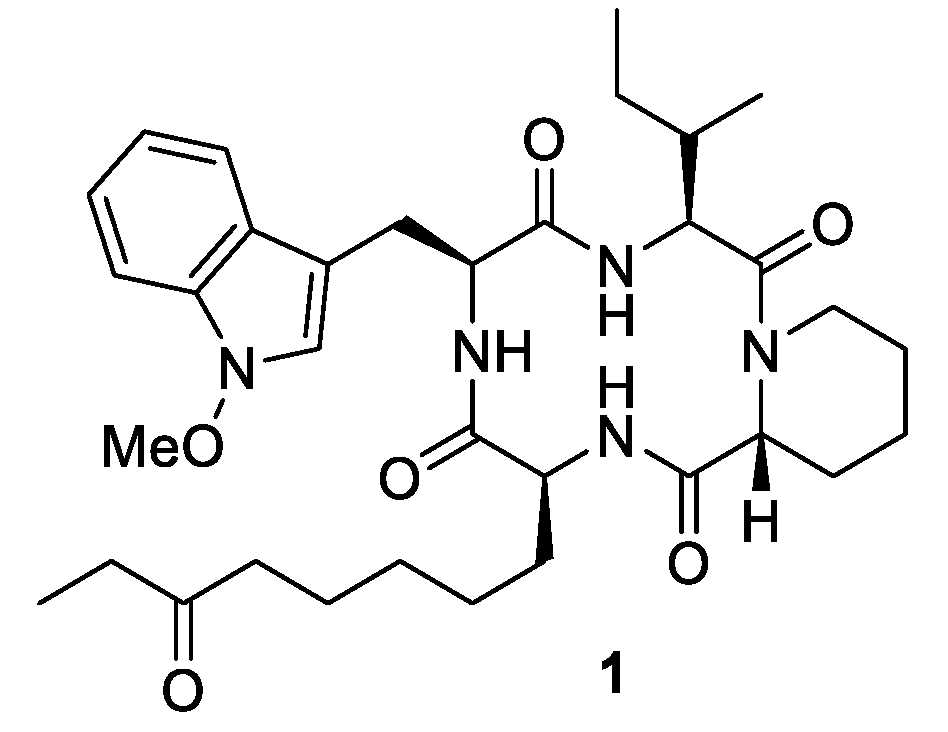
Our aim was the design of apicidin derivatives capable of the inhibition of the Wnt/
β-catenin signaling pathway. Thus, our structures lacked the characteristic
l-2-amino-8-oxodecanoic acid (Aoda), which is critical for the HDAC activity of apicidin (
1) [
15]. While analyzing the novel structures, we have observed that replacing individual amino acids with peptoid monomers has an interesting influence on the spatial structure of the macrocycles. Herein, we report our findings based on selecting apicidin congeners with different peptide to peptoid ratios.
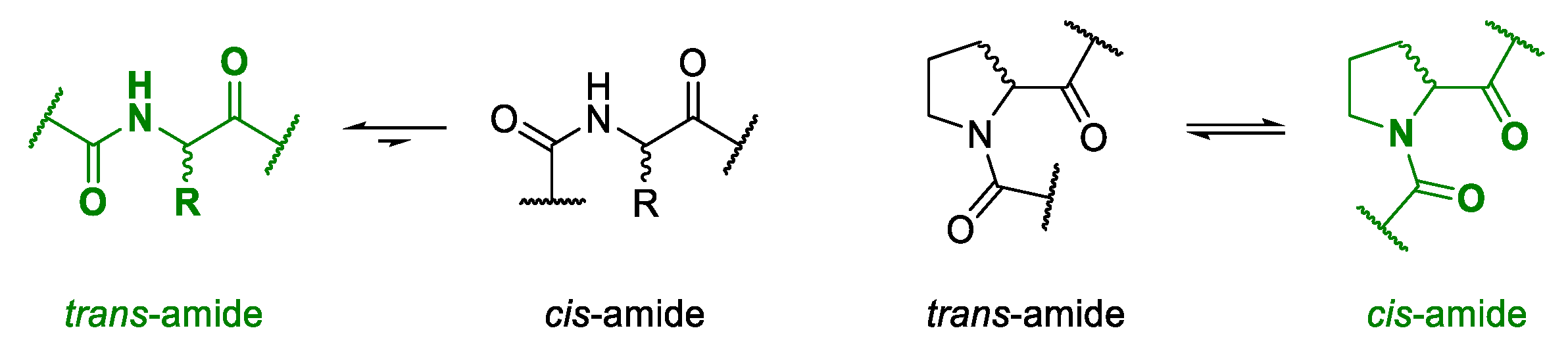
Due to the steric hindrance of their side chains, most amino acids form
trans-conformations with high energy barriers for
cis-
trans-isomerism [
67,
68]. The unusual structure of proline results in an equimolar distribution of both the
cis- and the
trans-conformation when incorporated into a polypeptide [
64,
65,
66]. In nature, isomers of proline are known as loop inducers due to
cis-bond formation [
69,
70]. As it is assumed that backbone
cis-conformations can facilitate the ring closure of tense cyclic tetramers [
8,
71,
72], proline was the building block of choice for the design of different macrocycles.
