
Molecular Dynamics Simulations in Designing DARPins as Phosphorylation-Specific Protein Binders of ERK2

 ,
,
Abstract
:1. Introduction
2. Results
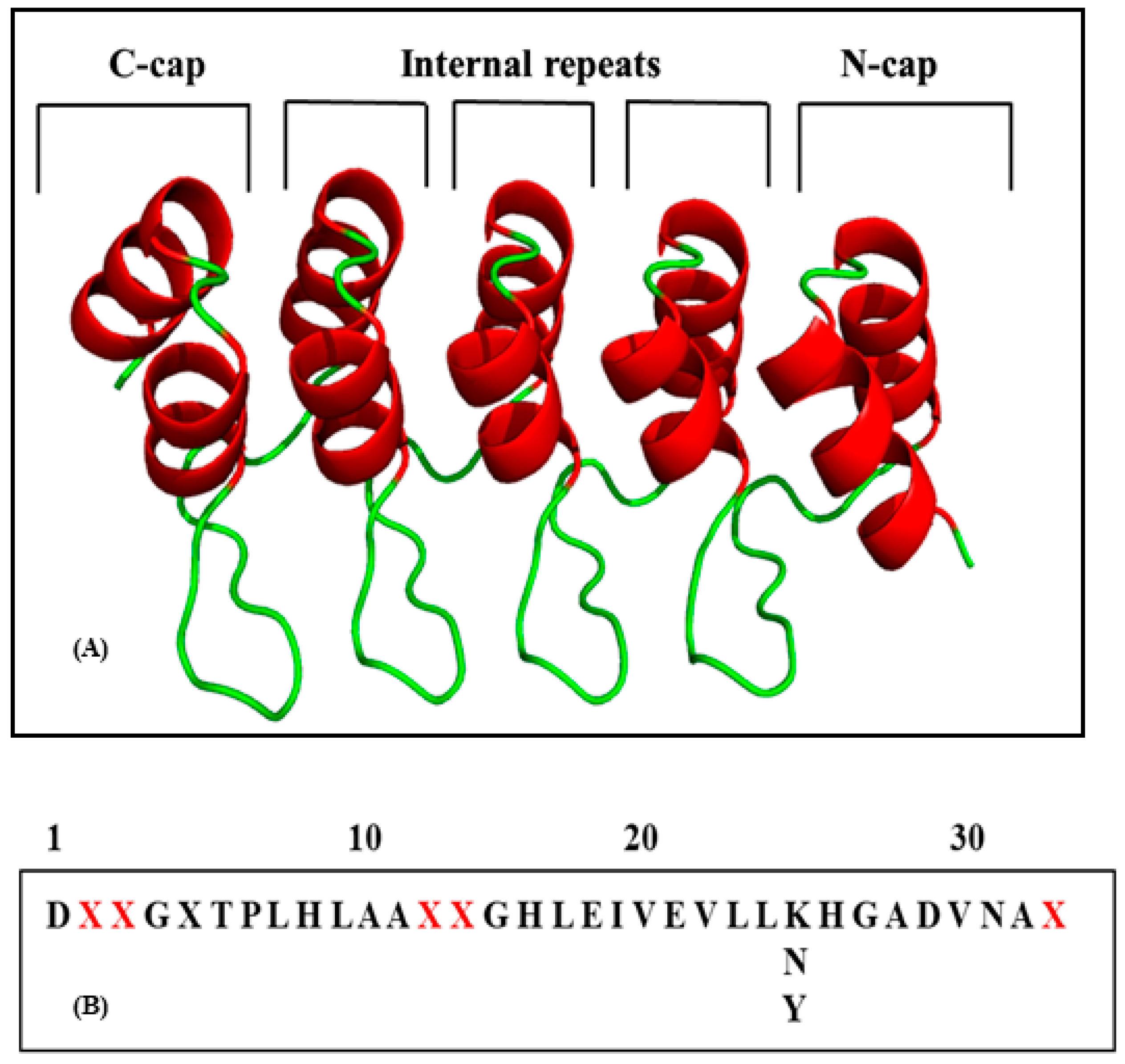
2.1. Design and Prediction of New Inhibitors
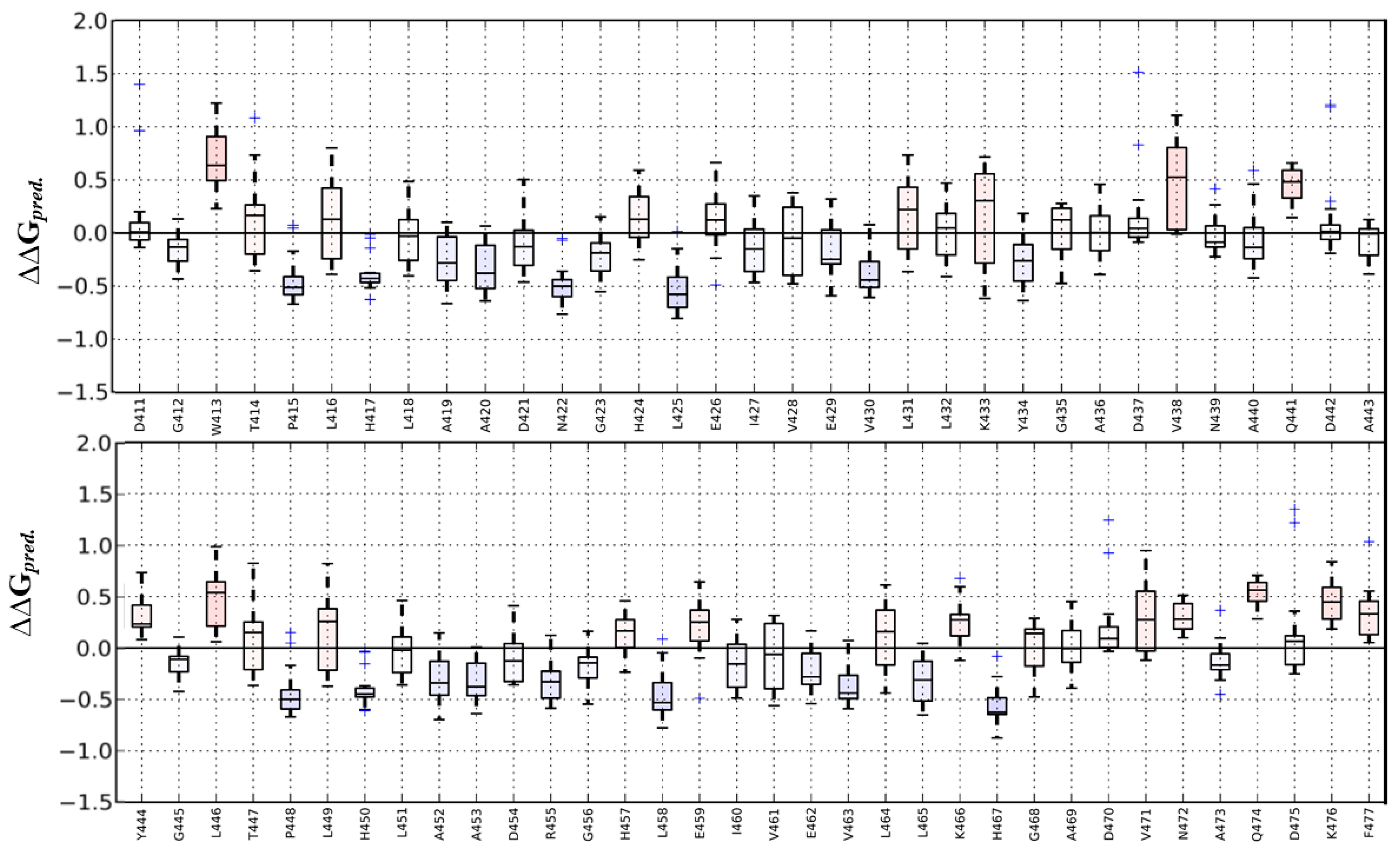
2.1.1. Evaluation of Specific Mutations
2.1.2. Evaluation of Selected Mutants by MDS
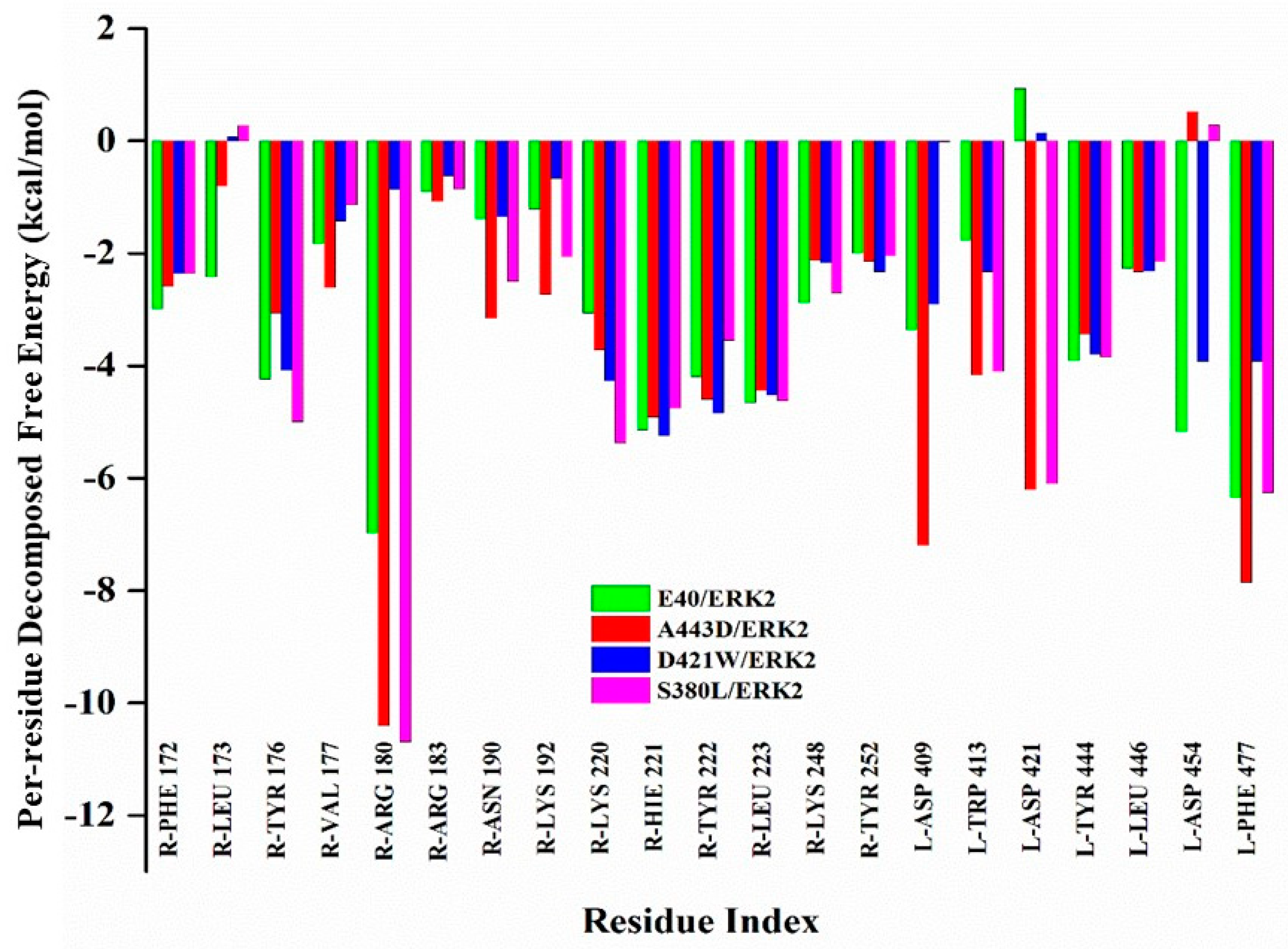
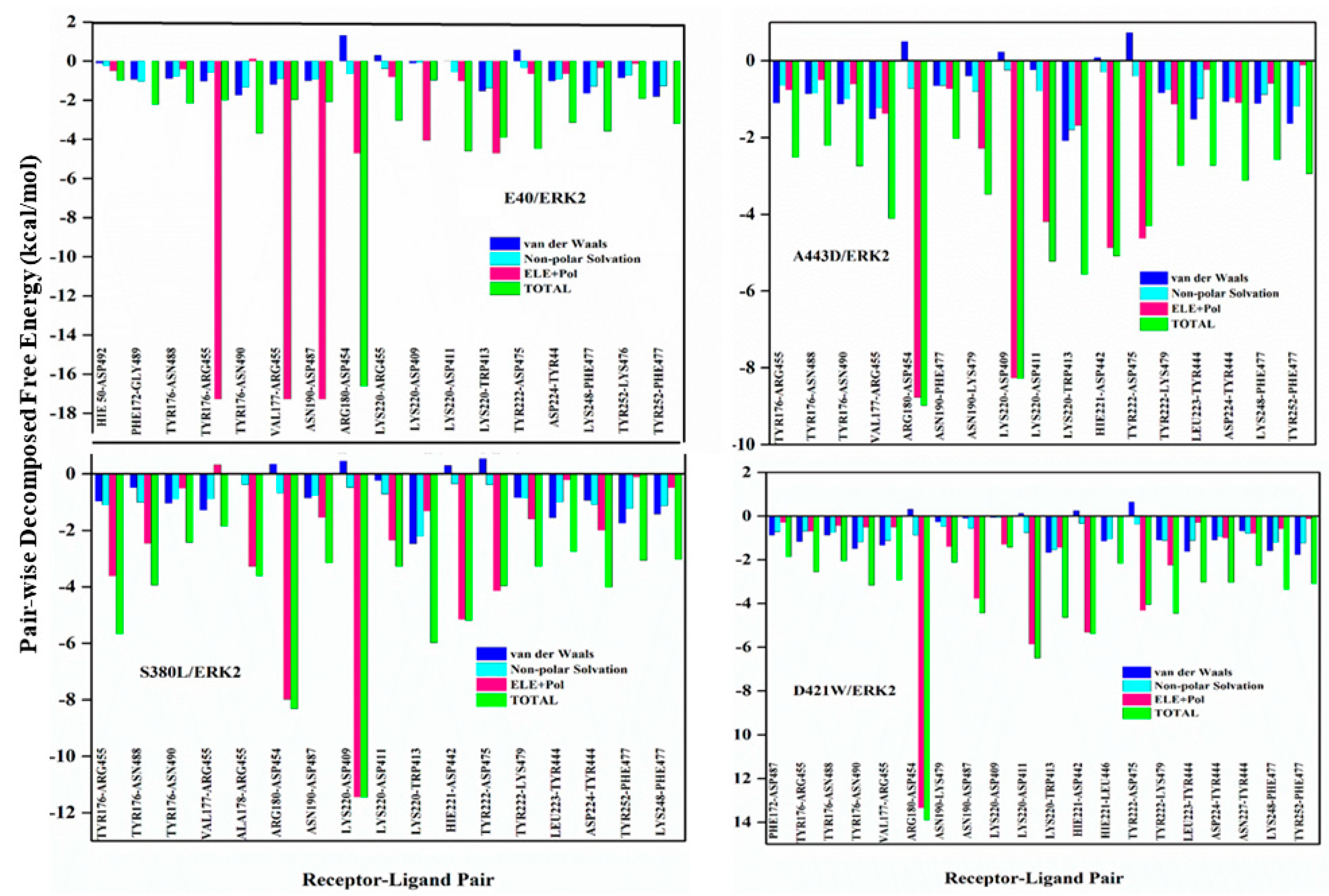
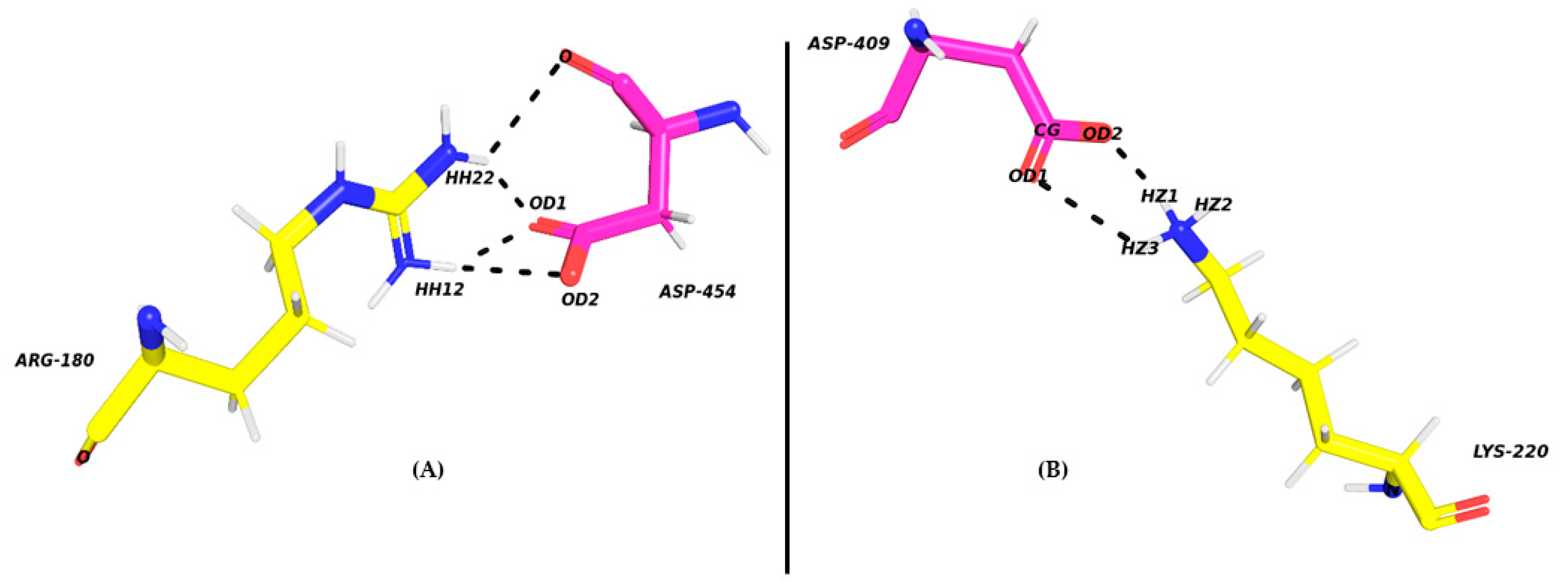
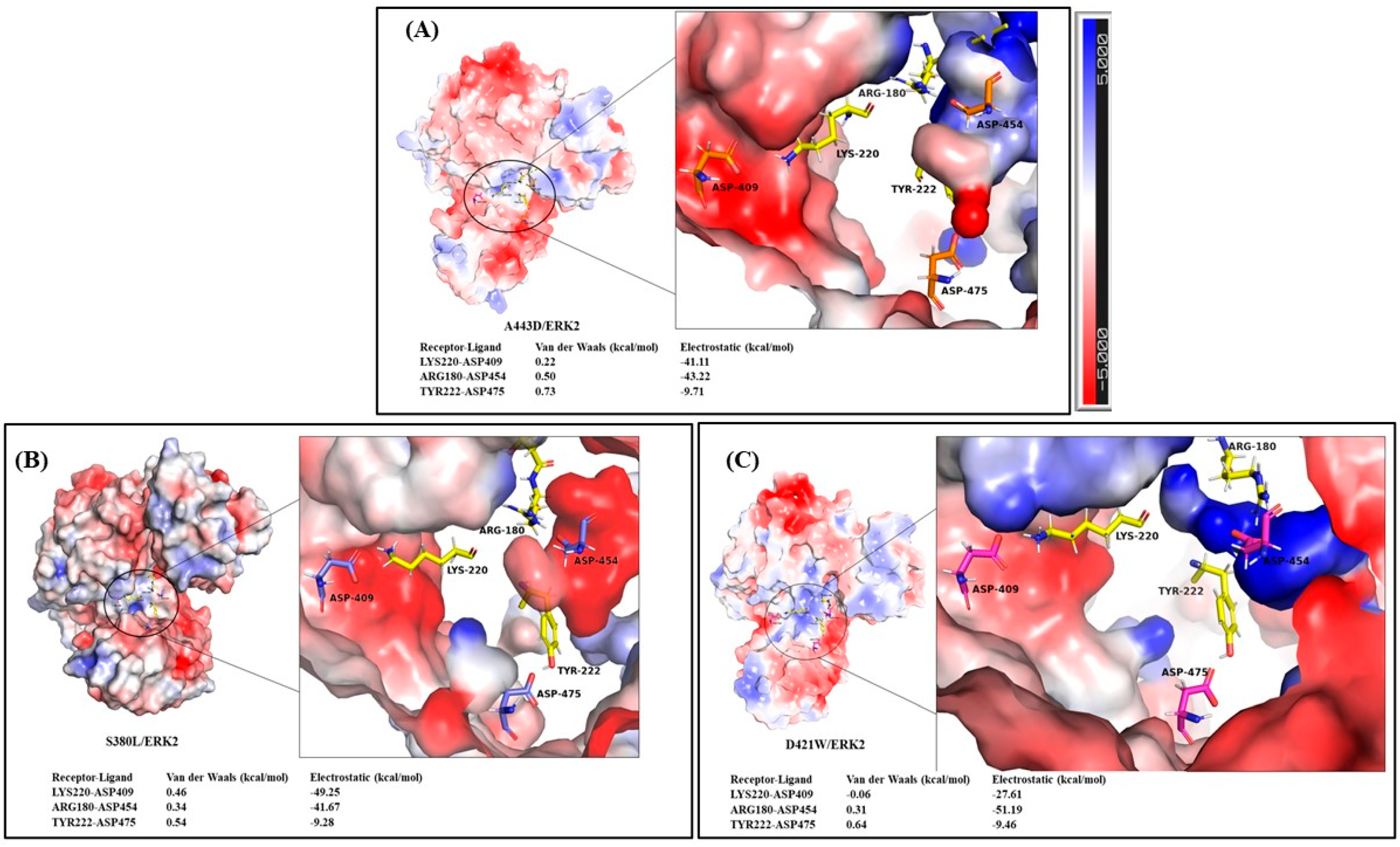
2.2. Exploring the Binding Mechanism of DARPins with ERK2
3. Discussion
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Roskoski, R. ERK1/2 MAP kinases: Structure, function, and regulation. Pharmacol. Res. 2012, 66, 105–143. [Google Scholar] [CrossRef]
- Pearson, G.; Robinson, F.; Gibson, T.B.; Xu, B.E.; Karandikar, M.; Berman, K.; Cobb, M.H. Mitogen-activated protein (MAP) kinase pathways: Regulation and physiological functions. Endocr. Rev. 2001, 22, 153–183. [Google Scholar]
- Bhullar, K.S.; Lagarón, N.O.; McGowan, E.M.; Parmar, I.; Jha, A.; Hubbard, B.P.; Rupasinghe, H.P.V. Kinase-targeted cancer therapies: Progress, challenges and future directions. Mol. Cancer 2018, 17, 1–20. [Google Scholar] [CrossRef]
- Dreier, B.; Plückthun, A. Rapid Selection of High-Affinity Antibody scFv Fragments Using Ribosome Display. Methods Mol. Biol. 2018, 1827, 235–268. [Google Scholar] [PubMed]
- Steiner, D.; Forrer, P.; Plückthun, A. Efficient selection of DARPins with sub-nanomolar affinities using SRP phage display. J. Mol. Biol. 2008, 382, 1211–1227. [Google Scholar] [CrossRef]
- Hartmann, J.; Münch, R.C.; Freiling, R.T.; Schneider, I.C.; Dreier, B.; Samukange, W.; Koch, J.; Seeger, M.A.; Plückthun, A.; Buchholz, C.J. A Library-Based Screening Strategy for the Identification of DARPins as Ligands for Receptor-Targeted AAV and Lentiviral Vectors. Mol. Ther. Methods Clin. Dev. 2018, 10, 128–143. [Google Scholar] [CrossRef] [PubMed]
- Smithwick, E.; Stewart, M.W. Designed Ankyrin Repeat Proteins: A Look at their Evolving Use in Medicine with a Focus on the Treatment of Chorioretinal Vascular Disorders. Antiinflamm. Antiallergy Agents Med. Chem. 2017, 16, 33–45. [Google Scholar] [CrossRef]
- Proshkina, G.; Deyev, S.; Ryabova, A.; Tavanti, F.; Menziani, M.C.; Cohen, R.; Katrivas, L.; Kotlyar, A. DARPin_9-29-Targeted Mini Gold Nanorods Specifically Eliminate HER2-Overexpressing Cancer Cells. ACS Appl. Mater Interfaces 2019, 11, 34645–34651. [Google Scholar] [CrossRef] [PubMed]
- Shilova, O.N.; Deyev, S.M. DARPins: Promising Scaffolds for Theranostics. Acta. Nat. 2019, 11, 42–53. [Google Scholar] [CrossRef]
- Stumpp, M.T.; Dawson, K.M.; Binz, H.K. Beyond Antibodies: The DARPin® Drug Platform. BioDrugs 2020, 34, 423–433. [Google Scholar] [CrossRef]
- Boersma, Y.L.; Plückthun, A. DARPins and other repeat protein scaffolds: Advances in engineering and applications. Curr. Opin. Biotechnol. 2011, 22, 849–857. [Google Scholar] [CrossRef]
- Forrer, P.; Binz, H.K.; Stumpp, M.T.; Plückthun, A. Consensus Design of Repeat Proteins. ChemBioChem 2004, 5, 183–189. [Google Scholar] [CrossRef]
- Forrer, P.; Stumpp, M.T.; Binz, H.K.; Plückthun, A. A novel strategy to design binding molecules harnessing the modular nature of repeat proteins. FEBS Lett. 2003, 39, 2–6. [Google Scholar] [CrossRef]
