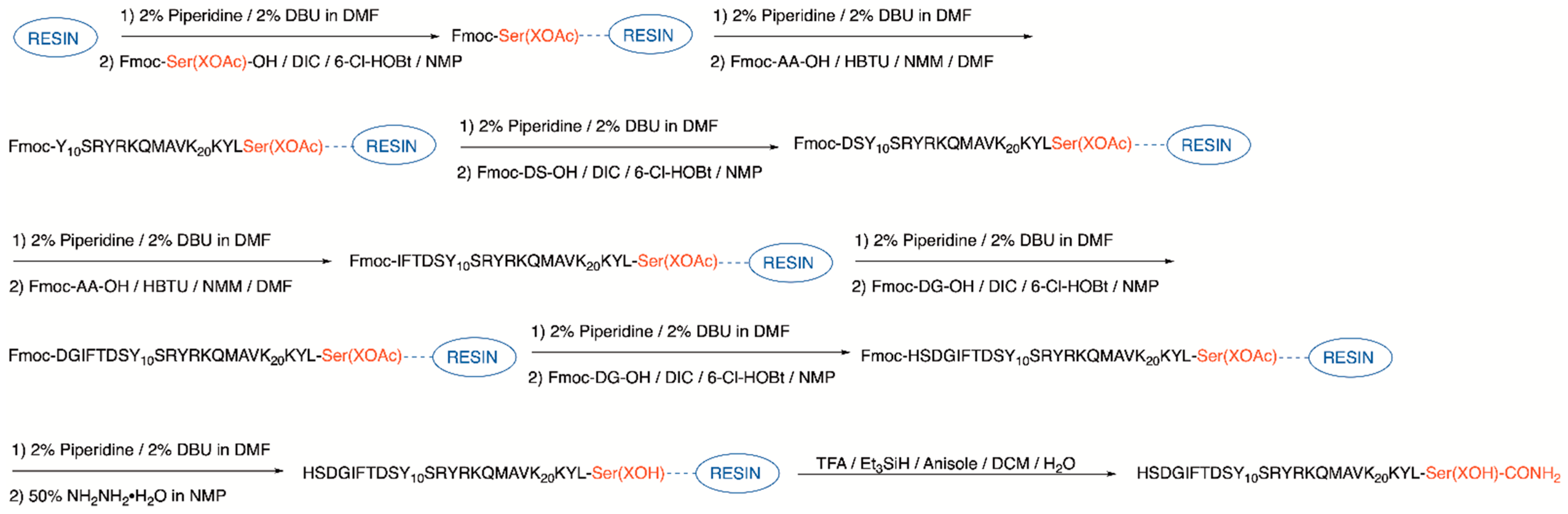
2.1. Synthesis
2.2. In Vitro Characterization
3. Discussion
4. Materials and Methods
4.1. Glycopeptide Synthesis and Purification
4.1.1. General
4.1.2. Rink Amide Resin Preparation
4.1.3. Serine or Glycosyl Amino Acid Loading
4.1.4. Loading of Additional Fmoc-Ser(Glc(OAc)4)-OH (3 Only)
4.1.5. Prelude® Automated Synthesis
4.1.6. Manual Loading of DS Dipeptide
4.1.7. Automated Addition of IFT
4.1.8. Manual Loading of DG Dipeptide
4.1.9. Manual Loading of Fmoc-Ala-OH (5) or Fmoc-Sar-OH (6)
4.1.10. Manual Loading of Remaining Amino Acids Fmoc-Asp(tBu)-OH (5 and 6 Only), Fmoc-Ser(tBu)-OH, and Fmoc-His(Trt)-OH
4.1.11. Acetyl Cleavage
4.1.12. Cleavage from the Resin and Global Side Chain Deprotection
4.1.13. HPLC Purification and Characterization of Peptides
4.2. Cell Culture
4.3. cAMP Accumulation Assay
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Marras, C.; Beck, J.C.; Bower, J.H.; Roberts, E.; Ritz, B.; Ross, G.W.; Abbott, R.D.; Savica, R.; Eeden, S.K.V.D.; Willis, A.W.; et al. Prevalence of Parkinson’s disease across North America. npj Park. Dis. 2018, 4, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Alzheimer’s Association; 2017 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2017, 13, 325–373. [CrossRef]
- Dewan, M.C.; Rattani, A.; Gupta, S.; Baticulon, R.; Hung, Y.-C.; Punchak, M.; Agrawal, A.; Adeleye, A.O.; Shrime, M.G.; Rubiano, A.M.; et al. Estimating the global incidence of traumatic brain injury. J. Neurosurg. 2019, 130, 1080–1097. [Google Scholar] [CrossRef] [Green Version]
- Feigin, V.L.; Norrving, B.; Mensah, G.A. Global Burden of Stroke. Circ. Res. 2017, 120, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Smith, Y.; Wichmann, T.; Factor, S.A.; DeLong, M.R. Parkinson’s Disease Therapeutics: New Developments and Challenges Since the Introduction of Levodopa. Neuropsychopharmacology 2011, 37, 213–246. [Google Scholar] [CrossRef]
- Tsang, K.K.-T.; Whitfield, P.C. Traumatic brain injury: Review of current management strategies. Br. J. Oral Maxillofac. Surg. 2012, 50, 298–308. [Google Scholar] [CrossRef]
- Streit, W.J.; Mrak, R.E.; Griffin, W.S.T. Microglia and neuroinflammation: A pathological perspective. J. Neuroinflammation 2004, 1, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirsch, E.C.; Hunot, S. Neuroinflammation in Parkinson’s disease: A target for neuroprotection? Lancet Neurol. 2009, 8, 382–397. [Google Scholar] [CrossRef]
- Becher, B.; Spath, S.; Goverman, J. Cytokine networks in neuroinflammation. Nat. Rev. Immunol. 2016, 17, 49–59. [Google Scholar] [CrossRef]
- Ransohoff, R.M. How neuroinflammation contributes to neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef]