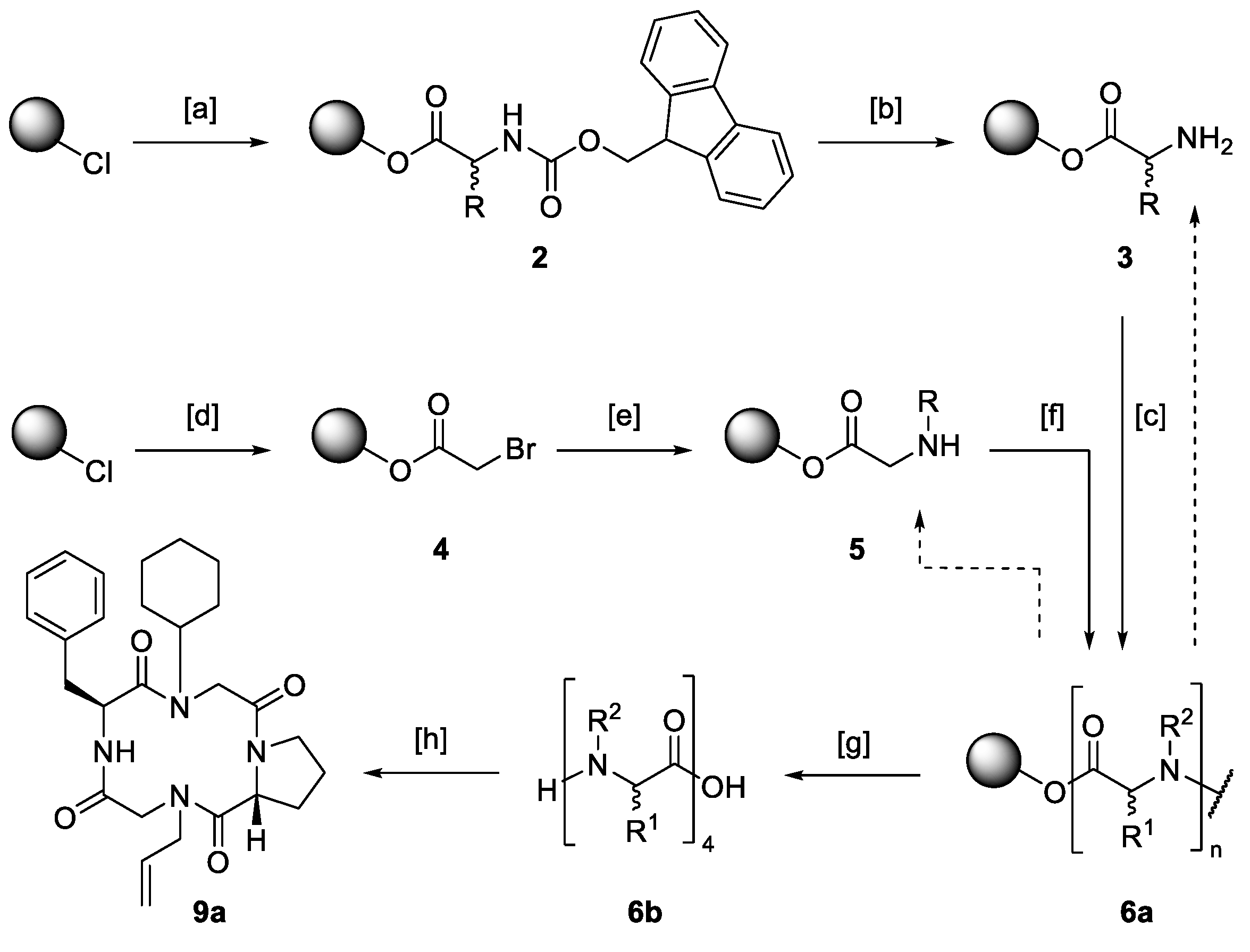
2.1. Synthesis of Macrocyclic Tetramers
Hybrid structures consisting of amino acids and peptoid monomers were built upon solid support. The synthetic protocol involved the well-known solid-phase peptide synthesis described by Merrifield [
73] as well as the submonomer method for the assembly of peptoids published by Zuckermann [
33] (
Scheme 2).