- Kohl, A.; Binz, H.K.; Forrer, P.; Stumpp, M.T.; Plückthun, A.; Grütter, M.G. Designed to be stable: Crystal structure of a consensus ankyrin repeat protein. Proc. Natl. Acad. Sci. USA 2003, 100, 1700–1705. [Google Scholar] [CrossRef] [PubMed]
- Gorina, S.; Pavletich, N.P. Structure of the p53 tumor suppressor bound to the ankyrin and SH3 domains of 53BP2. Science 1996, 274, 1001–1005. [Google Scholar] [CrossRef]
- Sedgwick, S.G.; Smerdon, S.J. The ankyrin repeat: A diversity of interactions on a common structural framework. Trends Biochem. Sci. 1999, 24, 311–316. [Google Scholar] [CrossRef]
- Binz, H.K.; Stumpp, M.T.; Forrer, P.; Amstutz, P.; Plückthun, A. Designing repeat proteins: Well-expressed, soluble and stable proteins from combinatorial libraries of consensus ankyrin repeat proteins. J. Mol. Biol. 2003, 332, 489–503. [Google Scholar] [CrossRef]
- Leader, B.; Baca, Q.J.; Golan, D.E. Protein therapeutics: A summary and pharmacological classification. Nat. Rev. Drug Discov. 2008, 7, 21–39. [Google Scholar] [CrossRef]
- Gautieri, A.; Beeg, M.; Gobbi, M.; Rigoldi, F.; Colombo, L.; Salmona, M. The Anti-Amyloidogenic Action of Doxycycline: A Molecular Dynamics Study on the Interaction with Aβ42. Int. J. Mol. Sci. 2019, 20, 4641. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Jensen, M.Ø.; Jogini, V.; Stein, R.A.; Lee, C.H.; Mchaourab, H.S.; Shaw, D.E.; Gouaux, E. Mechanism of NMDA receptor channel block by MK-801 and memantine. Nature 2018, 556, 515–519. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Yuan, C.; Ma, X.; Wang, Y.; Gu, X.; Fu, W. Molecular Mechanism of Action of RORγt Agonists and Inverse Agonists: Insights from Molecular Dynamics Simulation. Molecules 2018, 23, 3181. [Google Scholar] [CrossRef]
- Wang, H.; Gao, Z.; Song, P.; Hu, B.; Wang, J.; Cheng, M. Molecular dynamics simulation and QM/MM calculation reveal the selectivity mechanism of type I 1/2 kinase inhibitors: The effect of intramolecular H-bonds and conformational restriction for improved selectivity. Phys. Chem. Chem. Phys. 2019, 21, 24147–24164. [Google Scholar] [CrossRef]
- Vila-Viçosa, D.; Victor, B.L.; Ramos, J.; Machado, D.; Viveiros, M.; Switala, J.; Loewen, P.C.; Leitão, R.; Martins, F.; Machuqueiro, M. Insights on the Mechanism of Action of INH-C10 as an Antitubercular Prodrug. Mol. Pharm. 2017, 14, 4597–4605. [Google Scholar] [CrossRef]
- Barros, E.P.; Schiffer, J.M.; Vorobieva, A.; Dou, J.; Baker, D.; Amaro, R.E. Improving the Efficiency of Ligand-Binding Protein Design with Molecular Dynamics Simulations. J. Chem. Theory Comput. 2019, 15, 5703–5715. [Google Scholar] [CrossRef] [PubMed]
- Chew, T.A.; Orlando, B.J.; Zhang, J.; Latorraca, N.R.; Wang, A.; Hollingsworth, S.A.; Chen, D.H.; Dror, R.O.; Liao, M.; Feng, L. Structure and mechanism of the cation-chloride cotransporter NKCC1. Nature 2019, 572, 488–492. [Google Scholar] [CrossRef]
- Ferreira, J.V.; Capello, T.M.; Siqueira, L.J.; Lago, J.H.; Caseli, L. Mechanism of Action of Thymol on Cell Membranes Investigated through Lipid Langmuir Monolayers at the Air-Water Interface and Molecular Simulation. Langmuir 2016, 32, 3234–3241. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Zhang, W.; Cheng, J.; Nie, Y.; Xin, Q.; Yuan, S.; Dou, Y. Formation Mechanism of Ion Channel in Channelrhodopsin-2: Molecular Dynamics Simulation and Steering Molecular Dynamics Simulations. Int. J. Mol. Sci. 2019, 20, 3780. [Google Scholar] [CrossRef] [PubMed]