- Peixoto, C.A.; de Oliveira, W.H.; Araújo, S.M.D.R.; Nunes, A.K.S. AMPK activation: Role in the signaling pathways of neuroinflammation and neurodegeneration. Exp. Neurol. 2017, 298, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Perry, V.H.; Holmes, C. Microglial priming in neurodegenerative disease. Nat. Rev. Neurol. 2014, 10, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Block, M.L.; Zecca, L.; Hong, J.-S. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat. Rev. Neurosci. 2007, 8, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Vaudry, D.; Rousselle, C.; Basille, M.; Falluel-Morel, A.; Pamantung, T.F.; Fontaine, M.; Fournier, A.; Vaudry, H.; Gonzalez, B.J. Pituitary adenylate cyclase-activating polypeptide protects rat cerebellar granule neurons against ethanol-induced apoptotic cell death. Proc. Natl. Acad. Sci. USA 2002, 99, 6398–6403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Botia, B.; Jolivel, V.; Burel, D.; Le Joncour, V.; Roy, V.; Naassila, M.; Bénard, M.; Fournier, A.; Vaudry, H.; Vaudry, D. Neuroprotective Effects of PACAP Against Ethanol-Induced Toxicity in the Developing Rat Cerebellum. Neurotox. Res. 2010, 19, 423–434. [Google Scholar] [CrossRef]
- Farkas, O.; Tamás, A.; Zsombok, A.; Reglodi, R.; Pal, J.; Buki, A.; Lengvári, I.; Povlishock, J.T.; Doczi, T. Effects of pituitary adenylate cyclase activating polypeptide in a rat model of traumatic brain injury. Regul. Pept. 2004, 123, 69–75. [Google Scholar] [CrossRef]
- Reglodi, D.; Tamás, A.; Lubics, A.; Szalontay, L.; Lengvári, I. Morphological and functional effects of PACAP in 6-hydroxydopamine-induced lesion of the substantia nigra in rats. Regul. Pept. 2004, 123, 85–94. [Google Scholar] [CrossRef]
- Dejda, A.; Seaborn, T.; Bourgault, S.; Touzani, O.; Fournier, A.; Vaudry, H.; Vaudry, D. PACAP and a novel stable analog protect rat brain from ischemia: Insight into the mechanisms of action. Peptides 2011, 32, 1207–1216. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Samal, B.; Hamelink, C.R.; Xiang, C.C.; Chen, Y.; Chen, M.; Vaudry, D.; Brownstein, M.J.; Hallenbeck, J.M.; Eiden, L.E. Neuroprotection by endogenous and exogenous PACAP following stroke. Regul. Pept. 2006, 137, 4–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kojro, E.; Postina, R.; Buro, C.; Meiringer, C.; Gehrig-Burger, K.; Fahrenholz, F. The neuropeptide PACAP promotes ?-secretase pathway for processing Alzheimer amyloid precursor protein. FASEB J. 2006, 20, 512–514. [Google Scholar] [CrossRef]
- Liao, C.; De Molliens, M.P.; Schneebeli, S.T.; Brewer, M.; Song, G.; Chatenet, D.; Braas, K.M.; May, V.; Li, J. Targeting the PAC1 Receptor for Neurological and Metabolic Disorders. Curr. Top. Med. Chem. 2019, 19, 1399–1417. [Google Scholar] [CrossRef] [PubMed]