The attachment of the C-terminal amino acid (→ 2) or bromoacetic acid as the first submonomer of a peptoid building block (→ 4) to a 2-chlorotrityl chloride polystyrene resin was performed under basic conditions. In the case of amino acids, the Fmoc-protection group was cleaved using a mixture of 20% piperidine in DMF, resulting in the free primary amine 3. To build up peptoids, bromoacetic acid was substituted by any desired amine (→ 5). Depending on the sequence, free amines were either coupled to an amino acid or bromoacetic acid. Diisopropylcarbodiimide was used as a coupling agent in both cases. To avoid racemization, hydroxybenzotriazole was added for the attachment of amino acids.
Acetylation and substitution and amino acid coupling and deprotection were carried out until the desired linear precursor
6 was constructed. Cleavage was performed under mildly acidic conditions releasing a linear tetramer capable of a head-to-tail cyclization. The ring closure was carried out following a protocol by Aldrich [
74] with the help of the potent coupling reagent [dimethylamino(triazolo[4,5-
b]pyridin-3-yloxy)methylidene]-di-methylazanium hexafluoro-phosphate (HATU). This iminium salt is known for its potency in energetically unfavorable couplings, cyclizing constrained tetrapeptides [
8,
28,
75]. To avoid favored side reactions like cyclodimerizations [
72,
76,
77], a 5.00 m
M solution of the respective linear precursor was added dropwise to a 2.40 m
m solution of HATU.
Reactive moieties of side chains were masked with protecting groups. Deprotection was performed immediately after the cyclization step. After ten or eleven reaction steps, respectively, the synthetic protocol yielded cyclic tetramers, which required only a single purification step at the end of the reaction sequence. Purification was carried out via preparative reversed-phase high performance liquid chromatography (HPLC), and product formation was confirmed via matrix assisted laser desorption ionization-time of flight (MALDI-TOF) mass spectrometry.
In an initial library, several macrocyclic tetrapeptides of general structure
7 were synthesized. To match the model structure apicidin (
1), proline was incorporated in its
d-configuration. The remaining amino acids were applied in their
l-configuration. To avoid diketopiperazine formation [
78,
79],
d-proline was incorporated as the third building block during the modular solid-phase synthesis. Approaches with proline as
N-terminal building block yielded low amounts of the desired macrocycles (data not shown). It was assumed that the low nucleophilicity of the secondary amine prevented cyclization.
For this reason, the sequence of the linear precursors was changed in such a way that a primary amine was in the
N-terminal position (R
1). Cyclization of these precursors then led to moderate yields of the corresponding macrocycles. Nine derivatives with structural similarity were selected to represent the library of macrocyclic tetramers (
Table 1).
The N-terminus of the linear precursors (position R1) consisted of branched aliphatic or aromatic amino acids. In position R2, different alkyl side chains were incorporated. The C-terminus (position R3) was built by either l-phenylalanine or l-tryptophan. Ring closure was carried out by amidation of the N-terminal amine (R1) with the carboxyl function of the C-terminal, aromatic amino acid (R3). The use of different building blocks did not influence the overall yield of the reaction. Even additional deprotection steps (compounds 7b and 7g) had no clear effect on the yields of the macrocyclic tetrapeptides. On average, the cyclic tetramers were isolated in 42% ± 13 overall yield.