- Behmard, E.; Najafi, A.; Ahmadi, A. Understanding the resistance mechanism of penicillin binding protein 1a mutant against cefotaxime using molecular dynamic simulation. J. Biomol. Struct. Dyn. 2019, 37, 741–749. [Google Scholar] [CrossRef]
- Chen, Q.; Cheng, X.; Wei, D.; Xu, Q. Molecular dynamics simulation studies of the wild type and E92Q/N155H mutant of Elvitegravir-resistance HIV-1 integrase. Interdiscip. Sci. 2015, 7, 36–42. [Google Scholar] [PubMed]
- Cloete, R.; Kapp, E.; Joubert, J.; Christoffels, A.; Malan, S.F. Molecular modelling, and simulation studies of the Mycobacterium tuberculosis multidrug efflux pump protein Rv1258c. PLoS ONE 2018, e0207605. [Google Scholar] [CrossRef]
- Ge, Y.; Wu, J.; Xia, Y.; Yang, M.; Xiao, J.; Yu, J. Molecular dynamics simulation of the complex PBP-2x with drug cefuroxime to explore the drug resistance mechanism of Streptococcus suis R61. PLoS ONE 2012, 7, e35941. [Google Scholar] [CrossRef] [PubMed]
- Ul Haq, F.; Abro, A.; Raza, S.; Liedl, K.R.; Azam, S.S. Molecular dynamics simulation studies of novel β-lactamase inhibitor. J. Mol. Graph. Model. 2017, 74, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Aci-Sèche, S.; Ziada, S.; Braka, A.; Arora, R.; Bonnet, P. Advanced molecular dynamics simulation methods for kinase drug discovery. Future Med. Chem. 2016, 8, 545–566. [Google Scholar] [CrossRef] [PubMed]
- Ahuja, L.G.; Aoto, P.C.; Kornev, A.P.; Veglia, G.; Taylor, S.S. Dynamic allostery-based molecular workings of kinase:peptide complexes. Proc. Natl. Acad. Sci. USA 2019, 116, 15052–15061. [Google Scholar] [CrossRef]
- Bao, Y.; Zhou, L.; Dai, D.; Zhu, X.; Hu, Y.; Qiu, Y. Discover potential inhibitors for PFKFB3 using 3D-QSAR, virtual screening, molecular docking and molecular dynamics simulation. J. Recept. Signal Transduct. Res. 2018, 38, 413–431. [Google Scholar] [CrossRef]
- Li, K.; Zhu, J.; Xu, L.; Jin, J. Rational Design of Novel Phosphoinositide 3-Kinase Gamma (PI3Kγ) Selective Inhibitors: A Computational Investigation Integrating 3D-QSAR, Molecular Docking and Molecular Dynamics Simulation. Chem. Biodivers. 2019, 16, e1900105. [Google Scholar] [CrossRef]
- Liu, J.; Lu, Y.; Li, G.; Xiao, M.; Yang, G.; Pan, Y. Elucidation the binding mechanism of Nelumbo nucifera-derived isoquinoline alkaloids as Rho-kinase 1 inhibitors by molecular docking and dynamic simulation. J. Biomol. Struct Dyn. 2021, 39, 379–394. [Google Scholar] [CrossRef]
- Zou, Y.; Ewalt, J.; Ng, H.L. Recent Insights from Molecular Dynamics Simulations for G Protein-Coupled Receptor Drug Discovery. Int. J. Mol. Sci. 2019, 20, 4237. [Google Scholar] [CrossRef]
- Ma, C.; Hu, B.; Zhang, L.; Zhao, Y.; Wang, M.; Wang, J.; Cheng, M. Computational investigation of the antagonism effect towards GluN2B-Containing NMDA receptor: Combined ligand-based and target-based approach. J. Mol. Graph. Model. 2019, 86, 95–105. [Google Scholar] [CrossRef]
- Michael, E.; Polydorides, S.; Simonson, T.; Archontis, G. Hybrid MC/MD for protein design. J. Chem. Phys. 2020, 153, 054113. [Google Scholar] [CrossRef]