- Dejda, A.; Sokołowska, P.; Nowak, J.Z. Neuroprotective potential of three neuropeptides PACAP, VIP and PHI. Pharmacol. Rep. 2005, 57, 307–320. [Google Scholar]
- Vaudry, D.; Falluel-Morel, A.; Bourgault, S.; Basille, M.; Burel, D.; Wurtz, O.; Fournier, A.; Chow, B.K.C.; Hashimoto, H.; Galas, L.; et al. Pituitary Adenylate Cyclase-Activating Polypeptide and Its Receptors: 20 Years after the Discovery. Pharmacol. Rev. 2009, 61, 283–357. [Google Scholar] [CrossRef]
- Pal, K.; Melcher, K.; Xu, H.E. Structure and mechanism for recognition of peptide hormones by Class B G-protein-coupled receptors. Acta Pharmacol. Sin. 2012, 33, 300–311. [Google Scholar] [CrossRef]
- Hollenstein, K.; de Graaf, C.; Bortolato, A.; Wang, M.-W.; Marshall, F.H.; Stevens, R.C. Insights into the structure of class B GPCRs. Trends Pharmacol. Sci. 2013, 35, 12–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramos-Álvarez, I.; Mantey, S.A.; Nakamura, T.; Nuche-Berenguer, B.; Moreno, P.; Moody, T.W.; Maderdrut, J.L.; Coy, D.H.; Jensen, R.T. A structure–function study of PACAP using conformationally restricted analogs: Identification of PAC1 receptor-selective PACAP agonists. Peptides 2015, 66, 26–42. [Google Scholar] [CrossRef] [Green Version]
- Doan, N.-D.; Bourgault, S.; Dejda, A.; Létourneau, M.; Detheux, M.; Vaudry, D.; Vaudry, H.; Chatenet, D.; Fournier, A. Design and in vitro characterization of PAC1/VPAC1-selective agonists with potent neuroprotective effects. Biochem. Pharmacol. 2011, 81, 552–561. [Google Scholar] [CrossRef] [PubMed]
- Bourgault, S.; Chatenet, D.; Wurtz, O.; Doan, N.D.; Leprince, J.; Vaudry, H.; Fournier, A.; Vaudry, D. Strategies to Convert PACAP from a Hypophysiotropic Neurohormone Into a Neuroprotective Drug. Curr. Pharm. Des. 2011, 17, 1002–1024. [Google Scholar] [CrossRef]
- Bourgault, S.; Vaudry, D.; Ségalas-Milazzo, I.; Guilhaudis, L.; Couvineau, A.; Laburthe, M.; Vaudry, H.; Fournier, A. Molecular and Conformational Determinants of Pituitary Adenylate Cyclase-Activating Polypeptide (PACAP) for Activation of the PAC1 Receptor. J. Med. Chem. 2009, 52, 3308–3316. [Google Scholar] [CrossRef]
- Robberecht, P.; Gourlet, P.; Neef, P.; Woussen-Colle, M.-C.; Vandermeers-Piret, M.-C.; Vandermeers, A.; Christophe, J. Structural requirements for the occupancy of pituitary adenylate-cyclase-activating-peptide (PACAP) receptors and adenylate cyclase activation in human neuroblastoma NB-OK-1 cell membranes. Discovery of PACAP(6-38) as a potent antagonist. JBIC J. Biol. Inorg. Chem. 1992, 207, 239–246. [Google Scholar] [CrossRef]
