
Perturbating Intramolecular Hydrogen Bonds through Substituent Effects or Non-Covalent Interactions



Departamento de Química (Módulo 13, Facultad de Ciencias) and Institute of Advanced Chemical Sciences (IadChem), Universidad Autónoma de Madrid, Campus de Excelencia UAM-CSIC, Cantoblanco, 28049 Madrid, Spain
*
Authors to whom correspondence should be addressed.
Molecules 2021, 26(12), 3556; https://doi.org/10.3390/molecules26123556
Submission received: 27 May 2021
/
Revised: 7 June 2021
/
Accepted: 8 June 2021
/
Published: 10 June 2021
(This article belongs to the Special Issue Intramolecular Hydrogen Bonding 2021)
Abstract
:An analysis of the effects induced by F, Cl, and Br-substituents at the α-position of both, the hydroxyl or the amino group for a series of amino-alcohols, HOCH2(CH2)nCH2NH2 (n = 0–5) on the strength and characteristics of their OH···N or NH···O intramolecular hydrogen bonds (IMHBs) was carried out through the use of high-level G4 ab initio calculations. For the parent unsubstituted amino-alcohols, it is found that the strength of the OH···N IMHB goes through a maximum for n = 2, as revealed by the use of appropriate isodesmic reactions, natural bond orbital (NBO) analysis and atoms in molecules (AIM), and non-covalent interaction (NCI) procedures. The corresponding infrared (IR) spectra also reflect the same trends. When the α-position to the hydroxyl group is substituted by halogen atoms, the OH···N IMHB significantly reinforces following the trend H < F < Cl < Br. Conversely, when the substitution takes place at the α-position with respect to the amino group, the result is a weakening of the OH···N IMHB. A totally different scenario is found when the amino-alcohols HOCH2(CH2)nCH2NH2 (n = 0–3) interact with BeF2. Although the presence of the beryllium derivative dramatically increases the strength of the IMHBs, the possibility for the beryllium atom to interact simultaneously with the O and the N atoms of the amino-alcohol leads to the global minimum of the potential energy surface, with the result that the IMHBs are replaced by two beryllium bonds.
1. Introduction
The name “hydrogen bonding” was used for the first time by Linus Pauling in 1929 [1], although the interactions associated with a hydrogen bond had been described for the first time more than a century ago, by W.M. Latimer and W.H. Rodebush, in a paper published in 1920 [2]. Since 1934, the year in which Pauling and Brockway experimentally confirmed that carboxylic acids could indeed form hydrogen bonds [3], hydrogen bonding became one of the most fruitful concepts in chemistry, being behind a huge amount of stabilizing intermolecular interactions [4,5,6,7,8,9,10,11,12,13,14,15]. In addition, in the 1920s, the existence of intramolecular interactions that, nowadays, we know as intramolecular hydrogen bonds (IHBs), were described in the literature. Indeed, the first paper on the coordination of hydrogen to explain the abnormal solubilities of some benzene derivatives was published in 1924 [16]. The intramolecular hydrogen bond was actually described in a 1926 publication by H.E. Amstrong [17], making reference to a paper of T.M. Lowry on the anomalies of the optical rotatory dispersion of tartaric acid published the same year [18], as a “bigamous hydrogen”. Almost a century has elapsed since the publication of these articles, and the presence of IHBs is very often shown to be behind many different phenomena in chemistry, physics, in particular in photophysics, material science, and biochemistry, to the point that IUPAC dedicated a specific paper to the definition of this chemical interaction [19]. Let us mention here a few suitable examples, drawn from a huge number of publications, such as the substituent effects on the IHB of malonaldehyde [20], or the role of IMHBs on: protonation and deprotonation processes [21], NMR chemical shifts [22], bond dissociation enthalpies [23,24], or their influence on the role of aromaticity in chemical reactions [25]. They are also crucial to understand excited-state intramolecular proton transfers [26,27,28], or the control of photosubstitution phenomena in metal complexes [29], or the control of emissive properties of different organic compounds [30]. IMHBs are also behind the high chemical stability of metal-organic frameworks [31] and the excellent electroluminescence of some organic light-emitting diodes (OLEDs) [32], and related with linear and nonlinear electric properties [33] or with the thermodynamic properties of compounds like pyridinol derivatives [34]. In biochemistry, they are responsible for proton shuttle mechanisms in certain lipase-catalyzed reactions [35]. IMHBs between hydroxyl and amino groups of a serinol function, characteristic of an important lipid, sphingosine, seem to be responsible for its existence, as neutral at the physiological pH [36], IMHBs allow also for designing novel peptide inhibitors [37], and play an important role as far as the molecular conformation of amino acids is concerned [38]. In addition, important properties are associated with the so-called intramolecular charge-inverted hydrogen bonds [39,40].
Since the pioneering studies by microwave spectroscopy on 2-aminoethanol [41] and by infrared spectroscopy on trans-8a and -8b-decahydroquinilinol reveal the important role of IMHBs in their stability, the interest in amino-alcohols increased significantly. The first ab initio calculations showed that 2-aminoethanol, 3-aminopropanol, and 4-aminobutanol were indeed stabilized by OH···N IMHBs, this stabilization being larger the longer the chain [42]. Very recent microwave experiments in parallel with MP2 ab initio calculations confirmed that, in both 3-aminopropanol [43] and 4-aminobutanol [44], the ground state is stabilized through the formation of an OH···N IMHB with O-N internuclear distance of 2.856 and 2.954 Å, respectively. In addition, a recent and rather complete study [45] using FTIR measurements, 1H NMR spectroscopy, density functional theory (DFT) calculations, and molecular dynamics (MD) on 3-aminopropan-1-ol, 3-methylaminopropan-1-ol, and 3-dimethylaminopropan-1-ol, unambiguously showed that the methylation at the N atom results in a systematic enhancement of the OH···N IMHB, reflecting a decrease of the s-character of the nitrogen lone pair orbital.
These results clearly indicate that the effects of substituents directly attached to the active sites of the OH···N IMHB have a significant effect in its strength, but, to the best of our knowledge, not much is known on the effects induced by substituents that are at the α-position of both the hydroxyl or the amino group, or what the effects are when any of these two groups are also actively participating in a non-covalent interaction with a second body. Hence, one of the aims of this paper is to investigate, through the use of high-level ab initio calculations, the characteristics of the OH···N IMHB in the series of HOCHX(CH2)nCH2NH2 and HOCH2(CH2)nCHXNH2 (n = 0–5) with substituted amino- alcohols being X = H, F, Cl, Br. As mentioned above, there is an alternative way in which the strength of an IMHB can be altered, and this is through non-covalent interactions in which either the proton donor or the proton acceptor is engaged. Among the many possible non-covalent interactions capable of interacting with IMHBs, the so-called beryllium bonds [46] are particularly interesting. A good and rather complete compilation on the coordination chemistry of beryllium was reported recently by Perera et al. [47]. Beryllium bonds have been found to compete with dihydrogen bonds [48], and they are able to modulate the strength of tetrel bonds [49]. They can also be formed when interacting with other electron deficient systems, such as boron derivatives [50] or with noble gases [51]. Although there are previous studies on the mutual interaction of intermolecular hydrogen bonds and beryllium bonds [52,53], very little has been done dealing with IMHBs. In this respect, a recent theoretical study must be mentioned describing how intramolecular hydrogen bonds are able to enhance tetrel bonds [54], or the competition between pnictogen bonds and intramolecular hydrogen bonds in protonated systems [55]. Here, we have decided to investigate this phenomenon when the system interacting with the amino-alcohol is an electron deficient compound. To achieve this goal, we will investigate the characteristics of the complexes between HOCH2(CH2)nCH2NH2 (n = 0–3) when they interact, either through the amino group or the hydroxyl group, with BeF2 molecules, yielding the corresponding beryllium bonds [46].
2. Computational Details
In order to ensure the reliability of the theoretical characterization of the IMHB stabilizing the compounds under investigation, we have decided to use the Gaussian-4 (G4) theory [56]. The G4 theory is a high-level ab initio composite method based on DFT optimized geometries and thermochemical corrections obtained at the B3LYP/6-31G(2df,p) level [56]. Correlation effects are accounted for by using the Moller–Plesset perturbation theory up to the fourth-order and CCSD(T) coupled cluster theory. A further correction, to account for the Hartree–Fock basis set limit, is added using a linear two-point extrapolation scheme and quadruple-zeta and quintuple-zeta basis sets.
However, it would be impossible to apply the G4 scheme to characterize all possible conformers associated with these two series of substituted amino-alcohols HOCHX(CH2)nCH2NH2 and HOCH2(CH2)nCHXNH2 (n = 0–5, X = H, F, Cl, Br) under scrutiny because this number is huge in particular when n ≥ 3, and it would be applied only to the most stable ones. In order to make the selection of the most stable conformers, we first located the ensemble of them by using the conformer–rotamer ensemble sampling tool (CREST) recently developed by Grimme [57,58]. This method is based on the semiempirical tight-binding based quantum chemistry method GFN2-xTB [59] in the framework of meta-dynamics (MTD). Once the conformers ensemble is obtained, the script ENSO (Energetic Sorting of Crest ensembles) [60] is applied to classify them in a three-step process. The first one consists of a single point calculation at pbeh-3c/Def2-SVP [61] level of theory of the different structures of the ensemble. In a second step, the conformers in the range of 25 kJ/mol are selected for optimization at the same level of theory. In the last step, the range of energy is reduced to about 8 kJ/mol, the final energy, and the percentage of Boltzmann distribution being obtained from a single point wb97x/Def2-TZVPP calculations [62]. The conformers with a percentage higher than 1% were chosen to carry out the G4 calculations. It should also be mentioned that, for all the complexes including Beryllium bonds, final energies were also calculated at the G4 level, but, in these cases, instead of using the standard geometry optimization procedure within the G4 formalism, the structures were optimized using a larger basis set expansion, aug-cc-pVTZ, that includes diffuse functions, very often critical to correctly describe non-covalent interactions.