- De Vivo, M.; Masetti, M.; Bottegoni, G.; Cavalli, A. Role of Molecular Dynamics and Related Methods in Drug Discovery. J. Med. Chem. 2016, 59, 4035–4061. [Google Scholar] [CrossRef]
- Parthiban, V.; Gromiha, M.M.; Schomburg, D. CUPSAT: Prediction of protein stability upon point mutations. Nucleic Acids Res. 2006, 1, W239–W242. [Google Scholar] [CrossRef] [PubMed]
- Worth, C.L.; Preissner, R.; Blundell, T.L. SDM—A server for predicting effects of mutations on protein stability and malfunction. Nucleic Acids Res. 2011, 39, W215–W222. [Google Scholar] [CrossRef] [PubMed]
- Dehouck, Y.; Kwasigroch, J.M.; Gilis, D.; Rooman, M. PoPMuSiC 2.1: A web server for the estimation of protein stability changes upon mutation and sequence optimality. BMC Bioinformat. 2011, 13, 151. [Google Scholar] [CrossRef]
- De Baets, G.; Van Durme, J.; Reumers, J.; Maurer-Stroh, S.; Vanhee, P.; Dopazo, J.; Schymkowitz, J.; Rousseau, F. SNPeffect 4.0: On-line prediction of molecular and structural effects of protein-coding variants. Nucleic Acids Res. 2012, 40, D935–D939. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 76, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.; Ascher, D.B.; Blundell, T.L. mCSM: Predicting the effects of mutations in proteins using graph-based signatures. Bioinformatics 2014, 30, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Laimer, J.; Hofer, H.; Fritz, M.; Wegenkittl, S.; Lackner, P. MAESTRO-multi agent stability prediction upon point mutations. BMC Bioinformat. 2015, 16, 116. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, C.H.; Pires, D.E.; Ascher, D.B. DynaMut: Predicting the impact of mutations on protein conformation, flexibility and stability. Nucleic Acids Res. 2018, 2, W350–W355. [Google Scholar] [CrossRef]
- Rodrigues, C.H.M.; Myung, Y.; Pires, D.E.V.; Ascher, D.B. mCSM-PPI2: Predicting the effects of mutations on protein-protein interactions. Nucleic Acids Res. 2019, 2, W338–W344. [Google Scholar] [CrossRef]
- Birolo, G.; Benevenuta, S.; Fariselli, P.; Capriotti, E.; Giorgio, E.; Sanavia, T. Protein Stability Perturbation Contributes to the Loss of Function in Haploinsufficient Genes. Front. Mol. Biosci. 2021, 1, 620793. [Google Scholar] [CrossRef]
- Gerasimavicius, L.; Liu, X.; Marsh, J.A. Identification of pathogenic missense mutations using protein stability predictors. Sci. Rep. 2020, 21, 15387. [Google Scholar] [CrossRef] [PubMed]
- Powers, E.T.; Morimoto, R.I.; Dillin, A.; Kelly, J.W.; Balch, W.E. Biological and chemical approaches to diseases of proteostasis deficiency. Annu. Rev. Biochem. 2009, 78, 959–991. [Google Scholar] [CrossRef]
- Karmakar, M.; Rodrigues, C.H.M.; Holt, K.E.; Dunstan, S.J.; Denholm, J.; Ascher, D.B. Empirical ways to identify novel Bedaquiline resistance mutations in AtpE. PLoS ONE 2019, 29, 14. [Google Scholar] [CrossRef]
- Pines, G.; Fankhauser, R.G.; Eckert, C.A. Predicting Drug Resistance Using Deep Mutational Scanning. Molecules 2020, 25, 2265. [Google Scholar] [CrossRef]
- Portelli, S.; Phelan, J.E.; Ascher, D.B.; Clark, T.G.; Furnham, N. Understanding molecular consequences of putative drug resistant mutations in Mycobacterium tuberculosis. Sci. Rep. 2018, 8, 15356. [Google Scholar] [CrossRef]
- Fischer, A.; Seitz, T.; Lochner, A.; Sterner, R.; Merkl, R.; Bocola, M. A fast and precise approach for computational saturation mutagenesis and its experimental validation by using an artificial (βα)8-barrel protein. Chembiochem 2011, 12, 1544–1550. [Google Scholar] [CrossRef] [PubMed]