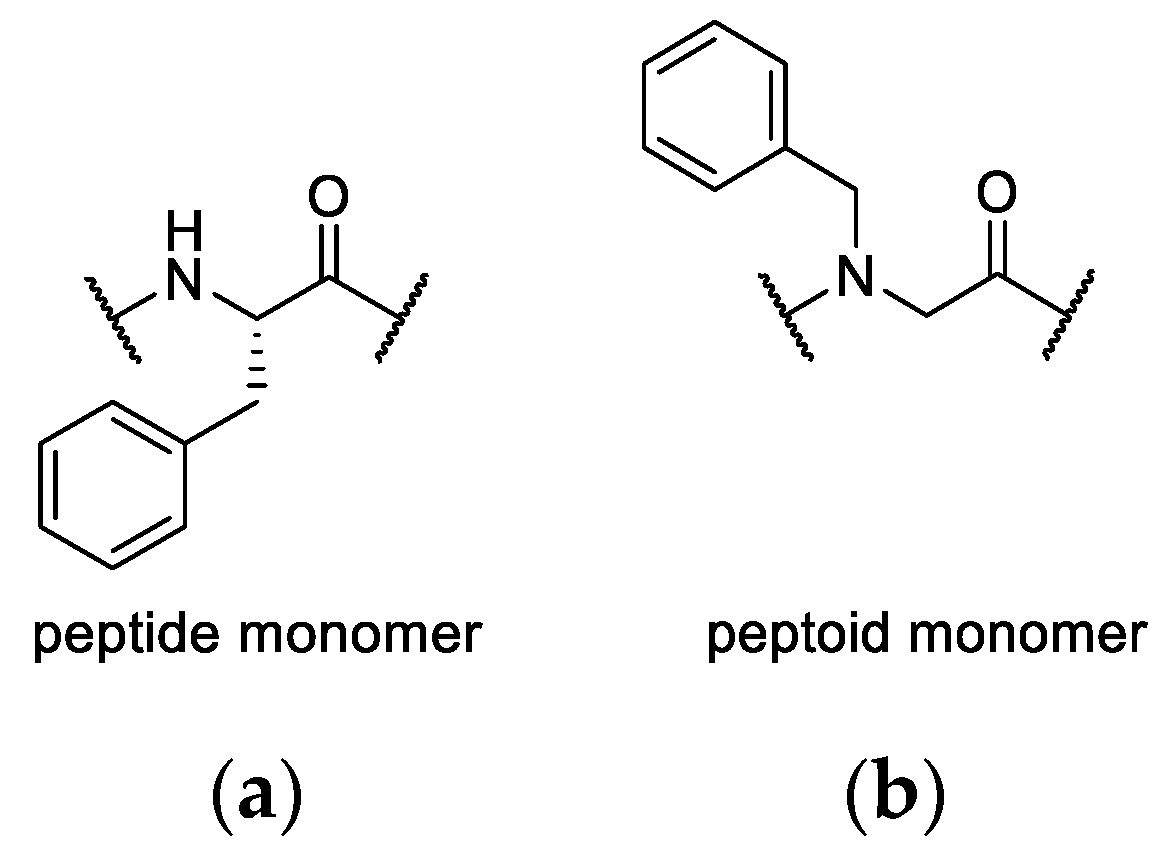
In a second library, individual amino acids were replaced by a peptoid monomer. Peptoids are peptidomimetics that promise high metabolic stability and outstanding biological activity [
33,
34,
35]. Compared to peptides, the side chain is formally shifted from the
α-carbon to the backbone nitrogen atom. This comes with high conformational flexibility as the amide nitrogen loses its capability to serve as a hydrogen bond donor. Moreover, the modification of the amide nitrogen lowers the energy barrier of
cis/trans isomerization [
36,
37,
80,
81]. However, a beneficial effect of this enhanced flexibility on the cyclization reaction was not observed. The nine macrocyclic hybrids representing a library composed of tetramers with three amino acids and one peptoid monomer were isolated in 31% ± 14 overall yields (
Table 2).
To resemble the model structure apicidin (1), the nine macrocycles 8a–i have an aromatic amino acid at the C-terminal end (R5) and an adjacent linear alkyl side chain in common (R3 or R4). The peptoid monomer was inserted at the C- or N-terminal position of d-proline (R1 or R3). In the latter case, cyclization was performed on the secondary amine of a peptoid building block, causing a lower yield on average (19% ± 8, 8a–c). Incorporating a peptoid monomer in the middle of the sequence resulted in overall yields similar to those obtained for cyclic tetrapeptides (36% ± 13, 8d–i).
Further peptoid building blocks were incorporated into the macrocycles to enhance structural diversity, resulting in the general structure
9.
Table 3 shows a selection of nine structurally similar apicidin congeners with both aromatic and aliphatic side chains (
Table 3).
The macrocyclic hybrids 9a–i are composed of two peptoid monomers (R1 and R2) and two amino acids (d-proline and R3) located in alternating order on opposite sides of the backbone ring system. Aromatic and linear, and cyclic aliphatic peptoid monomers built the N-terminus of the linear precursors (R1). The individual building blocks did not influence the overall yields, similar to the yields obtained for hybrids 8a–c that were also cyclized on a secondary amine (24% ± 11).