- Robberecht, P.; Gourlet, P.; De Neef, P.; Woussen-Colle, M.C.; Vandermeers-Piret, M.C.; Vandermeers, A.; Christophe, J. Receptor occupancy and adenylate cyclase activation in AR 4-2J rat pancreatic acinar cell membranes by analogs of pituitary adenylate cyclase-activating peptides amino-terminally shortened or modified at position 1, 2, 3, 20, or 21. Mol. Pharmacol. 1992, 42, 347–355. [Google Scholar]
- Gourlet, P.; Vandermeers, A.; Vandermeers-Piret, M.-C.; Rathé, J.; De Neef, P.; Robberecht, P. C-Terminally shortened pituitary adenylate cyclase-activating peptides (PACAP) discriminate PACAP I, PACAP II-VIP1 and PACAP II-VIP2 recombinant receptors. Regul. Pept. 1996, 62, 125–130. [Google Scholar] [CrossRef]
- Lamine, A.; De Molliens, M.P.; Létourneau, M.; Hébert, T.; Vaudry, D.; Fournier, A.; Chatenet, D. The amidated PACAP1–23 fragment is a potent reduced-size neuroprotective agent. Biochim. et Biophys. Acta (BBA)-Gen. Subj. 2019, 1863, 129410. [Google Scholar] [CrossRef]
- Fizanne, L.; Sigaudo-Roussel, D.; Saumet, J.L.; Fromy, B. Evidence for the involvement of VPAC1 and VPAC2 receptors in pressure-induced vasodilatation in rodents. J. Physiol. 2004, 554, 519–528. [Google Scholar] [CrossRef] [Green Version]
- Banks, W.A.; Kastin, A.; Komaki, G.; Arimura, A. Passage of pituary adenylate cyclase activating polypeptide1-27 and pituary adenylate cyclase activating polypeptide1-38 across the blood-brain barrier. J. Pharmacol. Exp. Ther. 1993, 267, 690–696. [Google Scholar]
- Banks, W.A. Peptides and the blood–brain barrier. Peptides 2015, 72, 16–19. [Google Scholar] [CrossRef] [Green Version]
- Muttenthaler, M.; King, G.F.; Adams, D.J.; Alewood, P.F. Trends in peptide drug discovery. Nat. Rev. Drug Discov. 2021, 20, 309–325. [Google Scholar] [CrossRef]
- Lau, J.L.; Dunn, M.K. Therapeutic peptides: Historical perspectives, current development trends, and future directions. Bioorganic Med. Chem. 2018, 26, 2700–2707. [Google Scholar] [CrossRef]
- Kaspar, A.A.; Reichert, J.M. Future directions for peptide therapeutics development. Drug Discov. Today 2013, 18, 807–817. [Google Scholar] [CrossRef]
- Apostol, C.R.; Hay, M.; Polt, R. Glycopeptide drugs: A pharmacological dimension between “Small Molecules” and “Bio-logics”. Peptides 2020, 131, 170369. [Google Scholar] [CrossRef]
- Varamini, P.; Toth, I. Lipid- and sugar-modified endomorphins: Novel targets for the treatment of neuropathic pain. Front. Pharmacol. 2013, 4, 155. [Google Scholar] [CrossRef] [Green Version]
- Moradi, S.V.; Hussein, W.M.; Varamini, P.; Simerska, P.; Toth, I. Glycosylation, an effective synthetic strategy to improve the bioavailability of therapeutic peptides. Chem. Sci. 2016, 7, 2492–2500. [Google Scholar] [CrossRef] [Green Version]