The bonding in all the systems investigated was analyzed through the use of four different approaches, namely the quantum theory of atoms in molecules (QTAIM) [63,64], the electron localization function (ELF) [65], the natural bond orbital (NBO) analysis [66], and the non-covalent interaction (NCI) formalism [67]. In the QTAIM approach, a topological analysis of the electron density of the systems permits the location of its critical points and the paths of minimum gradients connecting them, which leads to an unambiguous definition of chemical bonds, and/or ring and cage structures. The ELF procedure allows a partition of the molecular space in monosynaptic and disynaptic (or polysynaptic) basins in which the electrons of the system are distributed. The monosynaptic ones are associated with core electrons and/or lone pairs, whereas the disynaptic (or polysynaptic) correspond to bonding regions. The NBO method is based on the generation of localized hybrid orbitals which would correspond to Lewis-type representations of the molecular structures. The calculation of second-order orbital interaction energies between occupied and empty orbitals permit quantitatively characterizing donations and backdonations among occupied and empty localized molecular orbitals involved in inter and/or intramolecular interactions. The NCI formalism is an alternative analysis of the electron density, based on the fact that regions of small reduced-density gradient at low electronic densities are associated with the presence of non-covalent interactions. This analysis leads to rather visual representations if a colored scale is used to plot the isosurfaces of the reduced density, in both two- and three-dimensional spaces.
3. Results and Discussion
In our theoretical survey, we have investigated, for each one of the species under consideration, the possibility of having O–H···N or N–H···O IMHBs, though, in almost all cases, the former are stronger than the latter, and, therefore, in most of the cases, the global minima are characterized by the existence of an O–H···N IMHB.
The number of stable conformers of HOCHX(CH2)nCH2NH2 and HOCH2(CH2)nCHXNH2 (n = 0–5, X = H, F, Cl, Br) compounds obviously increases as the number of carbon atoms increases along the series, reaching for the larger values of n several hundred. In what follows, we will analyze the six more stable unsubstituted HOCH2(CH2)nCH2NH2 (n = 0–5) amino-alcohols first, and, after that, we will pay attention to the effect of the X substituent at the α-position with respect to the CH2OH group and with respect to the CH2NH2 group.
3.1. HOCH2(CH2)nCH2NH2 (n = 0–5) Compounds
We will start our analysis by looking at the effect that the length of the carbon chain has on the strength of the IMHB between the alcohol and the amino functional groups. As expected, the conformer stabilized by the formation of a O–H···N IMHB is more stable (see Figure S1) than that exhibiting a N–H···O IMHB because the hydroxy group is a better proton donor but a weaker base than the amino group, so, in what follows, we will discuss only the systems exhibiting a O–H···N IMHB.
The structures corresponding to the most stable conformer for each value of n are shown in Figure 1. It can be seen that the longest O–H···N IMHB corresponds to the first member of family, 2-amino ethanol (n = 0) which should be the amino-alcohol with the weakest bond along the series. This bond length reaches its minimum for n = 2, which in principle should be the member of the series with the strongest O–H···N IMHB, its strength decreasing for larger values of n (n = 0–5).
Unfortunately, although the calculation of the energy of an intermolecular hydrogen bond is straightforward, this is not so when dealing with IMHBs, though some procedures to estimate it have been proposed [68]. We propose, however, the use of the isodesmic reaction [69] (1) as a suitable method to have, at least, a reasonably good estimate of the relative stabilization gained in these compounds when the IMHB is formed. The isodesmic process we have used corresponds to the first reaction shown in Scheme 1.
Reaction (2) in Scheme 1, in which the carbon chain is fully deployed, was used just to check whether, in the isodesmic reactions proposed, the repulsive interaction between the terminal methyl groups may significantly affect the isodesmic energy obtained with the first reaction. The ideal situation should correspond to that in which the second reaction is thermoneutral. The results obtained for these two reactions at the G4 level of theory are summarized in Table 1. It can be observed that, indeed, the reactions (2) for the set of derivatives investigated are nearly thermoneutral, which is an indication of the reliability of our isodesmic reaction (1) to provide a good estimation of the stabilization energy associated with each IMHB. Not surprisingly, the largest deviation for thermoneutrality of reaction (2) is obtained for the first member of the series, as a consequence of its higher rigidity. Nevertheless, it should be taken into account that, in these isodesmic reactions, and, mainly, when the carbon chain is sufficiently large and very flexible, the number of possible conformers for the three compounds that do not have IMHB is very large, and, in many cases, the energy difference between them is very small, even smaller than 1 kJ·mol−1.
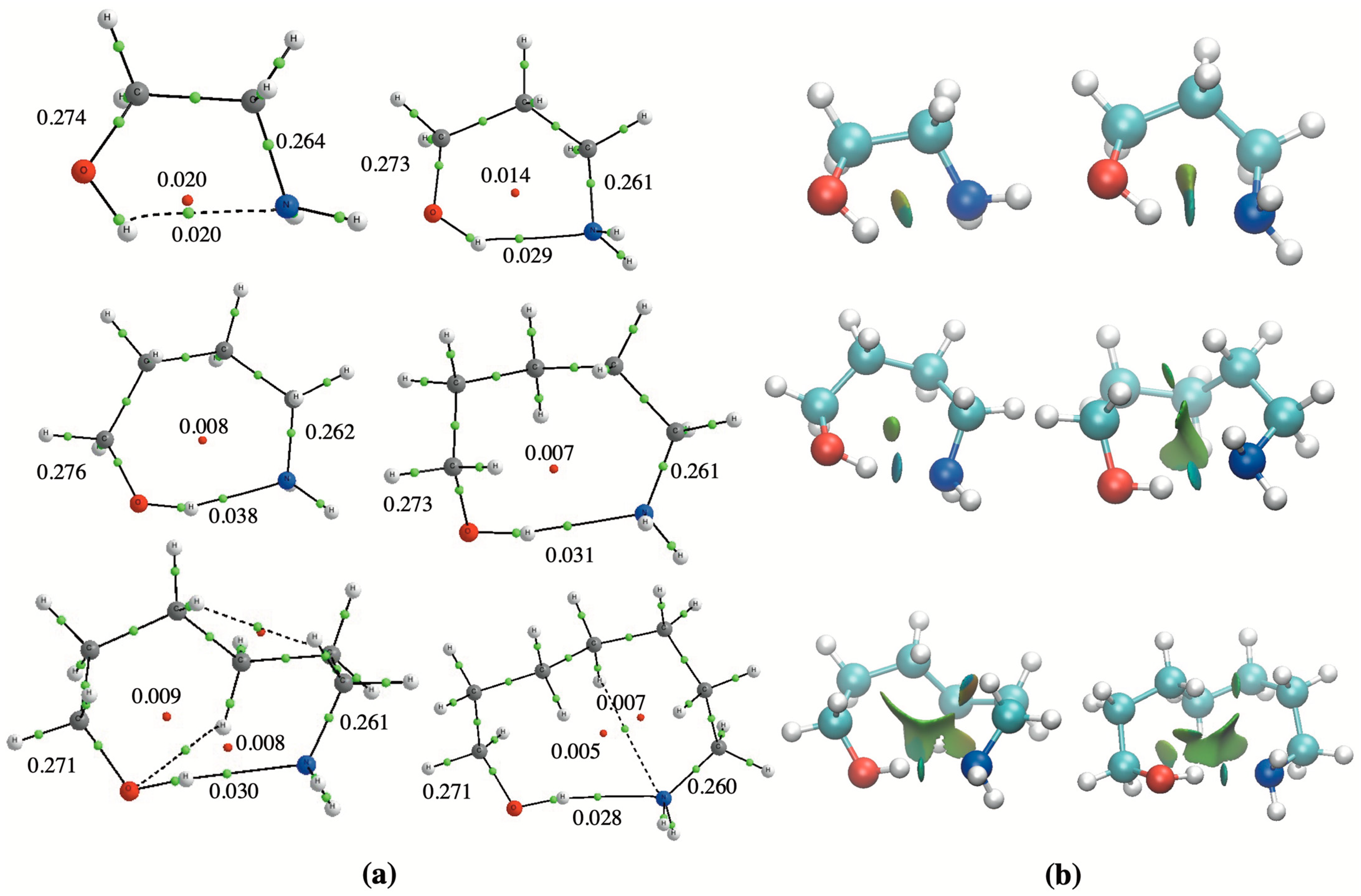
The values in Table 1 show, in agreement with the IMHB length reported in Figure 1, that the strongest isodesmic stabilization interaction energy corresponds to n = 2, being the weakest one corresponding to n = 0. These energetic trends are also consistent with the characteristics of the corresponding molecular graphs drawn in Figure 2a, showing that, in all cases, the electron density at the BCP located between the H atom of the hydroxyl group and the N atom of the amino group goes through a maximum for n = 2.
The 3D-NCI plots included in Figure 2b also show the existence of an isosurface between the OH and the NH2 groups, which denotes the existence of a NCI whose strength increases from n = 0 (greenish) up to n = 2 (blueish). It is also worth noting that, already for n = 1, a second greenish lobe appears attached to the blueish one associated to the van der Waals interactions range involving the chain of carbon atoms. Indeed, for n = 2, the two isosurfaces are now independent and, for the remaining systems (n = 3–5), the extension of these secondary interactions increases with the length of the chain of carbons.