2.2. Multiconformational Equilibrium Detected by NMR
Often multiple signal sets are detected in the nuclear magnetic resonance (NMR) spectra of macrocycles depending on the dielectric properties of the solvent [
11,
82,
83,
84]. This could be due to different conformers present or conformational equilibrium [
38,
85]. Influencing factors are i.e., side chains, solvent effects, and temperature [
71,
72]. For the model structure apicidin (
1), as an example, it is known that multiple conformations stem from
cis-trans isomerism of the pipecolic acid building block [
83,
86].
HPLC purification of the compounds resulted in sharp peaks indicating that one predominant isomer was synthesized [
87,
88,
89]. However, NMR-spectra of the cyclic tetramers corroborated the formation of several conformers for almost every macrocycle (
supplemental, Table S6). This was most prominent for cyclic tetrapeptides
7a–i, which tended to assemble in multiconformational equilibria due to multiple degrees of freedom.
The same applied to the macrocycles with one peptoid monomer. Macrocycles
8a–i revealed multiple signal sets in solution, indicating different conformers’ formation (
supplemental, Table S6). Nuclear Overhauser and exchange spectroscopy (NOESY) spectra of macrocycle
8f, for example, led to the identification of five separate conformers which interconverted on the NMR timescale. Surprisingly, the complexity of the spectra of the hybrids
8a–d was significantly reduced compared to spectra of structures
8e–i. The peptoid monomer was inserted at the
N-terminus in the former ones, resulting in one dominant structure next to another isomer in approximately 5:1. Therefore, incorporating a peptoid building block in this position could stabilize distinct isomers, decisive for biological applications.
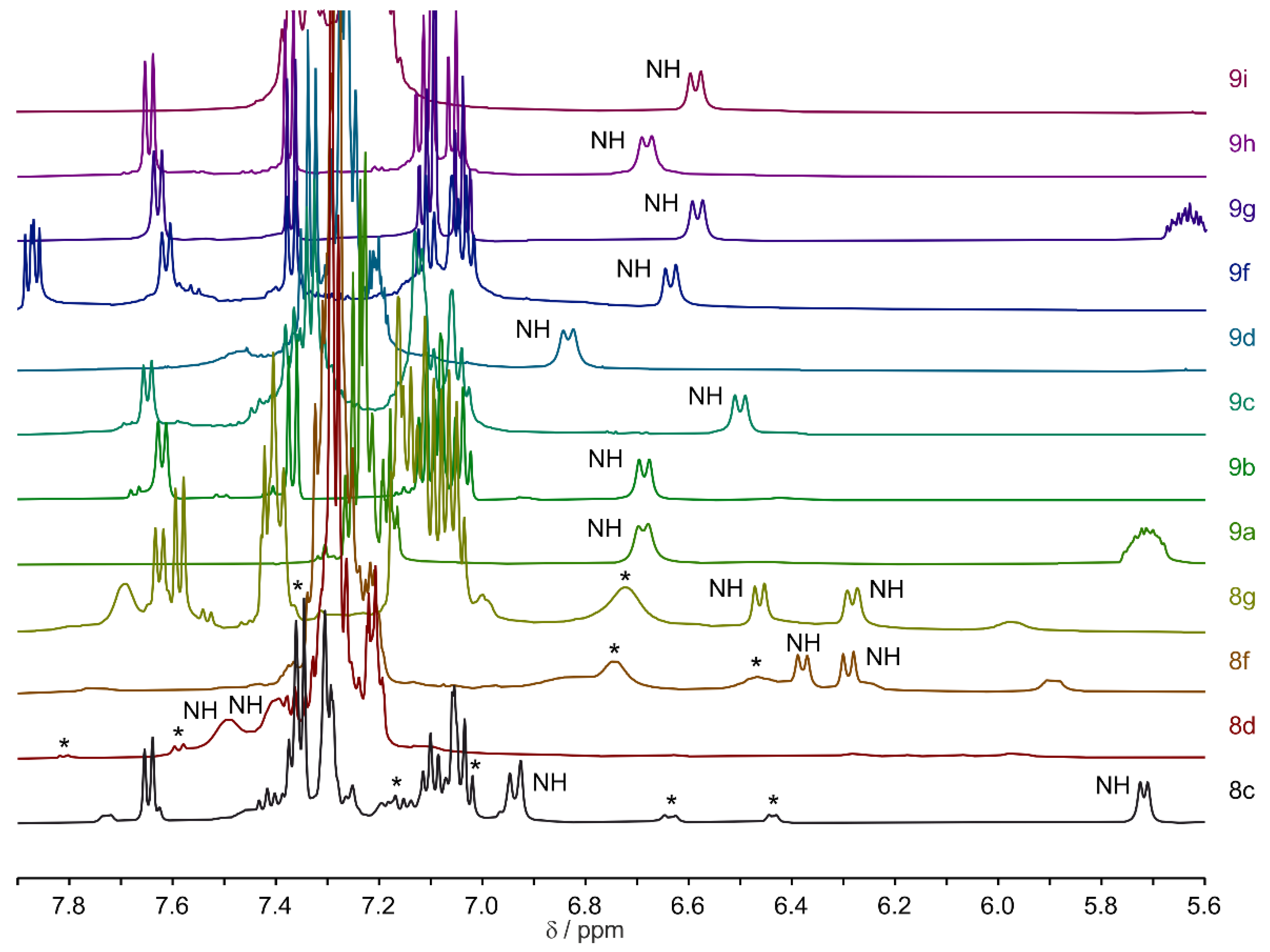
For macrocycles
9a–i, one dominant signal set was mostly observed (
supplemental, Table S6). To illustrate this,