- Jones, E.M.; Polt, R. CNS active O-linked glycopeptides. Front. Chem. 2015, 3, 40. [Google Scholar] [CrossRef] [Green Version]
- Mabrouk, O.S.; Falk, T.; Sherman, S.J.; Kennedy, R.T.; Polt, R. CNS penetration of the opioid glycopeptide MMP-2200: A microdialysis study. Neurosci. Lett. 2012, 531, 99–103. [Google Scholar] [CrossRef] [Green Version]
- Bartlett, M.J.; Mabrouk, O.S.; Szabò, L.; Flores, A.J.; Parent, K.L.; Bidlack, J.M.; Heien, M.L.; Kennedy, R.T.; Polt, R.; Sherman, S.J.; et al. The Delta-Specific Opioid Glycopeptide BBI-11008: CNS Penetration and Behavioral Analysis in a Preclinical Model of Levodopa-Induced Dyskinesia. Int. J. Mol. Sci. 2020, 22, 20. [Google Scholar] [CrossRef] [PubMed]
- Carmo, G.P.D.; Polt, R.; Bilsky, E.J.; Rice, K.C.; Negus, S.S. Behavioral Pharmacology of the μ/δ Opioid Glycopeptide MMP2200 in Rhesus Monkeys. J. Pharmacol. Exp. Ther. 2008, 326, 939–948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Louis, L.S.; Knapp, B.I.; Muthu, D.; Anglin, B.; Giuvelis, D.; Bidlack, J.M.; Bilsky, E.J.; Polt, R. Can Amphipathic Helices Influence the CNS Antinociceptive Activity of Glycopeptides Related to β-Endorphin? J. Med. Chem. 2014, 57, 2237–2246. [Google Scholar] [CrossRef] [PubMed]
- Hay, M.; Polt, R.; Heien, M.L.; Vanderah, T.W.; Largent-Milnes, T.M.; Rodgers, K.; Falk, T.; Bartlett, M.J.; Doyle, K.P.; Konhilas, J. A Novel Angiotensin-(1-7) Glycosylated Mas Receptor Agonist for Treating Vascular Cognitive Impairment and Inflammation-Related Memory Dysfunction. J. Pharmacol. Exp. Ther. 2019, 369, 9–25. [Google Scholar] [CrossRef] [Green Version]
- Lowery, J.J.; Yeomans, L.; Keyari, C.M.; Davis, P.; Porreca, F.; Knapp, B.I.; Bidlack, J.M.; Bilsky, E.J.; Polt, R. Glycosylation Improves the Central Effects of DAMGO. Chem. Biol. Drug Des. 2007, 69, 41–47. [Google Scholar] [CrossRef]
- Li, Y.; Lefever, M.R.; Muthu, D.; Bidlack, J.M.; Bilsky, E.; Polt, R. Opioid glycopeptide analgesics derived from endogenous enkephalins and endorphins. Futur. Med. Chem. 2012, 4, 205–226. [Google Scholar] [CrossRef] [Green Version]
- Bernard, K.; Bartlett, M.J.; Liu, C.; Molnar, G.; Apostol, C.R.; Szabò, L.Z.; Sherman, S.J.; Madhavan, L.; Streicher, J.M.; Polt, R.; et al. Evaluation of a neuroprotective PACAP glycopeptide as systemically delivered CNS active drug to treat Parkinson’s disease. In Proceedings of the Society for Neuroscience, Chicago, IL, USA, 11–13 January 2021; Program No. 117.08. 2021 Neuroscience Meeting Planner. Available online: https://www.sfn.org/meetings/virtual-events/sfn-global-connectome-a-virtual-event/abstracts (accessed on 10 August 2021).