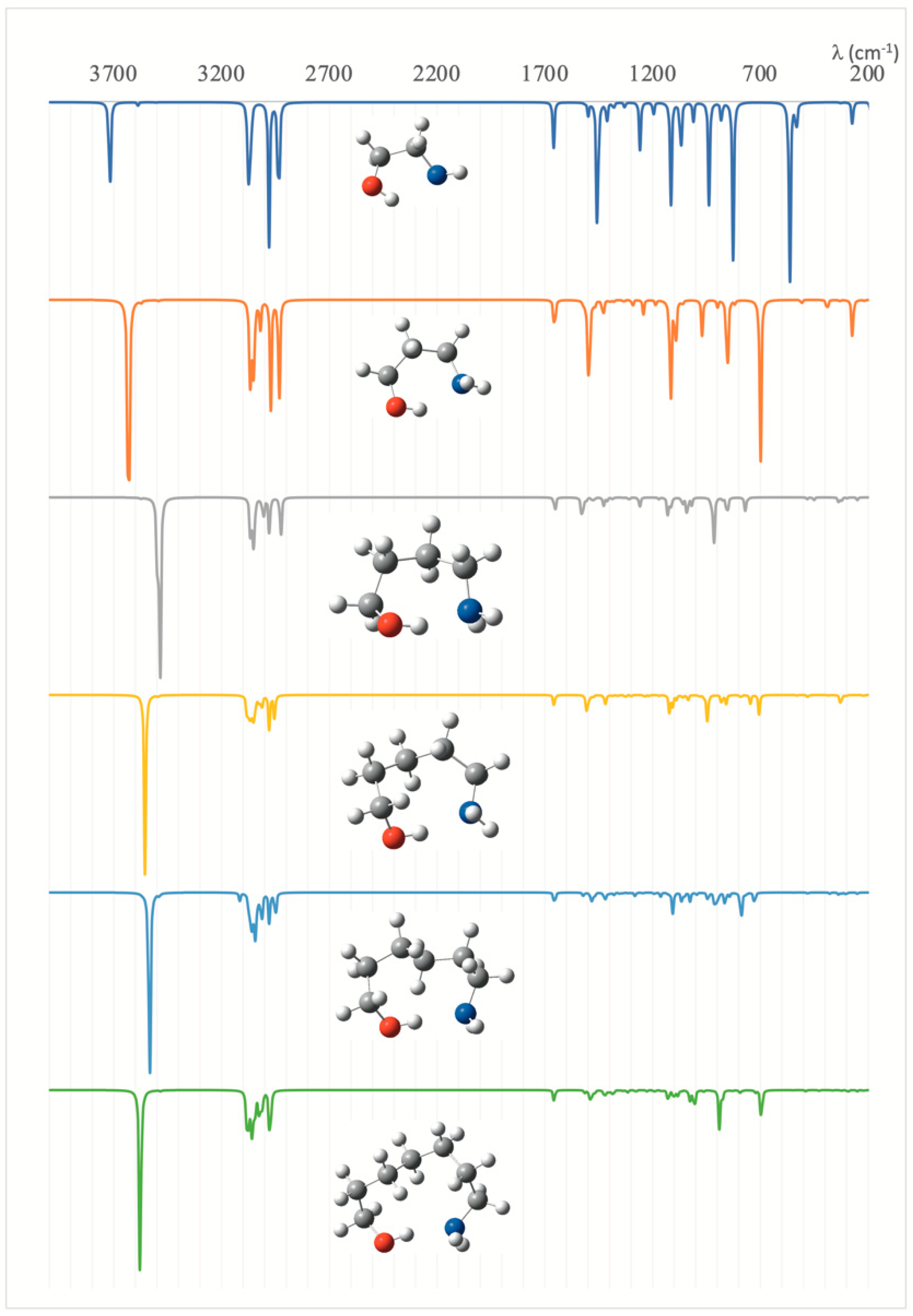
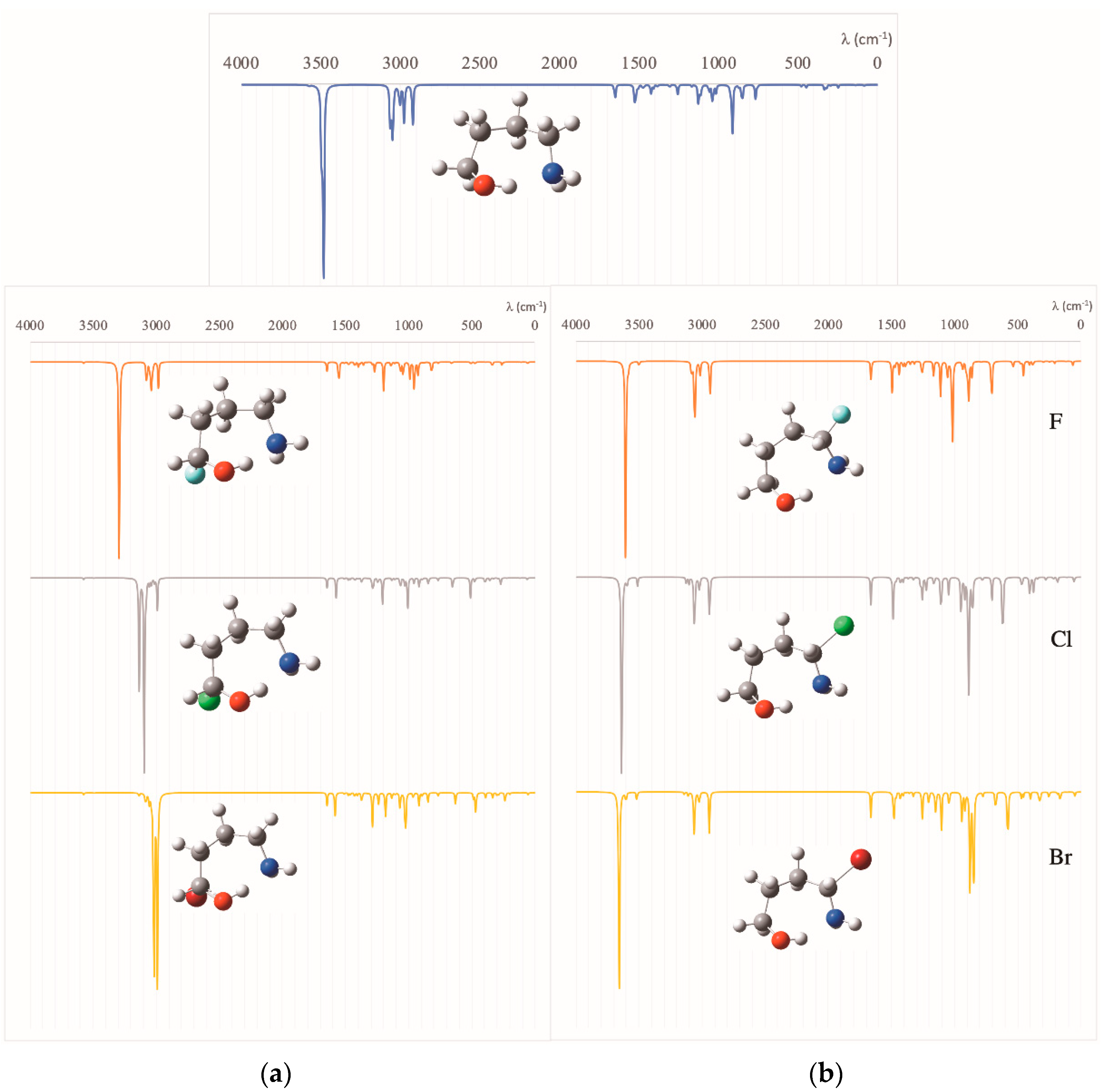
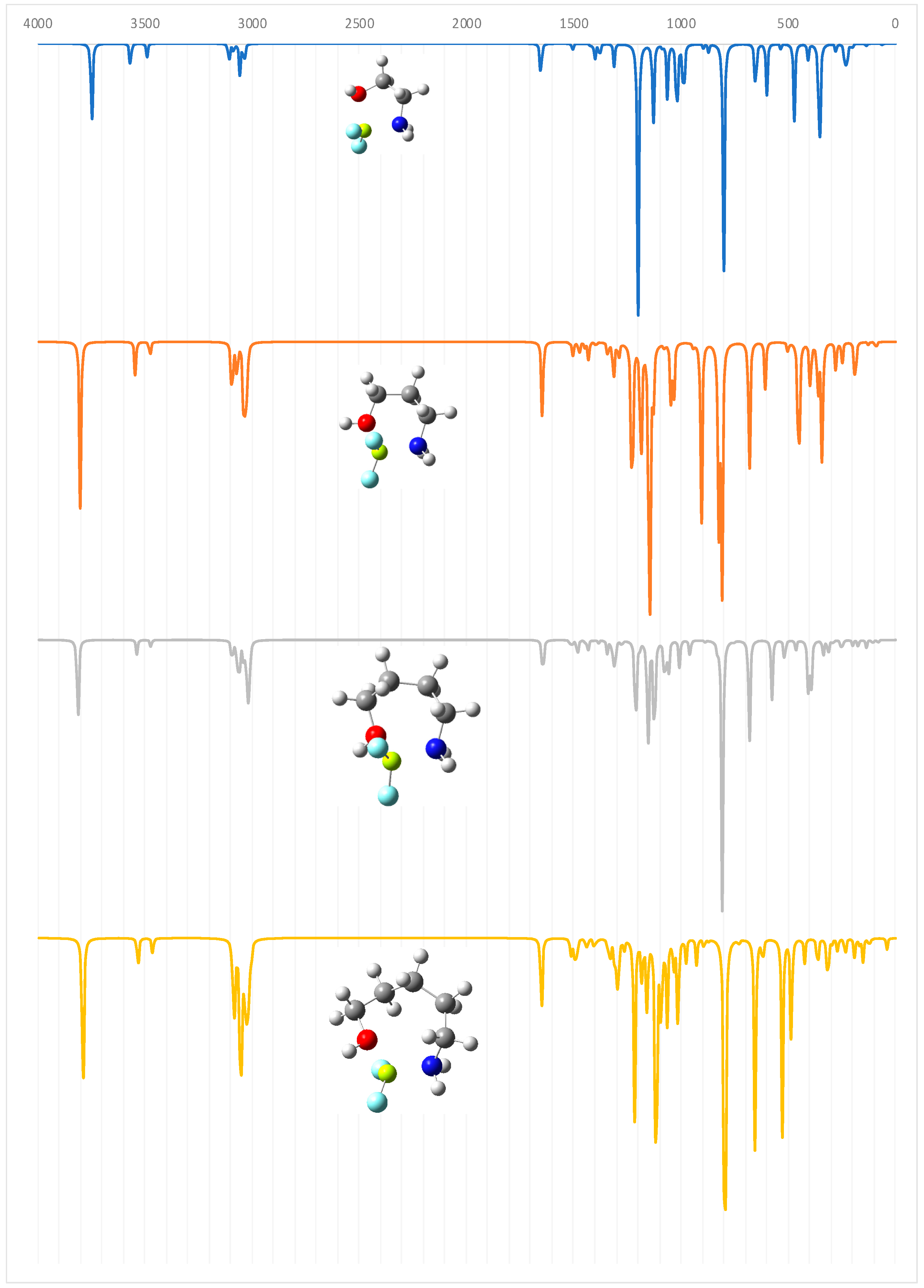
The evolution of the characteristics of the IMHB along the series is nicely reflected in the IR spectra of the different species. As shown in Figure 3, the absorption band associated with the O–H stretching of the alcoholic function, which, for the first compound (n = 0), is predicted to appear at 3715 cm−1, is clearly redshifted when moving to larger compounds. This red-shifting is maximum (233 cm−1) for n = 2, but even for n = 5 the red-shifting with respect to n = 0 is significant (141 cm−1), indicating, in agreement with the other indices that the IMHB for n = 5, though weaker than that for n = 2 is still stronger than for n = 0.