- Munir, A.; Vedithi, S.C.; Chaplin, A.K.; Blundell, T.L. Genomics, Computational Biology and Drug Discovery for Mycobacterial Infections: Fighting the Emergence of Resistance. Front. Genet. 2020, 11, 965. [Google Scholar] [CrossRef] [PubMed]
- Xin, L.; Yu, H.; Hong, Q.; Bi, X.; Zhang, X.; Zhang, Z.; Kong, Z.; Zheng, Q.; Gu, Y.; Zhao, Q.; et al. Identification of Strategic Residues at the Interface of Antigen-Antibody Interactions by In Silico Mutagenesis. Interdiscip. Sci. 2018, 10, 438–448. [Google Scholar] [CrossRef]
- BIOVIA, D.S. Dassault Systèmes BIOVIA, (Version 9.24); Dassault Systèmes: San Diego, CA, USA, 2016. [Google Scholar]
- Gautam, V.; Chong, W.L.; Chin, S.P.; Zain, S.M.; Rahman, N.A.; Vao-soongnern, V.; Lee, V.S. Loop dynamics behind the affinity of DARPins towards ERK2: Molecular dynamics simulations (MDs) and elastic network model (ENM). J. Mol. Liq. 2019, 274, 612–620. [Google Scholar] [CrossRef]
- Vascon, F.; Gasparotto, M.; Giacomello, M.; Cendron, L.; Bergantino, E.; Filippini, F.; Righetto, I. Protein electrostatics: From computational and structural analysis to discovery of functional fingerprints and biotechnological design. Comput. Struct. Biotechnol. J. 2020, 18, 1774–1789. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC.
- Yachdav, G.; Kloppmann, E.; Kajan, L.; Hecht, M.; Goldberg, T.; Hamp, T.; Hönigschmid, P.; Schafferhans, A.; Roos, M.; Bernhofer, M.; et al. PredictProtein—an open resource for online prediction of protein structural and functional features. Nucleic Acids Res. 2014, 42, W337–W343. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Randall, A.Z.; Sweredoski, M.J.; Baldi, P. SCRATCH: A protein structure and structural feature prediction server. Nucleic Acids Res. 2005, 33, W72–W76. [Google Scholar] [CrossRef]
- Kummer, L.; Parizek, P.; Rube, P.; Millgramm, B.; Prinz, A.; Mittl, P.R.; Kaufholz, M.; Zimmermann, B.; Herberg, F.W.; Plückthun, A. Structural and functional analysis of phosphorylation-specific binders of the kinase ERK from designed ankyrin repeat protein libraries. Proc. Natl. Aacd. Sci. USA 2012, 109, E2248–E2257. [Google Scholar] [CrossRef]
- Case, D.A.; Babin, V.; Berryman, J.T.; Betz, R.M.; Cerutti, D.; Cheatham, T.E., III; Darden, T.A.; Duke, R.E.; Kollman, P.A. AMBER 14; University of California: San Francisco, CA, USA, 2014. [Google Scholar]
- Pérez, A.; Marchán, I.; Svozil, D.; Sponer, J.; Cheatham, T.E., III; Laughton, C.A.; Orozco, M. Refinement of the AMBER force field for nucleic acids: Improving the description of alpha/gamma conformers. Biophys. J. 2007, 92, 3817–3829. [Google Scholar] [CrossRef] [PubMed]
- MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Karplus, M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 1998, 102, 3586–3616. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E., III. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Linse, B.; Linse, P. Tuning the smooth particle mesh Ewald sum: Application on ionic solutions and dipolar fluids. J. Chem. Phys. 2014, 141, 184114. [Google Scholar] [CrossRef]