- Apostol, C.R.; Bernard, K.; Molnar, G.; Bartlett, M.J.; Szabò, L.; Liu, C.; Ortiz, J.B.; Saber, M.; Giordano, K.R.; Green, T.F.R.; et al. Design and Synthesis of Brain Penetrant Glycopeptide Analogues of PACAP with Neuroprotective Potential for Stroke, Traumatic Brain Injury, and Parkinsonism. Unpublished work.
- Lalatsa, A.; Butt, A.M. Physiology of the Blood–Brain Barrier and Mechanisms of Transport Across the BBB. In Nanotechnology-Based Targeted Drug Delivery Systems for Brain Tumors; Kesharwani, P., Gupta, U., Eds.; Elsevier Academic Press: Amsterdam, The Netherlands, 2018; pp. 49–74. [Google Scholar] [CrossRef]
- Lefever, M.R.; Szabò, L.Z.; Anglin, B.; Ferracane, M.; Hogan, J.; Cooney, L.; Polt, R. Glycosylation of α-amino acids by sugar acetate donors with InBr3. Minimally competent Lewis acids. Carbohydr. Res. 2012, 351, 121–125. [Google Scholar] [CrossRef] [Green Version]
- Paradís-Bas, M.; Puche, J.T.; Albericio, F. The road to the synthesis of “difficult peptides”. Chem. Soc. Rev. 2015, 45, 631–654. [Google Scholar] [CrossRef] [PubMed]
- Dölling, R.; Beyermann, M.; Haenel, J.; Kernchen, F.; Krause, E.; Francke, P.; Brudel, M.; Bienert, M. Piperidine–mediated Side Product Formation for Asp(OBut)–containing Peptides. J. Chem. Soc. Chem. Commun. 1994, 7, 853–854. [Google Scholar] [CrossRef]
- Subirós-Funosas, R.; El-Faham, A.; Albericio, F. Aspartimide formation in peptide chemistry: Occurrence, prevention strategies and the role of N-hydroxylamines. Tetrahedron 2011, 67, 8595–8606. [Google Scholar] [CrossRef]
- Yang, Y.; Sweeney, W.V.; Schneider, K.; Thörnqvist, S.; Chait, B.T.; Tam, J.P. Aspartimide formation in base-driven 9-fluorenylmethoxycarbonyl chemistry. Tetrahedron Lett. 1994, 35, 9689–9692. [Google Scholar] [CrossRef]
- Cardona, V.; Eberle, I.; Barthelemy, S.; Beythien, J.; Doerner, B.; Schneeberger, P.; Keyte, J.; White, P.D. Application of Dmb-Dipeptides in the Fmoc SPPS of Difficult and Aspartimide-Prone Sequences. Int. J. Pept. Res. Ther. 2008, 14, 285–292. [Google Scholar] [CrossRef]
- Sampson, W.R.; Patsiouras, H.; Ede, N.J. The synthesis of “difficult” peptides using 2-hydroxy-4- methoxybenzyl or pseu-doproline amino acid building blocks: A comparative study. J. Pept. Sci. 1999, 5, 403–409. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Structure 1 |
|---|---|
| PACAP1–38 | HSDGIFTDSY10SRYRKQMAVK20KYLAAVLGKRYKQRVKNK-CONH2 |