3.2. HOCHX(CH2)nCH2NH2 and HOCH2(CH2)nCHXNH2 (n = 0–5, X = F, Cl, Br) Derivatives
Let us look now at the effects of halogen substituents at the α-position of both the -CH2OH and the -CH2NH2 functional groups. As indicated above, the number of possible conformers is huge, so, in Figures S2 and S3 of the Supplementary Materials, we present only the optimized geometries of the most stable conformers stabilized by an O–H···N or a N–H···O IMHB. As it was also the case for the unsubstituted amino alcohols, and regardless of whether the halogen substituent is at the α-position of the CH2OH group or the CH2NH2 group, the conformers with a N–H···O IMHB are less stable than those with a O–H···N IMHB, with the only exception being the HOCH2CHXNH2 (X = F, Cl, Br) derivatives (see the first row of Figure S3), where the conformer with a N–H···O IMHB is predicted to be, at the G4-level of theory, slightly more stable than the conformer exhibiting a O–H···N IMHB regardless of the nature of the substituent X.
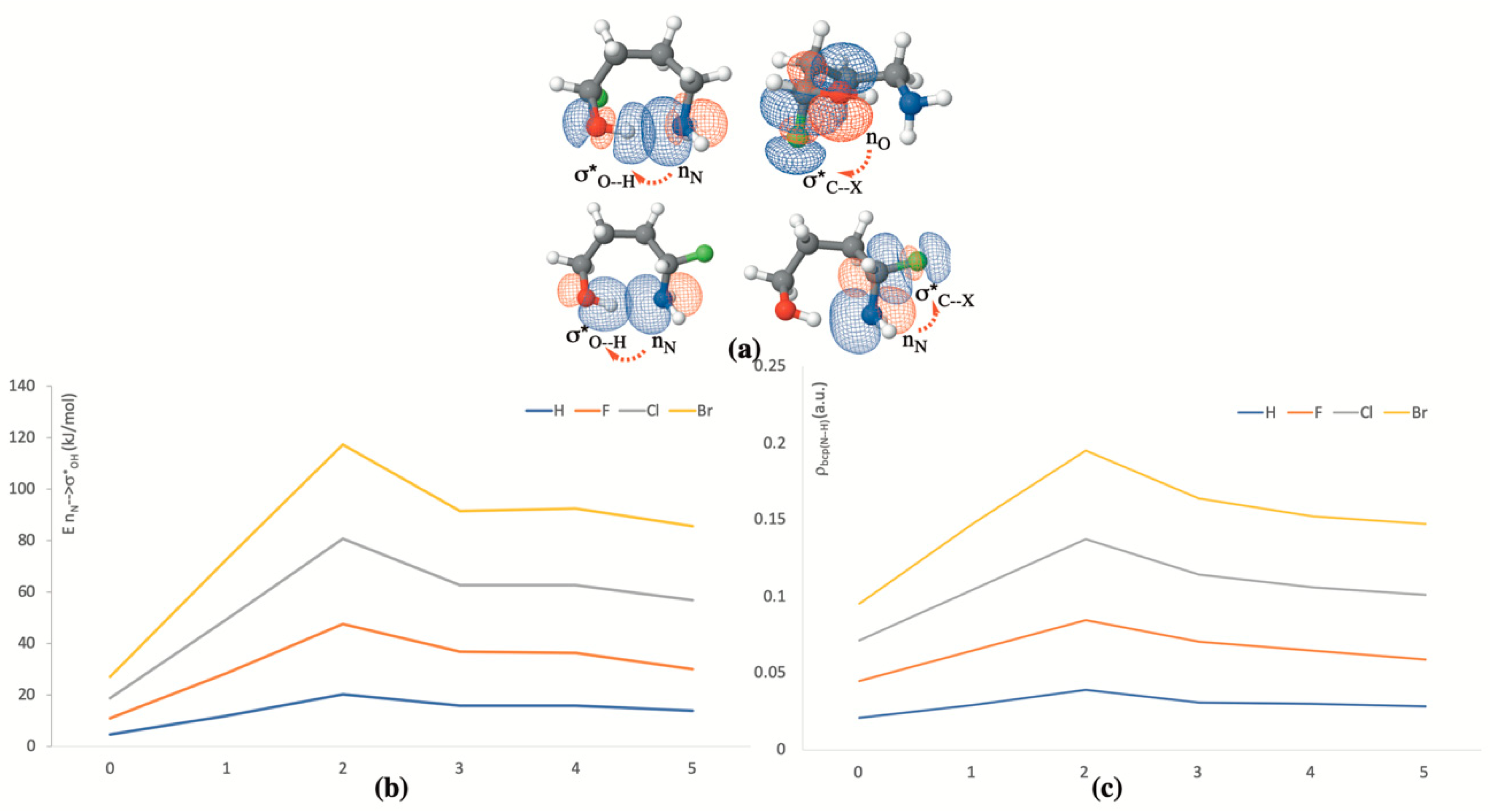
Focusing our attention then on the compounds stabilized by O–H···N IMHB, it can be observed that, as it was the case for the unsubstituted amino-alcohols, when the halogen substituent (F, Cl or Br) is at the α-position of the CH2OH group, the IMHB length decreases from n = 0 to n = 2, where it reaches its minimum value. An NBO analysis of these compounds not only indicates (see Figure 4a) that, as expected, the most significant orbital interactions involve the occupied lone pair of the amino group and the empty antibonding σ*O–H orbital, which necessarily results in a weakening of the O–H bond, but also (see Figure 4b) that this interaction reaches its maximum, regardless of the nature of the substituent (F, Cl or Br) for n = 2. It is also very important to note that the same picture shows that this effect increases following the sequence: H < F < Cl < Br, parallel to the energy of the antibonding σ*O–H orbital decreases, favoring the charge donation from the nitrogen lone-pair. Consistently, the electron densities at the O–H BCP decrease, but those at the IMHB BCP (See Figure 4c) increase following the sequence H < F < Cl < Br (see Figure 4c).
The effects on the IMHB characteristics by introducing the halogen substituent (F, Cl or Br) at the α-position of the CH2NH2 group are just the opposite as those just discussed for α-substitution with respect to the CH2OH group. Indeed, the presence of an electronegative atom at α-position of the CH2NH2 group results in a reduction of the intrinsic basicity of the amino group, which accordingly becomes a poorer donor towards the σ*O–H orbital, leading to O–H···N IMHB weaker than in the unsubstituted compounds. Accordingly, this IMHB becomes about 10% longer. Again, the substituent effect increases as H < F < Cl < Br, and, therefore, whereas for substituents α to the CH2OH group the strongest O–H···N IMHB is observed for the Br derivative, when the substituent is α to the CH2NH2 group, the Br derivative exhibits the weaker IMHB. This behavior is in perfect agreement with the values of the electron densities at the BCP associated with the IMHB, as shown in Table 2 for the particular case n = 2 being taken as a suitable example.
These different trends are also nicely reflected in the characteristics of the corresponding IR spectra. We are going to illustrate this point again using the case n = 2 as a suitable example. As shown in Figure 5a, for the case in which the substitution takes place at the α-position with respect to the CH2OH group, the absorption band associated with the O–H stretching frequency is clearly red shifted upon F, Cl, and Br substitution. If the substitution takes place at the α-position with respect to the CH2NH2 group (see Figure 5b), the O–H stretching band is now blue shifted when H is replaced by F, Cl, Br.
3.3. Complexes between HOCH2(CH2)nCH2NH2 (n = 0–3) and BeF2
In this section, we will examine the effect that the interaction of the amino-alcohols HOCH2(CH2)nCH2NH2 (n = 0–3) with a strong Lewis acid as BeF2 will have on the IMHBs stabilizing these compounds and characterized in Section 3.1. However, the presence of the beryllium derivative opens up new scenarios in which new IMHBs enter into play as well as the possibility of having beryllium bonds replacing the IMHBs which characterize the isolated amino-alcohols.