Figure 3 displays the NH regions of selected macrocycles from series
8 and
9, which were soluble in pure acetonitrile. In the NH region of series
9 macrocycles, only one peptide bond amide signal is visible. For macrocycles
8c,
8d,
8f, and
8g, one main signal set was accompanied by a second or third signal set of lesser intensity. Macrocycles from series
7 are not shown here, as the molecules were primarily soluble in dimethyl sulfoxide (DMSO) (see
supplemental, Table S6).
2.3. Spatial Structure in Solid-State
X-ray diffraction is a highly reliable method to determine the spatial structure of molecules in the solid-state [
90,
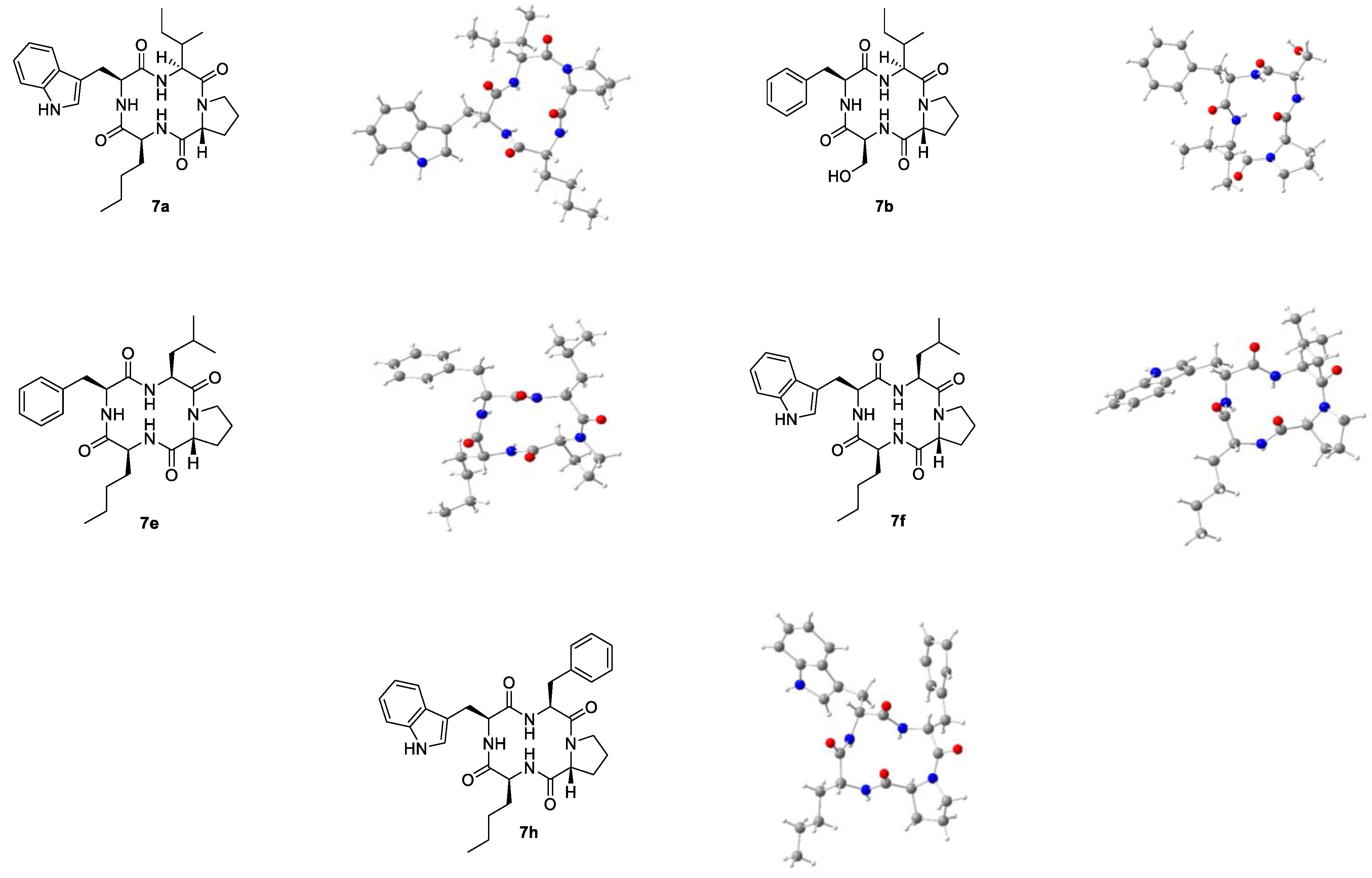
91]. Crystallization of the macrocyclic hybrids was attempted via evaporation of acetonitrile, isopropanol, and methanol. Most macrocycles aggregated into amorphous powders during this process, some became viscous oils, and others produced polycrystalline needle-shaped structures. However, some single crystals were obtained from multiple attempts for each of the five similar cyclic tetrapeptides
7a,
7b,
7e,
7f and
7h (
Figure 4).
Macrocycle 7b represents the only tetrapeptide with a polar building block crystallized in sufficient quality for X-ray diffraction. In contrast to the other structures, 7b is equipped with l-serine instead of the alkyl side chain l-norleucine. Thus, the macrocycle 7b is the only member of the apicidin tetrapeptide library without l-norleucine that crystallized upon vapor diffusion.
For both tetrapeptides
7a and
7b containing an
N-terminal
l-isoleucyl residue, the structure of one isomer was determined via X-ray diffraction. As for the model structure apicidin (
1) [
84], at least three independent structures (I, II, III etc.) each were obtained for macrocycles
7e,
7f and
7h. Their dihedral angles differ slightly from each other but show the same
cis-trans arrangement (
Table 4).