| PACAP1–27 | HSDGIFTDSY10SRYRKQMAVK20KYLAAVL-CONH2 |
| PACAP1–23 | HSDGIFTDSY10SRYRKQMAVK20KYL-CONH2 |
| VIP | HSDAVFTDNY10TRLRKQMAVK20KYLNSILN-CONH2 |
| 1 | HSDGIFTDSY10SRYRKQÑAVK20KYL-Ser(OH)-CONH2 |
| 2 | HSDGIFTDSY10SRYRKQÑAVK20KYL-Ser(Glc)-CONH2 |
| 3 | HSDGIFTDSY10SRYRKQÑAVK20KYL-Ser(Glc)-Ser(Glc)-CONH2 |
| 4 | HSDGIFTDSY10SRYRKQÑAVK20KYL-Ser(Lac)-CONH2 |
| 5 | HSDAIFTDSY10SRYRKQÑAVK20KYL-Ser(Lac)-CONH2 |
| 6 | HSD∑IFTDSY10SRYRKQÑAVK20KYL-Ser(Lac)-CONH2 |
| Compound | Molecular Formula | Calculated Mass | 1 Experimental Mass → | 2 HPLC Ret. Time (min) | |
|---|---|---|---|---|---|
| 1 | C128H199N37O37 | 2846.48 | 570.71 (M+5H)5+ | 2848.55 | 13.98 |
| 2 | C134H209N37O42 | 3008.54 | 603.09 (M+5H)5+ | 3010.45 | 13.60 |
| 3 | C143H224N38O49 | 3257.62 | 652.76 (M+5H)5+ | 3258.80 | 13.24 |
| 4 | C140H219N37O47 | 3170.59 | 793.93 (M+4H)4+ | 3171.72 | 13.59 |
| 5 | C141H221N37O47 | 3184.60 | 638.09 (M+5H)5+ | 3185.45 | 13.48 |
| 6 | C141H221N37O47 | 3184.60 | 638.01 (M+5H)5+ | 3185.45 | 13.42 |
| Drug Candidate | PAC1 | VPAC1 | VPAC2 | |||
|---|---|---|---|---|---|---|
| EC50 (nM) | EMAX (%) | EC50 (nM) | EMAX (%) | EC50 (nM) | EMAX (%) | |
| PACAP1–27 | 20, 9.0 | 100, 100 | 30, 6.5 | 100, 100 | 71, 29 | 100, 100 |
| 1 | 2.2 | 198 | 5.2 | 42 | 571 | 111 |
| 2 | 0.64 | 181 | 37 | 104 | 567 | 110 |
| 3 | 1.9 | 152 | 26 | 147 | 7369 | 128 |
| 4 | 1.8 | 174 | 21 | 118 | 1943 | 169 |
| 5 | 7.0 | 181 | 38 | 124 | 321 | 139 |
| 6 | 67 | 134 | 195 | 107 | NC | NC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Apostol, C.R.; Tanguturi, P.; Szabò, L.Z.; Varela, D.; Gilmartin, T.; Streicher, J.M.; Polt, R. Synthesis and In Vitro Characterization of Glycopeptide Drug Candidates Related to PACAP1–23. Molecules 2021, 26, 4932. https://doi.org/10.3390/molecules26164932
Apostol CR, Tanguturi P, Szabò LZ, Varela D, Gilmartin T, Streicher JM, Polt R. Synthesis and In Vitro Characterization of Glycopeptide Drug Candidates Related to PACAP1–23. Molecules. 2021; 26(16):4932. https://doi.org/10.3390/molecules26164932
Chicago/Turabian StyleApostol, Christopher R., Parthasaradhireddy Tanguturi, Lajos Z. Szabò, Daniel Varela, Thiago Gilmartin, John M. Streicher, and Robin Polt. 2021. "Synthesis and In Vitro Characterization of Glycopeptide Drug Candidates Related to PACAP1–23" Molecules 26, no. 16: 4932. https://doi.org/10.3390/molecules26164932