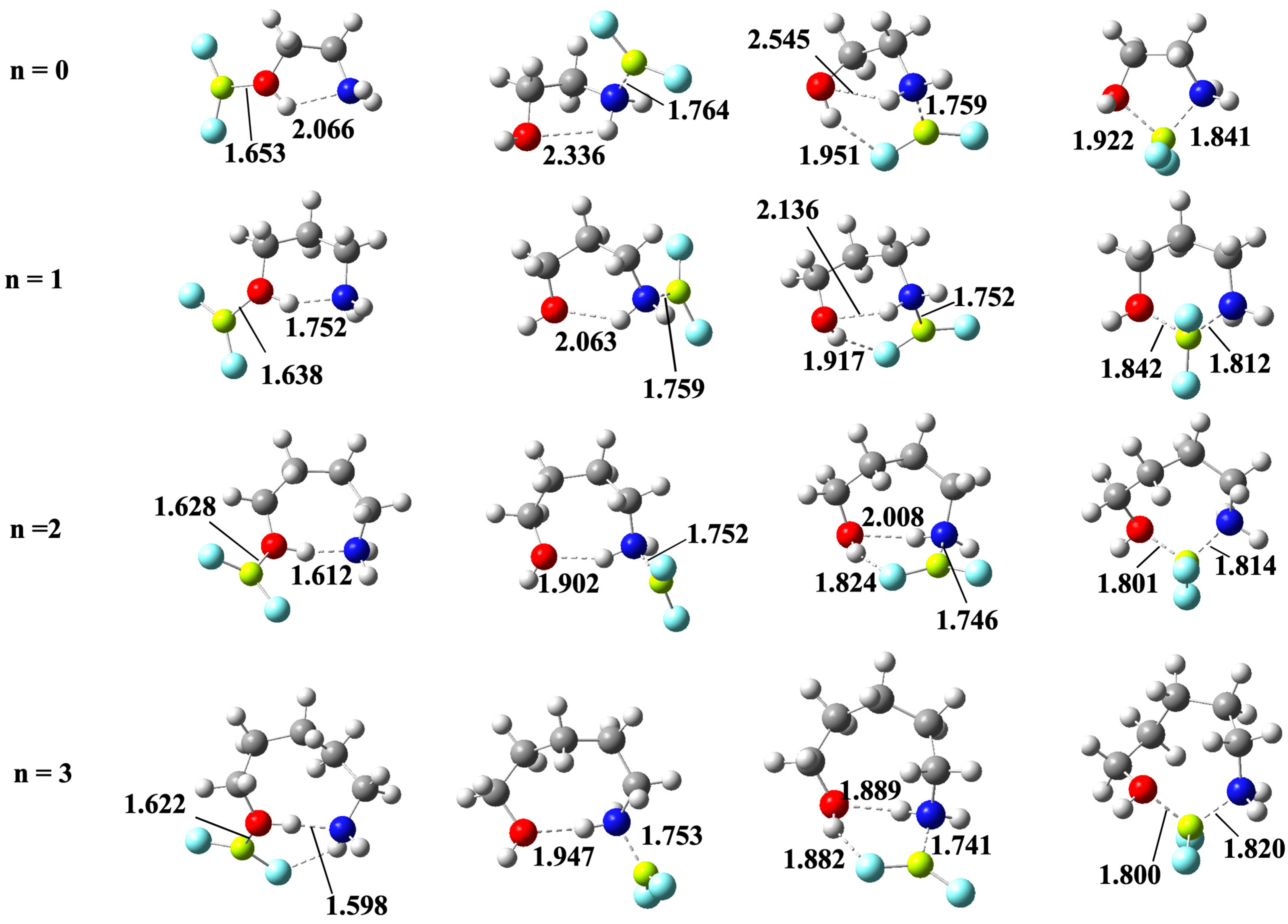
Indeed, the association of BeF2 to the alcohol functional group (see the first column of Figure 6) leads to the amino-alcohols moiety structures being very similar to those exhibited by the isolated amino-alcohol, though the bond length of the OH···N IMHB is much shorter than the one found in Section 3.1 for the isolated system.
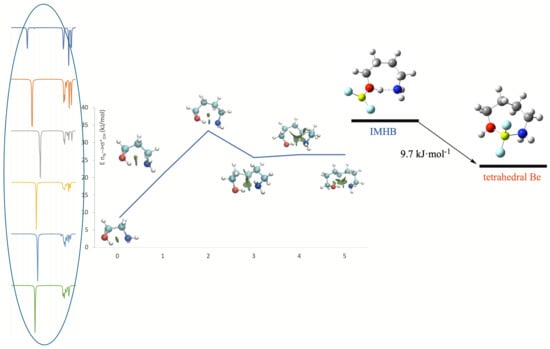
This reinforcement of the OH···N IMHB is the obvious consequence of the significant electron density transfer from the oxygen atom of the hydroxy group to the empty orbitals of the Be atom to form the corresponding beryllium bond, which necessarily increases the proton donor character of the OH group. Indeed, the second-order orbital interaction energies between the nitrogen lone-pair and the σ *O–H antibonding orbital, responsible for the formation of the O–H···N IMHB, are about four times larger for the BeF2 complexes than for the isolated amino-alcohol as shown in Table S1 of the Supplementary Materials. The effect is qualitatively similar when BeF2 interacts with the amino group (second column of Figure 6), which, upon beryllium association, also becomes a stronger proton donor, reinforcing the N–H···O IMHB, but necessarily to a lesser extent than when the group involved in the alcohol function, with the only exception of the amino-ethanol, in which case the conformer with a N–H···O IMHB is predicted to be 9 kJ·mol−1 more stable than the one stabilized by the formation of a O–H···N IMHB (see Table 3).
It is also worth noting that, when the complex is stabilized through the formation of a N–H···O IMHB, the possible formation of a second IMHB by the interaction of the O–H group with one of the fluorine atoms attached to beryllium is open, and, indeed, as shown in Table 3, this new conformer is systematically (from 6 to 11 kJ·mol−1) more stable (compare the third and the fourth column of Table 3). Nevertheless, the most important finding is that, in all cases, the global minimum (fifth column of Table 3) corresponds to a complex in which no IMHBs are formed because the interaction of the O and N atoms of the amino-alcohol with beryllium atom of BeF2 molecule forming the corresponding beryllium bonds is energetically more favorable. The propensity of Be to exhibit a tetrahedral coordination has been previously reported in the literature when competing with intermolecular hydrogen bonds [47,70]. Here, we find that this tendency of Be to behave like a “tetrahedral proton” [47,70] is also observed when competing with IMHBs. In addition, importantly, the enthalpy gap between these bridged structures and the most stable conformers exhibiting a O–H···N IMHB is big enough (8.7 to 22.2 kJ·mol−1) as to conclude, using a Boltzmann distribution function, that, at room temperature, practically 100% of the complexes are those stabilized through the formation of beryllium bonds.
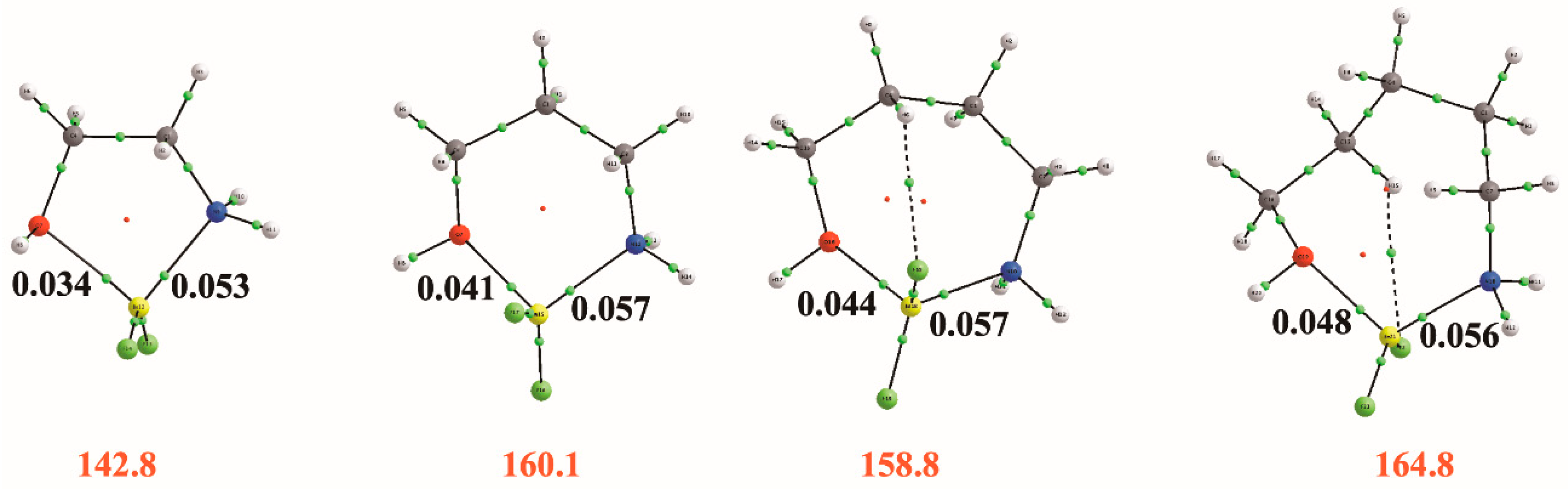
An inspection of the molecular graphs of these complexes (see Figure 7) confirms the tendency of Be to be tetracoordinated, the electron density at the N–Be beryllium bond being systematically larger than that at the O–Be beryllium bond, since the amino group is a better electron donor than the hydroxyl one. On the other hand, the intrinsic stability of the complex, measured by the corresponding binding energy, increases from the amino-ethanol to the amino-pentanol, but, for the amino-butanol, goes through a little sinkhole that is consistent with the fact that, as we have discussed before, the stabilization produced in the isolated compound by the O–H···N IMHB is maximum for amino-butanol.
Finally, it is interesting to highlight some of the peculiarities of the IR spectra of these complexes, showed in Figure 8.
The first conspicuous fact is that the stretching O–H absorption band appears significantly blue-shifted with respect to those in Figure 5a, as it corresponds to a free OH group that, in these systems, does not participate in hydrogen bonding. Indeed, the OH stretching frequency calculated for the bridge complexes in Figure 8 go from 3767 to 3832 cm−1, values rather similar to that obtained for methanol (3831 cm−1) at the same level of theory. However, for the amino-ethanol, the blue-shifting, with respect to the isomer in Figure 5a, is only 200 cm−1 because of the rather weak O–H···N IMHB in the isomer of Figure 5a. For amino-butanol and amino-pentanol, the shifting is greater than 1100 cm−1. Another common finger-print of the IR spectra of these complexes is the presence of a rather intense band always around 800 cm−1 corresponding to the symmetric stretching of the BeF2 moiety.
4. Conclusions
From our G4 calculations, we can conclude that, when F, Cl, or Br atoms are introduced as substituents at the α-position of both the hydroxyl or the amino group of amino-alcohols, there is a significant change in the strength of both the O–H···N or the N–H···O IMHBs that can be formed in these compounds. Substitution at the α-position of the hydroxyl group results in a significant reinforcement of the O–H···N, which follows the trend H < F < Cl < Br, the effects on the N–H···O IMHB being the opposite. Consistently, substitution at the α-position of the amino group also results in a reinforcement of the N–H···O IMHB, but the effect is weaker than the one affecting the O–H···N IMHB. Accordingly, the global minima for the unsubstituted and the substituted amino-alcohols are stabilized by the formation of O–H···N IMHBs. In addition, for both sets, the strength of the O–H···N IMHB, as shown by the characteristics of the corresponding molecular graphs, the NBO and NCI analyses, and the stabilization energy estimated through the use of isodesmic reactions, goes through a maximum for n = 2 (amino-butanol).