To elucidate the backbone conformation, dihedral angles of the individual macrocycles were measured. The dihedral angle ω describes the torsion angle of the axis between the
α- and the amide carbon atom of one amino acid and the axis between the amide nitrogen and the
α-carbon atom of the following building block. Due to the partial double-bond character of the peptide bond, this angle is forced into two distinct values: ω = 0° or ω = ±180°. Sterical hindrance can lead to a deviation of the dihedral angles from their ideal values, but an angle close to ω = 0° indicates a
cis-conformation while ω = ±180° indicates a
trans-peptide bond [
92].
All conformers of the five macrocycles
7a,
7b,
7e,
7f, and
7h showed a
cis-conformation between the nitrogen atom of their respective
d-prolyl residue and the amide carbon of the adjacent amino acid (ω
C). The
trans-trans-cis-trans sequence of the backbone has also been reported for the model structure apicidin (
1) [
83] and similar cyclotetrapeptides [
93]. The largest deviations from the ideal dihedral angle were measured between the nitrogen atoms of the large aromatic side chains
l-phenylalanine or
l-tryptophan and the amide carbon of the following building blocks (ω
A).
The measurements of the configurations of the α-carbon atoms showed the expected stereochemistry: the α-carbon of every d-proline building block was (R)-, the ones of the remaining amino acids were (S)-configurated. Furthermore, the macrocycles resembled each other in the location of their side chains: while the aliphatic ring of d-proline pointed above the ring level, the remaining side chains were located below.
Crystallization preparations of hybrids containing one peptoid monomer provided single crystals of two compounds:
8e and
8f (
Table 5). Both revealed strong structural similarities to the cyclic tetrapeptides
7a–i. Moreover, the macrocycles
8e and
8f are equivalent to each other in large parts of their structure but differ in their peptoid-based alkyl side chain length.