- Darden, T.; Perera, L.; Li, L.; Pedersen, L. New tricks for modelers from the crystallography toolkit: The particle mesh Ewald algorithm and its use in nucleic acid simulations. Structure 1999, 15, R55–R60. [Google Scholar] [CrossRef]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, B.; Kollman, P.A. Binding of a diverse set of ligands to avidin and streptavidin: An accurate quantitative prediction of their relative affinities by a combination of molecular mechanics and continuum solvent models. J. Med. Chem. 2000, 43, 3786–3791. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutants | ΔΔGpred | Cpred | Mutants | ΔΔGpred | Cpred | Mutants | ΔΔGpred | Cpred | Mutants | ΔΔGpred | Cpred | Mutants | ΔΔGpred | Cpred | Mutants | ΔΔGpred | Cpred | Mutants | ΔΔGpred | Cpred |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| S380(A) | 0.038 | 0.959 | N422(A) | −0.517 | 0.907 | A443(R) | 0.067 | 0.807 | D454(A) | −0.035 | 0.933 | R455(A) | −0.146 | 0.922 | D421(A) | −0.031 | 0.926 | I389(Y) | −0.709 | 0.861 |
| S380(R) | 0.402 | 0.76 | N422(R) | −0.05 | 0.851 | A443(N) | −0.232 | 0.92 | D454(R) | 0.173 | 0.801 | R455(N) | −0.27 | 0.915 | D421(A) | 0.145 | 0.808 | I389(W) | -0.672 | 0.848 |
| S380(N) | −0.007 | 0.946 | N422(D) | −0.697 | 0.835 | A443(D) | −0.325 | 0.947 | D454(N) | −0.094 | 0.906 | R455(D) | −0.171 | 0.934 | D421(A) | −0.115 | 0.909 | I389(V) | −0.45 | 0.855 |
| S380(D) | 0.084 | 0.944 | N422(E) | −0.739 | 0.832 | A443(E) | −0.385 | 0.948 | D454(Q) | −0.125 | 0.909 | R455(E) | −0.305 | 0.931 | D421(A) | −0.461 | 0.882 | I389(T) | −0.522 | 0.846 |
| S380(E) | 0.065 | 0.937 | N422(Q) | −0.441 | 0.872 | A443(Q) | −0.278 | 0.925 | D454(H) | 0.016 | 0.894 | R455(Q) | −0.439 | 0.895 | D421(A) | −0.128 | 0.913 | I389(S) | −0.378 | 0.84 |
| S380(Q) | −0.075 | 0.915 | N422(H) | −0.359 | 0.898 | A443(H) | 0.127 | 0.896 | D454(I) | −0.287 | 0.886 | R455(H) | −0.517 | 0.864 | D421(A) | 0.023 | 0.901 | I389(R) | −0.059 | 0.881 |
| S380(H) | −0.102 | 0.878 | N422(I) | −0.484 | 0.912 | A443(I) | 0.056 | 0.905 | D454(L) | −0.352 | 0.867 | R455(I) | −0.433 | 0.903 | D421(A) | −0.24 | 0.913 | I389(Q) | −0.644 | 0.858 |
| S380(M) | −0.227 | 0.896 | N422(L) | −0.412 | 0.902 | A443(L) | 0.041 | 0.902 | D454(K) | 0.411 | 0.79 | R455(L) | −0.401 | 0.92 | D421(A) | −0.255 | 0.908 | I389(N) | −0.556 | 0.84 |
| S380(F) | −0.346 | 0.898 | N422(K) | −0.067 | 0.909 | A443(K) | 0.028 | 0.848 | D454(M) | -0.335 | 0.876 | R455(K) | −0.534 | 0.881 | D421(A) | 0.503 | 0.795 | I389(M) | −0.589 | 0.871 |
| S380(W) | −0.466 | 0.86 | N422(M) | −0.484 | 0.895 | A443(M) | −0.133 | 0.922 | D454(F) | −0.347 | 0.888 | R455(M) | −0.473 | 0.885 | D421(A) | −0.256 | 0.894 | I389(L) | −0.449 | 0.874 |
| S380(T) | −0.002 | 0.944 | N422(F) | −0.512 | 0.879 | A443(F) | −0.052 | 0.914 | D454(S) | 0.072 | 0.93 | R455(F) | −0.502 | 0.889 | D421(A) | −0.351 | 0.891 | I389(K) | −0.054 | 0.893 |
| S380(Y) | −0.392 | 0.872 | N422(S) | −0.536 | 0.899 | A443(S) | −0.246 | 0.931 | D454(T) | −0.116 | 0.924 | R455(S) | −0.155 | 0.917 | D421(A) | 0.033 | 0.941 | I389(H) | −0.434 | 0.875 |
| S380(V) | −0.292 | 0.905 | N422(T) | −0.608 | 0.902 | A443(T) | 0.039 | 0.909 | D454(W) | −0.317 | 0.873 | R455(T) | −0.3 | 0.925 | D421(A) | −0.428 | 0.872 | I389(F) | −0.704 | 0.859 |
| S380(K) | 0.278 | 0.807 | N422(W) | −0.764 | 0.842 | A443(W) | −0.006 | 0.901 | D454(Y) | −0.352 | 0.888 | R455(W) | −0.568 | 0.879 | D421(A) | −0.021 | 0.955 | I389(E) | -0.793 | 0.83 |