The scenario changes completely when the HOCH2(CH2)nCH2NH2 (n = 0–3) amino-alcohols interact with a typical electro-deficient compound such as BeF2. The interaction of BeF2 with either the hydroxyl or the amino group of the amino-alcohol changes the strength of the IMHBs dramatically. In the first case, the O–H···N IMHB becomes much stronger, whereas, in the second case, it is the N–H···O IMHB that becomes very much reinforced, though the effect is still weaker than the one observed for the O–H···N IMHB. In this second case, interaction of BeF2 with the amino group, a new possibility is open through the formation of an additional IMHB between the O–H group of the alcoholic function and one of the F atoms of the BeF2 molecule. However, it is the possibility for the beryllium atom to interact simultaneously with the O and the N atoms of the amino-alcohol that leads to the global minimum of the potential energy surface, with the result that the IMHBs are replaced by two beryllium bonds. These complexes stabilized by the formation of two beryllium bonds, O···Be and N···Be, present very peculiar IR spectra, in which the OH stretching band appears in some cases more than 1100 cm−1 blue-shifted with respect to the α-substituted amino-alcohols, whereas a very intense band, around 800 cm−1, corresponding to the symmetric stretching of the BeF2 moiety, is another finger-print of these spectra.
Supplementary Materials
The following are available: Figure S1: Relative stability of the HOCH2(CH2)nCH2NH2 (n = 0–2) aminoalcohols stabilized by the formation of an O–H···N IMHB or a N-H···O IMHB, Figure S2: Molecular structures of the substituted HOCHX(CH2)nCH2NH2 (n = 0–5, X = H, F, Cl, Br) aminoalcohols, Figure S3: Molecular structures of the substituted HOCH2(CH2)nCHXNH2 (n = 0–5, X = F, Cl, Br) aminoalcohols, Table S1: Second-order orbital interactions energies between the nitrogen lone-pair and the s*OH antibonding orbital for the HOCH2(CH2)nCH2NH2 (n = 0–3) amino-alcohols and their O···BeF2 complexes.
Author Contributions
Conceptualization, M.Y. and A.M.L.; methodology, O.M. and A.M.L.; software, A.M.L.; validation, A.M.L., O.M. and M.Y.; formal analysis, A.M.L., O.M. and M.Y.; investigation, A.M.L. and M.Y.; resources, O.M. and M.Y.; data curation, A.M.L.; writing—original draft preparation, M.Y.; writing—review and editing, A.M.L., O.M. and M.Y.; visualization, O.M.; supervision, M.Y. and A.M.L.; project administration, O.M. and M.Y.; funding acquisition, O.M., A.M.L. and M.Y. All authors have read and agreed to the published version of the manuscript.
Funding
This work was carried out with financial support from the Ministerio de Ciencia, Innovación y Universidades of Spain (Project No. PGC2018-094644-B-C21 and PID2019-110091GB-I00 (MICINN), and the PRIES-CM project Ref: Y2020/EMT-6290 from the Comunidad Autónoma de Madrid.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available in the article and in the Supplementary Material.
Acknowledgments
This work was carried out with the financial support from the project PGC2018-094644-B-C21 and PID2019-110091GB-I00 (MICINN) of the Ministerio de Ciencia, Innovación y Universidades of Spain and the PRIES-CM project Ref: Y2020/EMT-6290 from the Comunidad Autónoma de Madrid. The authors would also like to thank the Centro de Computación Científica of the UAM (CCC-UAM) for the generous allocation of computer time and for their continued technical support.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds are not available from the authors.
References
- Pauling, L. The Shared-Electron Chemical Bond. Proc. Natl. Acad. Sci. USA 1928, 14, 359–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latimer, W.M.; Rodebush, W.H. Polarity and Ionization from the Standpoint of the Lewis Theory of Valence. J. Am. Chem. Soc. 1920, 42, 1419–1433. [Google Scholar] [CrossRef] [Green Version]
- Pauling, L.; Brockway, L.O. The Structure of the Carboxyl Group: I. The Investigation of Formic Acid by the Diffraction of Electrons. Proc. Natl. Acad. Sci. USA 1934, 20, 336–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pimentel, G.C.; McClelland, A.L. The Hydrogen Bond; W.H. Freeman: San Francisco, CA, USA, 1960. [Google Scholar]
- Kollman, P.A.; Allen, L.C. Theory of hydrogen bond.Electronic structure and properties of water dimer. J. Chem. Phys. 1969, 51, 3286. [Google Scholar] [CrossRef]
- Witkowski, A. Far-Infrared Spectrum of the Hydrogen Bond in Acetic Acid Dimers. J. Chem. Phys. 1970, 52, 4403. [Google Scholar] [CrossRef]
- Allen, L.C. Simple model of hydrogen bonding. J. Am. Chem. Soc. 1975, 97, 6921–6940. [Google Scholar] [CrossRef]
- Jeffrey, G.A.; Saenger, W. Hydrogen Bonding in Biological Structures; Springer Science and Business Media LLC: Berlin, Germany, 1991. [Google Scholar]
- Jeffrey, G.A. An Introduction to Hydrogen Bonding; Oxford University Press: New York, NY, USA, 1997. [Google Scholar]
- Desiraju, G.R.; Steiner, T. The Weak Hydrogen Bond in Structural Chemistry and Biology; Oxford University Press: Oxford, UK, 1999. [Google Scholar]
- Grabowski, S.J. Hydrogen Bonding—New Insights; Springer: Dordrecht, The Netherlands, 2006; Volume 3. [Google Scholar]
- Han, K.-L.; Zhao, G.-J. Hydrogen Bonding and Transfer in the Excited State; John Wiley& Sons: Chichester, UK, 2011. [Google Scholar]
- Kuo, S.-W. Hydrogen Bonding in Polymeric Materials; Wiley-VCH: Weinheim, Germany, 2018. [Google Scholar]
- Han, K.; Zhao, G. Hydrogen-Bonding Research in Photochemistry, Photobiology, and Optoelectronic Materials; World Scientific Pub. Co. Pte. Lt.: Singapore, 2018. [Google Scholar]
- Grabowski, S.J. Understanding Hydrogen Bonds: Theoretical and Experimental Views; CPI Group, UK Ltd.: Croydon, UK, 2020. [Google Scholar]
- Sidwick, N.V.; Callow, R.K. Abnormal benzene derivatives. J. Chem. Soc. 1924, 125, 527–538. [Google Scholar] [CrossRef]
- Amstrong, H.E. Bigamous Hydrogen—A Protest. Nature 1926, 117, 553–554. [Google Scholar] [CrossRef]
- Lowry, T.M. Optical Rotatory Dispersion. Nature 1926, 117, 271–275. [Google Scholar] [CrossRef] [Green Version]
- Arunan, E.; Desiraju, G.R.; Klein, R.A.; Sadlej, J.; Scheiner, S.; Alkorta, I.; Clary, D.; Crabtree, R.H.; Dannenberg, J.J.; Hobza, P.; et al. Definition of the hydrogen bond (IUPAC Recommendations 2011). Pure Appl. Chem. 2011, 83, 1637–1641. [Google Scholar] [CrossRef]
- Buemi, G.; Zuccarello, F. DFT study of the intramolecular hydrogen bonds in the amino and nitro-derivatives of malonaldehyde. Chem. Phys. 2004, 306, 115–129. [Google Scholar] [CrossRef]
- Gonzalez, L.; Mó, O.; Yáñez, M. Protonation and Deprotonation of Thiomalonaldehyde. The Role of the Intramolecular Hydrogen Bond. In Recent Theoretical and Experimental Advances in Hydrogen Bonded Clusters; Springer Science and Business Media LLC: Berlin, Germany, 2000; pp. 393–402. [Google Scholar]
- West-Nielsen, M.; Dominiak, P.; Wozniak, K.; Hansen, P.E. Strong intramolecular hydrogen bonding involving nitro- and acetyl groups. Deuterium isotope effects on chemical shifts. J. Mol. Struct. 2006, 789, 81–91. [Google Scholar] [CrossRef]
- Nazarparvar, E.; Zahedi, M.; Klein, E. Density Functional Theory (B3LYP) Study of Substituent Effects on O–H Bond Dissociation Enthalpies of trans-Resveratrol Derivatives and the Role of Intramolecular Hydrogen Bonds. J. Org. Chem. 2012, 77, 10093–10104. [Google Scholar] [CrossRef]
- Nazarparvar, E.; Zahedi, M.; Klein, E. Theoretical study of the substituent effects on O–H BDE of trans-resveratrol derivatives in water and benzene: NBO analysis of intramolecular hydrogen bonds. Struct. Chem. 2014, 26, 47–59. [Google Scholar] [CrossRef]
- Galvao, T.L.P.; Rocha, I.M.; da Silva, M.D.M.C.R.; da Silva, M.A.V.R. Energetic Study of 4(3H)-Pyrimidinone: Aromaticity of Reactions, Hydrogen Bond Rules, and Support for an Anomeric Effect. J. Phys. Chem. A 2014, 118, 3360–3366. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.-Y.; Hu, J.-W.; Huang, T.-H.; Chen, K.-Y.; Chou, P.-T. Excited-state intramolecular proton transfer in the kinetic-control regime. Phys. Chem. Chem. Phys. 2020, 22, 22271–22278. [Google Scholar] [CrossRef]
- Laner, J.N.; Junior, H.d.C.S.; Rodembusch, F.S.; Moreira, E.C. New insights on the ESIPT process based on solid-state data and state-of-the-art computational methods. Phys. Chem. Chem. Phys. 2021, 23, 1146–1155. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Ji, D.; Zhao, J. Theoretical insights into photochemical behavior and ESIPT mechanism for 2,6-dimethyl phenyl derivatives. Chem. Phys. Lett. 2021, 767, 138377. [Google Scholar] [CrossRef]
- Hirahara, M.; Nakano, H.; Uchida, K.; Yamamoto, R.; Umemura, Y. Intramolecular Hydrogen Bonding: A Key Factor Controlling the Photosubstitution of Ruthenium Complexes. Inorg. Chem. 2020, 59, 11273–11286. [Google Scholar] [CrossRef] [PubMed]