The dihedral angles of the peptide-peptoid hybrids resemble the ones measured for cyclic tetrapeptides. Again, a cis-conformation was measured between the nitrogen atom of d-proline and the amide carbon of the following building block. As for tetrapeptides of general structure 7, three residues were located on the same side of the ring plane while the alkyl ring of d-proline pointed towards the opposite direction.
We could not successfully crystallize any cyclic hybrid of compounds with two peptoid units (series 9). Thus, we decided to use NMR data for the structure elucidation of the exemplarily chosen macrocycle 9a.
2.4. Spatial Structure in Solution State
Structural information on
9a was obtained by recording NOESY spectra. Internuclear distances were calculated from NOE cross-peak intensities (see
supplemental, Table S2 for details). Using the internuclear distance data from NOESY spectra and dihedral angle information from
J-coupling constants, a 3D model for
9a was constructed with the molecular modeling software Avogadro [
94]. This model was further structurally optimized utilizing a discrete Fourier transform (DFT) approach using the quantum chemical calculation software Turbomole [
95]
.However, nuclear Overhauser effect (NOE) data of small molecules is often not sufficient to unambiguously select for one conformation, especially in structural backbone dynamics. Additional structural information regarding
9a was obtained by extraction of one- and two-bond residual dipolar couplings (RDCs) in a uniaxially stretched polyethylene glycol (PEG) gel [
96]. This was achieved by recording clean in-phase heteronuclear single quantum coherence (CLIP-HSQC) [
97] and P.E.HSQC [
98] spectra of the molecule in an isotropic environment and under anisotropic conditions in a uniaxially stretched polyethylene glycol (PEG) gel [
96]. The RDCs were then used to validate the NOE-derived structure. To assess whether the RDCs agree with the constructed model, they were analyzed using single value decomposition (SVD) in the MSpin
-RDC software [
99]. An SVD omitting the RDC data of the more mobile sidechains yielded acceptable results. The back-calculated and experimental RDCs were in good agreement, with 7 out of 8 RDCs fulfilled within the experimental error (
supplemental, Table S3). A full back-calculation including sidechain RDCs can be found in the
supporting information (
supplemental, Table S3). Although the deviation between experimental and back-calculated values was higher in this case, all RDC values were reasonably well reproduced. The constructed model is therefore largely in agreement with the experimental NOE,
J-coupling, and RDC data and can be seen to represent the dominant solution state structure of
9a.
The 3D model of
9a indicated interesting structural differences compared to the macrocycles with three or four amino acids (
Table 6).
The model of
9a displays an alternating
cis-
trans-configuration of the cyclic backbone and an overall oblong ring shape. Thereby, the torsion angles ω
A and ω
C indicate a
trans-peptide bond between the amino functions of both amino acids and the carbonyl moieties of the subsequent peptoid monomers. In contrast to previous structures, no
cis-bond was measured between the nitrogen atom of proline and the amide carbon of the following building block (ω
C). Instead, two
cis-bonds were detected between the nitrogen atoms of the peptoid monomers and the subsequent carbonyl carbon atoms (ω
B and ω
D). Likewise, the dihedral angle ω
A next to the sterically demanding side chains of
l-phenylalanine was no longer distorted from ideal values (ω
A = 179.3°). Previous studies on small cyclic peptoids have shown that the
cis-
trans-
cis-
trans arrangement represents the lowest energy conformation and forms during the crystallization process of different cyclic tetrapeptoids [
100,
101,
102]. Our data indicate that this characteristic backbone arrangement is also favored in cyclic hybrids of general structure
9. Thus, with an increase in the peptide-peptoid ratio, the backbone configuration of apicidin derivatives can be easily modified.
Besides backbone configuration, the side chains of
9a differed from previous derivatives: the side chains were located alternately above and below the ring plane. This characteristic orientation is also known for pure peptoid macrocycles of different ring sizes [
41,
100,
101] and various
N-alkylated tetrapeptides [
103,
104,
105,
106,
107,
108].
Our data indicate that the increase of the peptoid-to-peptide ratio leads to significant structural changes of the entire macrocycle, which must be considered when developing potential inhibitors of the Wnt/β-catenin pathway.


 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



























































