| S380(L) | −0.285 | 0.907 | N422(Y) | −0.65 | 0.866 | A443(Y) | −0.001 | 0.905 | D454(V) | −0.215 | 0.921 | R455(Y) | −0.586 | 0.879 | D421(A) | −0.401 | 0.876 | I389(D) | −0.827 | 0.834 |
| S380(I) | −0.209 | 0.904 | N422(V) | −0.479 | 0.899 | A443(V) | 0.054 | 0.909 | D454(E) | −0.357 | 0.891 | R455(V) | −0.328 | 0.928 | D421(A) | −0.163 | 0.919 | I389(A) | −0.294 | 0.854 |
| Mutants | MM-PBSA (kcal/mol) | MM-GBSA (kcal/mol) |
|---|---|---|
| A443D | −91.51 ± 0.42 | −59.86 ± 0.29 |
| S380L | −94.90 ± 0.34 | −56.74 ± 0.27 |
| A443N | −80.29 ± 0.34 | −51.59 ± 0.26 |
| N422A | −66.44 ± 0.39 | −38.87 ± 0.27 |
| N422I | −64.34 ± 0.34 | −37.49 ± 0.27 |
| N422T | −86.74 ± 0.33 | −52.86 ± 0.25 |
| S380I | −71.96 ± 0.41 | −40.69 ± 0.31 |
| D421I | −62.95 ± 0.39 | −45.20 ± 0.29 |
| I389D | −70.14 ± 0.31 | −47.45 ± 0.23 |
| I389W | −71.06 ± 0.34 | −45.55 ± 0.22 |
| I389T | −57.75 ± 0.39 | −42.92 ± 0.26 |
| D454W | −62.27 ± 0.37 | −44.12 ± 0.26 |
| D421W | −42.71 ± 0.37 | −41.08 ± 0.26 |
| E40/ERK2 | −75.64 ± 0.27 | −49.50 ± 0.2 |
| Mutants | A443D | A443N | S380L | S380I | N422A | N422I | N422T | |
|---|---|---|---|---|---|---|---|---|
| Energetics | VDWAALS | −97.75 ± 0.23 | −88.88 ± 0.22 | −91.76 ± 0.25 | −97.73 ± 0.22 | −94.84 ± 0.28 | −86.68 ± 0.26 | −91.07 ± 0.24 |
| EEL | −626.46 ± 2.63 | −589.4 ± 2.97 | −673.80 ± 1.87 | −521.631 ± 2.08 | −441.77 ± 1.77 | −457.6 9 ± 1.88 | −407.50 ± 2.08 | |
| EGB | 672.13 ± 2.41 | 634.15 ± 2.86 | 712.20 ± 1.78 | 582.05 ± 1.95 | 503.72 ± 1.76 | 512.63 ± 1.82 | 448.52 ± 1.97 | |
| ESURF | −7.78 ± 0.03 | −7.43 ± 3.01 | −3.38 ± 0.03 | −3.38 ± 0.04 | −5.97 ± 0.03 | −5.75 ± 0.04 | −2.81 ± 0.03 | |
| ∆Ggas | −724.22 ± 2.59 | −678.31 ± 3.01 | −765.56 ± 1.89 | −619.36 ± 2.07 | −536.62 ± 1.84 | −544.37 ± 1.92 | -498.58 ± 2.07 | |
| ∆Gsolvation | 664.35 ± 2.41 | 626.72 ± 2.86 | 708.82 ± 1.77 | 578.67 ± 1.94 | 497.75 ± 1.75 | 506.88 ±1.80 | 445.71 ± 1.97 | |
| ∆G TOTAL | −59.8 6 ± 0.29 | −51.59 ± 0.26 | −56.74 ± 0.27 | −40.69 ± 0.31 | −38.87 ± 0.27 | −37.49 ± 0.27 | −52.86 ± 0.25 | |
| Mutants | D421I | I389D | I389W | I389T | D454W | D421W | ||
| Energetics | VDWAALS | −102.03 ± 0.2 | −92.03 ± 0.22 | −93.21 ± 0.22 | −84.65 ± 0.22 | −104.74 ± 0.27 | −104.74 ± 0.27 | |
| EEL | −357.28 ± 2.08 | −409.02 ± 2.03 | −501.96 ± 1.85 | −341.38 ± 2.02 | −307.17 ± 2.28 | −274.95 ± 1.61 | ||
| EGB | 423.76 ± 1.81 | 463.06 ± 1.95 | 558.67 ± 1.79 | 390.69 ± 1.95 | 379.18 ± 2.29 | 348.28 ± 1.54 | ||
| ESURF | −9.65 ± 0.01 | −9.04 ± 0.01 | −9.04 ± 0.01 | −8.77 ± 0.01 | −10.18 ± 0.01 | −10.06 ± 0.01 | ||
| ∆Ggas | −459.31 ± 1.96 | −501.06 ± 2.01 | −595.18 ± 1.86 | −426.04 ± 1.97 | −411.92 ± 2.40 | −379.30 ± 1.62 | ||
| ∆Gsolvation | 414.11 ± 1.81 | 453.61 ± 1.95 | 549.62 ± 1.79 | 381.91 ± 1.95 | 368.99 ± 2.28 | 338.21 ± 1.53 | ||
| ∆G TOTAL | −45.20 ± 0.29 | −47.45 ± 0.23 | −45.55 ± 0.22 | −44.12 ± 0.26 | −42.92 ± 0.2 | −41.08 ± 0.26 | ||
| System | Acceptor | Donor | Occupancy (%) | Distance | Angle |
|---|---|---|---|---|---|
| A443D/ERK2 | ASP475-OD2 | TYR222-OH | 99.0 | 2.66 | 166.77 |
| ASP442-OD2 | HIE221-NE2 | 84.6 | 2.80 | 154.14 | |
| ASP454-OD1 | ARG180-NH1 | 52.5 | 2.78 | 159.34 | |
| VAL177-O | ARG455-NH1 | 50.7 | 2.81 | 154.93 | |
| ASP224-OD2 | TYR444-OH | 43.9 | 2.70 | 161.74 | |