- Listkowski, A.; Masiera, N.; Kijak, M.; Luboradzki, R.; Leśniewska, B.; Waluk, J. Controlling Emissive Properties by Intramolecular Hydrogen Bonds: Alkyl and Aryl meso-Substituted Porphycenes. Chem. A Eur. J. 2021, 27, 6324–6333. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Wang, Q.; Wang, X.; Wang, M.; Wang, Q.; Shen, H.-M.; Yang, Y.-F.; She, Y.-B. Intramolecular hydrogen bond-induced high chemical stability of metal–organic frameworks. Inorg. Chem. Front. 2020, 7, 3548–3554. [Google Scholar] [CrossRef]
- Li, W.; Chasing, P.; Benchaphanthawee, W.; Nalaoh, P.; Chawanpunyawat, T.; Kaiyasuan, C.; Kungwan, N.; Namuangruk, S.; Sudyoadsuk, T.; Promarak, V. Intramolecular hydrogen bond—Enhanced electroluminescence performance of hybridized local and charge transfer (HLCT) excited-state blue-emissive materials. J. Mater. Chem. C 2021, 9, 497–507. [Google Scholar] [CrossRef]
- Avramopoulos, A.; Jabłoński, M.; Papadopoulos, M.G.; Sadlej, A.J. Linear and nonlinear electric properties and their dependence on the conformation and intramolecular H-bonding: A model study. Chem. Phys. 2006, 328, 33–44. [Google Scholar] [CrossRef]
- Fallas, J.A.; Gonzalez, L.; Corral, I. Density functional theory rationalization of the substituent effects in trifluoromethyl-pyridinol derivatives. Tetrahedron 2009, 65, 232–239. [Google Scholar] [CrossRef]
- Syrén, P.-O.; Le Joubioux, F.; Henda, Y.B.; Maugard, T.; Hult, K.; Graber, M. Proton Shuttle Mechanism in the Transition State of Lipase-Catalyzed N-Acylation of Amino Alcohols. ChemCatChem 2013, 5, 1842–1853. [Google Scholar] [CrossRef] [Green Version]
- Loru, D.; Pena, I.; Alonso, J.L.; Sanz, M.E. Intramolecular interactions in the polarheadgroup of sphingosine: Serinol. Chem. Commun. 2016, 52, 3615–3618. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Zhong, J.; Zhang, Q.; Qian, J.; Song, K.; Ruan, C.; Xu, J.; Ding, K.; Zhang, J. Rational Design and Structure Validation of a Novel Peptide Inhibitor of the Adenomatous-Polyposis-Coli (APC)–Rho-Guanine-Nucleotide-Exchange-Factor-4 (Asef) Interaction. J. Med. Chem. 2018, 61, 8017–8028. [Google Scholar] [CrossRef]
- Giubertoni, G.; Sofronov, O.O.; Bakker, H.J. Effect of intramolecular hydrogen-bond formation on the molecular conformation of amino acids. Commun. Chem. 2020, 3, 1–6. [Google Scholar] [CrossRef]
- Jabłoński, M. Binding of X–H to the lone-pair vacancy: Charge-inverted hydrogen bond. Chem. Phys. Lett. 2009, 477, 374–376. [Google Scholar] [CrossRef]
- Jabłoński, M. QTAIM-Based Comparison of Agostic Bonds and Intramolecular Charge-Inverted Hydrogen Bonds. J. Phys. Chem. A 2015, 119, 4993–5008. [Google Scholar] [CrossRef]
- Penn, R.E.; Curl, R.F. Microwave Spectrum of 2-Aminoethanol: Structural Effects of the Hydrogen Bond. J. Chem. Phys. 1971, 55, 651–658. [Google Scholar] [CrossRef]
- Kelterer, A.-M.; Ramek, M. Intramolecular hydrogen bonding in 2-aminoethanol, 3-aminopropanol and 4-aminobutanol. J. Mol. Struct. Theochem 1991, 232, 189–201. [Google Scholar] [CrossRef]
- Khalil, A.S.; Lavrich, R.J. Intramolecular hydrogen bond stabilized conformation of 3-aminopropanol. J. Mol. Spectrosc. 2020, 370, 111279. [Google Scholar] [CrossRef]
- Khalil, A.S.; Duguay, T.M.; Lavrich, R.J. Conformation and hydrogen bonding in 4-Aminobutanol. J. Mol. Struct. 2017, 1138, 12–16. [Google Scholar] [CrossRef]
- Batista, P.R.; Karas, L.J.; Viesser, R.V.; De Oliveira, C.C.; Gonçalves, M.B.; Tormena, C.F.; Rittner, R.; Ducati, L.C.; De Oliveira, P.R. Dealing with Hydrogen Bonding on the Conformational Preference of 1,3-Aminopropanols: Experimental and Molecular Dynamics Approaches. J. Phys. Chem. A 2019, 123, 8583–8594. [Google Scholar] [CrossRef]
- Yáñez, M.; Sanz, P.; Mó, O.; Alkorta, I.; Elguero, J. Beryllium Bonds, do they exist? J. Chem Theor. Comput. 2009, 5, 2763–2771. [Google Scholar] [CrossRef]
- Perera, L.C.; Raymond, O.; Henderson, W.; Brothers, P.J.; Plieger, P.G. Advances in beryllium coordination chemistry. Coord. Chem. Rev. 2017, 352, 264–290. [Google Scholar] [CrossRef]
- Li, Q.Z.; Liu, X.F.; Li, R.; Cheng, J.B.; Li, W.Z. Competition between dihydrogen bond and beryllium bond in complexes between HBeH and HArF: A huge blue shift of distant H-Ar stretch. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2012, 90, 135–140. [Google Scholar] [CrossRef]
- Liu, M.; Yang, L.; Li, Q.; Cheng, J.; Xiao, B.; Yu, X. Modulating the strength of tetrel bonding through beryllium bonding. J. Mol. Model. 2016, 22, 10. [Google Scholar] [CrossRef] [PubMed]
- Arnold, T.; Braunschweig, H.; Ewing, W.; Kramer, T.; Mies, J.; Schuster, J.K. Beryllium bis(diazaborolyl): Old neighbors finally shake hands. Chem. Commun. 2014, 51, 737–740. [Google Scholar] [CrossRef]
- Zhang, Q.; Chen, M.; Zhou, M.; Andrada, D.M.; Frenking, G. Experimental and Theoretical Studies of the Infrared Spectra and Bonding Properties of NgBeCO3and a Comparison with NgBeO (Ng = He, Ne, Ar, Kr, Xe). J. Phys. Chem. A 2014, 119, 2543–2552. [Google Scholar] [CrossRef]
- Albrecht, L.; Boyd, R.J.; Mó, O.; Yáñez, M. Cooperativity between hydrogen bonds and beryllium bonds in (H2O)nBeX2 (n = 1–3, X = H, F) complexes. A new perspective. Phys. Chem. Chem. Phys. 2012, 14, 14540–14547. [Google Scholar] [CrossRef]
- Mó, O.; Yáñez, M.; Alkorta, I.; Elguero, J. Modulating the Strength of Hydrogen Bonds through Beryllium Bonds. J. Chem. Theory Comput. 2012, 8, 2293–2300. [Google Scholar] [CrossRef] [PubMed]
- Trujillo, C.; Alkorta, I.; Elguero, J.; Sánchez-Sanz, G. Cooperative Effects in Weak Interactions: Enhancement of Tetrel Bonds by Intramolecular Hydrogen Bonds. Molecules 2019, 24, 308. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Sanz, G.; Trujillo, C.; Alkorta, I.; Elguero, J. Competition between intramolecular hydrogen and pnictogen bonds in protonated systems. Theor. Chem. Accounts 2016, 135, 140. [Google Scholar] [CrossRef]
- Curtiss, L.A.; Redfern, P.C.; Raghavachari, K. Gaussian-4 theory. J. Chem. Phys. 2007, 126, 084108. [Google Scholar] [CrossRef]
- Pracht, P.; Bohle, F.; Grimme, S. Automated exploration of the low-energy chemical space with fast quantum chemical methods. Phys. Chem. Chem. Phys. 2020, 22, 7169–7192. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S. Exploration of Chemical Compound, Conformer, and Reaction Space with Meta-Dynamics Simulations Based on Tight-Binding Quantum Chemical Calculations. J. Chem. Theory Comput. 2019, 15, 2847–2862. [Google Scholar] [CrossRef]
- Bannwarth, C.; Ehlert, S.; Grimme, S. GFN2-xTB-An Accurate and Broadly Parametrized Self-Consistent Tight-Binding Quantum Chemical Method with Multipole Electrostatics and Density-Dependent Dispersion Contributions. J. Chem. Theory Comput. 2019, 15, 1652–1671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimme, S.; Bannwarth, C.; Dohm, S.; Hansen, A.; Pisarek, J.; Pracht, P.; Seibert, J.; Neese, F. Fully automated quantum-chemistry-based computation of spin–spin-coupled nuclear magnetic resonance spectra. Angew. Chem. Int. Ed. 2017, 56, 14763–14769. [Google Scholar] [CrossRef]
- Grimme, S.; Brandenburg, J.G.; Bannwarth, C.; Hansen, A. Consistent structures and interactions by density functional theory with small atomic orbital basis sets. J. Chem. Phys. 2015, 143, 054107. [Google Scholar] [CrossRef]
- Chai, J.-D.; Head-Gordon, M. Systematic optimization of long-range corrected hybrid density functionals. J. Chem. Phys. 2008, 128, 084106. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.F.W. Atoms in Molecules. A Quantum Theory; Clarendon Press: Oxford, UK, 1990. [Google Scholar]
- Popelier, P.L.A. Quantum Chemical Topology: On Bonds and Potentials. In Intermolecular Forces and Clusters I. Structure and Bonding; Wales, D.J., Ed.; Springer: Berlin/Heidelberg, Germany, 2005; Volume 115. [Google Scholar]
- Savin, A.; Nesper, R.; Wengert, S.; Fässler, T.F. ELF: The Electron Localization Function. Angew. Chem. Int. Ed. 1997, 36, 1808–1832. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Contreras-García, J.; Johnson, E.R.; Keinan, S.; Chaudret, R.; Piquemal, J.-P.; Beratan, D.N.; Yang, W. NCIPLOT: A Program for Plotting Noncovalent Interaction Regions. J. Chem. Theory Comput. 2011, 7, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Jabłoński, M.; Kaczmarek, A.; Sadlej, A.J. Estimates of the Energy of Intramolecular Hydrogen Bonds. J. Phys. Chem. A 2006, 110, 10890–10898. [Google Scholar] [CrossRef]
- Muller, P. Glossary of Terms Used in Physical Organic Chemistry. Pure Appl. Chem. 1994, 66, 1077–1184. [Google Scholar] [CrossRef] [Green Version]
- McCleskey, T.M.; Ehler, D.S.; Keizer, T.S.; Asthagiri, D.N.; Pratt, L.R.; Michalczyk, R.; Scott, B.L. Beryllium displacement of H+ from strong hydrogen bonds. Angew. Chem. Int. Edit. 2007, 46, 2669–2671. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
G4 optimized structures for HOCH2(CH2)nCH2NH2 (n = 0–5) amino alcohols, showing the IMHB length in Å.