| ASN488-O | TYR176-OH | 42.7 | 2.76 | 160.74 | |
| ASP409-OD2 | LYS220-NZ | 22.2 | 2.79 | 152.82 | |
| TYR176-O | ARG455-NH | 16.2 | 2.86 | 160.46 | |
| ASN190-OD1 | LYS479-NZ | 3.6 | 2.85 | 154.21 | |
| D421W/ERK2 | ASP475-OD2 | TYR222-OH | 99.0 | 2.66 | 166.77 |
| ASP442-OD2 | HIE221-NE2 | 84.6 | 2.81 | 154.13 | |
| VAL177-O | ARG455-NH1 | 50.7 | 2.81 | 154.92 | |
| ASP454-OD2 | ARG180-NH1 | 46.7 | 2.80 | 159.39 | |
| ASP224-OD1 | TYR444-OH | 43.9 | 2.71 | 161.74 | |
| ASN488-O | TYR176-OH | 42.7 | 2.75 | 163.75 | |
| ASP454-OD1 | ARG180-NH1 | 38.3 | 2.80 | 157.34 | |
| ASP409-OD2 | LYS220-NZ | 22.2 | 2.79 | 152.82 | |
| TYR176-O | ARG455-NH | 16.2 | 2.85 | 160.46 | |
| S380L/ERK2 | ASP475-OD2 | TYR222-OH | 98.9 | 2.65 | 167.16 |
| ASP442-OD2 | HIE221-NE2 | 84.4 | 2.80 | 153.80 | |
| ASP421-OD1 | ARG180-NH2 | 72.6 | 2.80 | 154.62 | |
| ASN488-O | TYR176-OH | 68.7 | 2.75 | 162.41 | |
| ASP454-OD1 | ARG180-NH1 | 55.1 | 2.77 | 160.96 | |
| ASP224-OD1 | TYR444-OH | 52.8 | 2.70 | 161.61 | |
| ASP421-OD1 | ARG180NH1 | 49.6 | 2.84 | 149.43 | |
| TYR176-O | ARG455-NH1 | 45.3 | 2.85 | 159.24 | |
| ASP409-OD2 | LYS220-NZ | 14.0 | 2.79 | 154.31 |
| System | Acceptor | Donor | Interaction |
|---|---|---|---|
| A443D/ERK2 | ASP475-OD2 | TYR222-OH | Cation-Π |
| ASP442-OD2 | HIE221-NE2 | Salt Bridge | |
| ASP454-OD1 | ARG180-NH1 | Salt Bridge | |
| VAL177-O | ARG455-NH1 | Salt Bridge | |
| ASP224-OD2 | TYR444-OH | Cation-Π | |
| ASN488-O | TYR176-OH | Amino-Π | |
| ASP421-OD1 | ARG455-NH | Salt Bridge | |
| ASP409-OD2 | LYS220-NZ | Salt Bridge | |
| TYR176-O | ARG455-NH | Cation-Π | |
| ASN190-OD1 | LYS479-NZ | Salt Bridge | |
| D421W/ERK2 | ASP475-OD2 | TYR222-OH | Cation-Π |
| ASP442-OD2 | HIE221-NE2 | Salt Bridge | |
| VAL177-O | ARG455-NH1 | Salt Bridge | |
| ASP454-OD2 | ARG180-NH1 | Salt Bridge | |
| ASP224-OD1 | TYR444-OH | Cation-Π | |
| ASN488-O | TYR176-OH | Amino-Π | |
| ASP454-OD1 | ARG180-NH1 | Salt Bridge | |
| ASP409-OD2 | LYS220-NZ | Salt Bridge | |
| TYR176-O | ARG455-NH | Cation-Π | |
| TRP421-O | ARG455-NE | Cation-Π | |
| S380L/ERK2 | ASP475-OD2 | TYR222-OH | Cation-Π |
| ASP442-OD2 | HIE221-NE2 | Salt Bridge | |
| ASP421-OD1 | ARG180-NH2 | Salt Bridge | |
| ASN488-O | TYR176-OH | Amino-Π | |
| ASP454-OD1 | ARG180-NH1 | Salt Bridge | |
| ASP224-OD1 | TYR444-OH | Cation-Π | |
| ASP421-OD1 | ARG180NH1 | Salt Bridge | |
| TYR176-O | ARG455-NH1 | Cation-Π | |
| ASP409-OD2 | LYS220-NZ2 | Salt Bridge |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gautam, V.; Nimmanpipug, P.; Zain, S.M.; Rahman, N.A.; Lee, V.S. Molecular Dynamics Simulations in Designing DARPins as Phosphorylation-Specific Protein Binders of ERK2. Molecules 2021, 26, 4540. https://doi.org/10.3390/molecules26154540
Gautam V, Nimmanpipug P, Zain SM, Rahman NA, Lee VS. Molecular Dynamics Simulations in Designing DARPins as Phosphorylation-Specific Protein Binders of ERK2. Molecules. 2021; 26(15):4540. https://doi.org/10.3390/molecules26154540
Chicago/Turabian StyleGautam, Vertika, Piyarat Nimmanpipug, Sharifuddin Md Zain, Noorsaadah Abd Rahman, and Vannajan Sanghiran Lee. 2021. "Molecular Dynamics Simulations in Designing DARPins as Phosphorylation-Specific Protein Binders of ERK2" Molecules 26, no. 15: 4540. https://doi.org/10.3390/molecules26154540