Figure 1.
G4 optimized structures for HOCH2(CH2)nCH2NH2 (n = 0–5) amino alcohols, showing the IMHB length in Å.

Scheme 1.
Isodesmic reactions to estimate the stabilization produced by the IMHB (reaction (1)) and the repulsive interaction between the terminal methyl groups (reaction (2)).
Scheme 1.
Isodesmic reactions to estimate the stabilization produced by the IMHB (reaction (1)) and the repulsive interaction between the terminal methyl groups (reaction (2)).

Figure 2.
(a) Molecular graphs for HOCH2(CH2)nCH2NH2 (n = 0–5) amino alcohols showing the electron density at the BCPs (green dots) and at the RCPs (red dots); (b) 3D-NCI plots for the same compounds showing the isosurfaces associated with the different intramolecular interactions.
Figure 2.
(a) Molecular graphs for HOCH2(CH2)nCH2NH2 (n = 0–5) amino alcohols showing the electron density at the BCPs (green dots) and at the RCPs (red dots); (b) 3D-NCI plots for the same compounds showing the isosurfaces associated with the different intramolecular interactions.

Figure 3.
Calculated IR spectra for the HOCH2(CH2)nCH2NH2 (n = 0–5) amino alcohols.

Figure 4.
(a) Dominant orbital interactions associated with the formation of O–H···N IMHBs when the substituents are at the α-position with respect to the CH2OH group (first row) and with respect to the CH2NH2 group (second row); (b) variation of the energy involved in these orbital interactions as a function of n for the different substituents considered; (c) variation of the electron density at the IMHB BCP as a function of n for the different substituents considered.
Figure 4.
(a) Dominant orbital interactions associated with the formation of O–H···N IMHBs when the substituents are at the α-position with respect to the CH2OH group (first row) and with respect to the CH2NH2 group (second row); (b) variation of the energy involved in these orbital interactions as a function of n for the different substituents considered; (c) variation of the electron density at the IMHB BCP as a function of n for the different substituents considered.

Figure 5.
Calculated IR spectra for HOCH2(CH2)2CH2NH2 and for compounds: (a) HOCHX(CH2)2CH2NH2 (X = H, F, Cl, Br) and (b) HOCH2(CH2)2CHXNH2 (X = F, Cl, Br).
Figure 5.
Calculated IR spectra for HOCH2(CH2)2CH2NH2 and for compounds: (a) HOCHX(CH2)2CH2NH2 (X = H, F, Cl, Br) and (b) HOCH2(CH2)2CHXNH2 (X = F, Cl, Br).

Figure 6.
B3LYP/aug-cc-pVTZ optimized structures for the complexes between HOCH2(CH2)nCH2NH2 (n = 0–3) amino-alcohols with BeF2 stabilized through OH···N IMHBs (first column), NH···O IMHBs (second column), NH···O and OH···F IMHBs, simultaneously (third column) and through the beryllium bonds involving both the OH and the NH2 groups (fourth column). The length of all these non-covalent interactions is in Å.
Figure 6.
B3LYP/aug-cc-pVTZ optimized structures for the complexes between HOCH2(CH2)nCH2NH2 (n = 0–3) amino-alcohols with BeF2 stabilized through OH···N IMHBs (first column), NH···O IMHBs (second column), NH···O and OH···F IMHBs, simultaneously (third column) and through the beryllium bonds involving both the OH and the NH2 groups (fourth column). The length of all these non-covalent interactions is in Å.

Figure 7.
Molecular graphs for the HOCH2(CH2)nCH2NH2–BeF2 (n = 0–3) bridged conformers showing the electron densities (a.u.) at the BCPs between Be and the two basic sites of the amino-alcohol. The values in red correspond to the binding energies (kJ·mol−1) defined as the energy for the reaction: HOCH2(CH2)nCH2NH2–BeF2 → HOCH2(CH2)nCH2NH2 + BeF2.
Figure 7.
Molecular graphs for the HOCH2(CH2)nCH2NH2–BeF2 (n = 0–3) bridged conformers showing the electron densities (a.u.) at the BCPs between Be and the two basic sites of the amino-alcohol. The values in red correspond to the binding energies (kJ·mol−1) defined as the energy for the reaction: HOCH2(CH2)nCH2NH2–BeF2 → HOCH2(CH2)nCH2NH2 + BeF2.

Figure 8.
Calculated IR spectra for the bridged HOCH2(CH2)nCH2NH2-BeF2 (n = 0–3).


{kind=link}
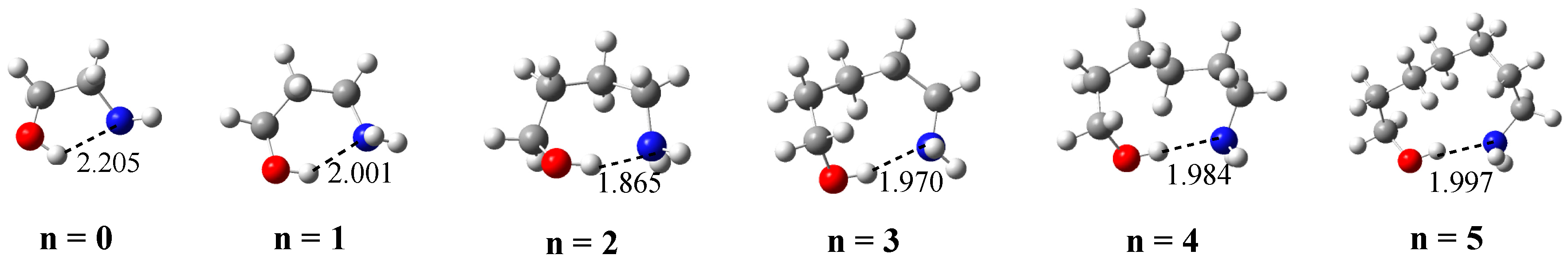{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
G4 calculated enthalpies (kJ·mol−1) for the isodesmic reactions included in Scheme 1.
Table 1.
G4 calculated enthalpies (kJ·mol−1) for the isodesmic reactions included in Scheme 1.
| n | ∆HHB | ∆HInt |
|---|---|---|
| 0 | −8.7 | 1.8 |
| 1 | −16.8 | −0.6 |
| 2 | −24.6 | −0.5 |
| 3 | −16.1 | 0.1 |
| 4 | −16.6 | 0.1 |
| 5 | −17.5 | 0.3 |
Table 2.
Electron densities (a.u.) at the O–H···N IMHB BCP in HOCHX(CH2)2CH2NH2 and HOCH2(CH2)2CHXNH2 (X = H, F, Cl, Br).
Table 2.
Electron densities (a.u.) at the O–H···N IMHB BCP in HOCHX(CH2)2CH2NH2 and HOCH2(CH2)2CHXNH2 (X = H, F, Cl, Br).
| X | HOCHX(CH2)2CH2NH2 | HOCH2(CH2)2CHXNH2 |
|---|---|---|
| H | 0.038 | 0.038 |
| F | 0.046 | 0.032 |
| Cl | 0.053 | 0.031 |
| Br | 0.057 | 0.030 |
Table 3.
Relative stability (kJ·mol−1) of the complexes between HOCH2(CH2)nCH2NH2 (n = 0–3) amino-alcohols and BeF2, stabilized by the different IMHBs indicated in the first row, or through the bridge resulting from the simultaneous interaction of the Be atom with the O and N atoms of the amino-alcohol.
Table 3.
Relative stability (kJ·mol−1) of the complexes between HOCH2(CH2)nCH2NH2 (n = 0–3) amino-alcohols and BeF2, stabilized by the different IMHBs indicated in the first row, or through the bridge resulting from the simultaneous interaction of the Be atom with the O and N atoms of the amino-alcohol.
| n | O–H···N | N–H···O | N–H···O, O–H···F | Bridged |
|---|---|---|---|---|
| 0 | 22.2 | 13.2 | 8.9 | 0.0 |
| 1 | 23.4 | 30.8 | 19.9 | 0.0 |
| 2 | 9.7 | 32.0 | 24.7 | 0.0 |
| 3 | 8.7 | 33.4 | 27.2 | 0.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Lamsabhi, A.M.; Mó, O.; Yáñez, M. Perturbating Intramolecular Hydrogen Bonds through Substituent Effects or Non-Covalent Interactions. Molecules 2021, 26, 3556. https://doi.org/10.3390/molecules26123556
AMA Style
Lamsabhi AM, Mó O, Yáñez M. Perturbating Intramolecular Hydrogen Bonds through Substituent Effects or Non-Covalent Interactions. Molecules. 2021; 26(12):3556. https://doi.org/10.3390/molecules26123556
Chicago/Turabian StyleLamsabhi, Al Mokhtar, Otilia Mó, and Manuel Yáñez. 2021. "Perturbating Intramolecular Hydrogen Bonds through Substituent Effects or Non-Covalent Interactions" Molecules 26, no. 12: 3556. https://doi.org/10.3390/molecules26123556