
Mechanochemical Aza-Vinylogous Povarov Reactions for the Synthesis of Highly Functionalized 1,2,3,4-Tetrahydroquinolines and 1,2,3,4-Tetrahydro-1,5-Naphthyridines



Abstract
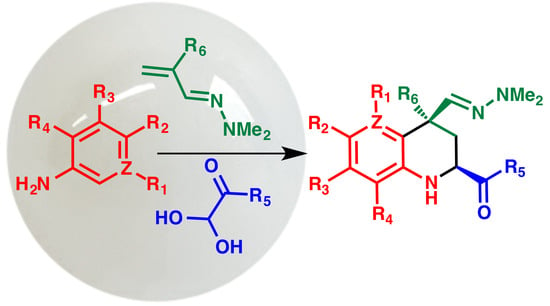
:
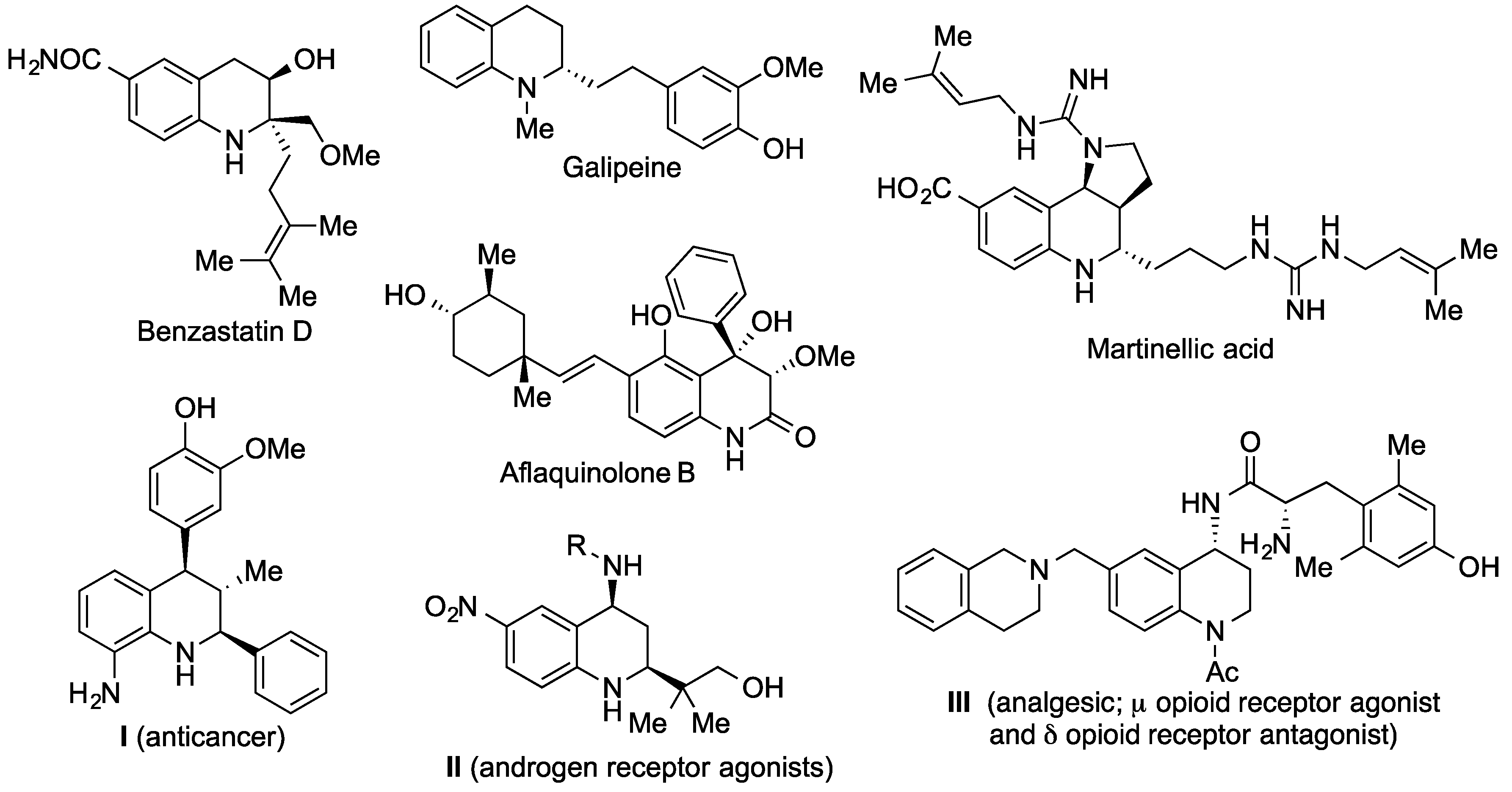
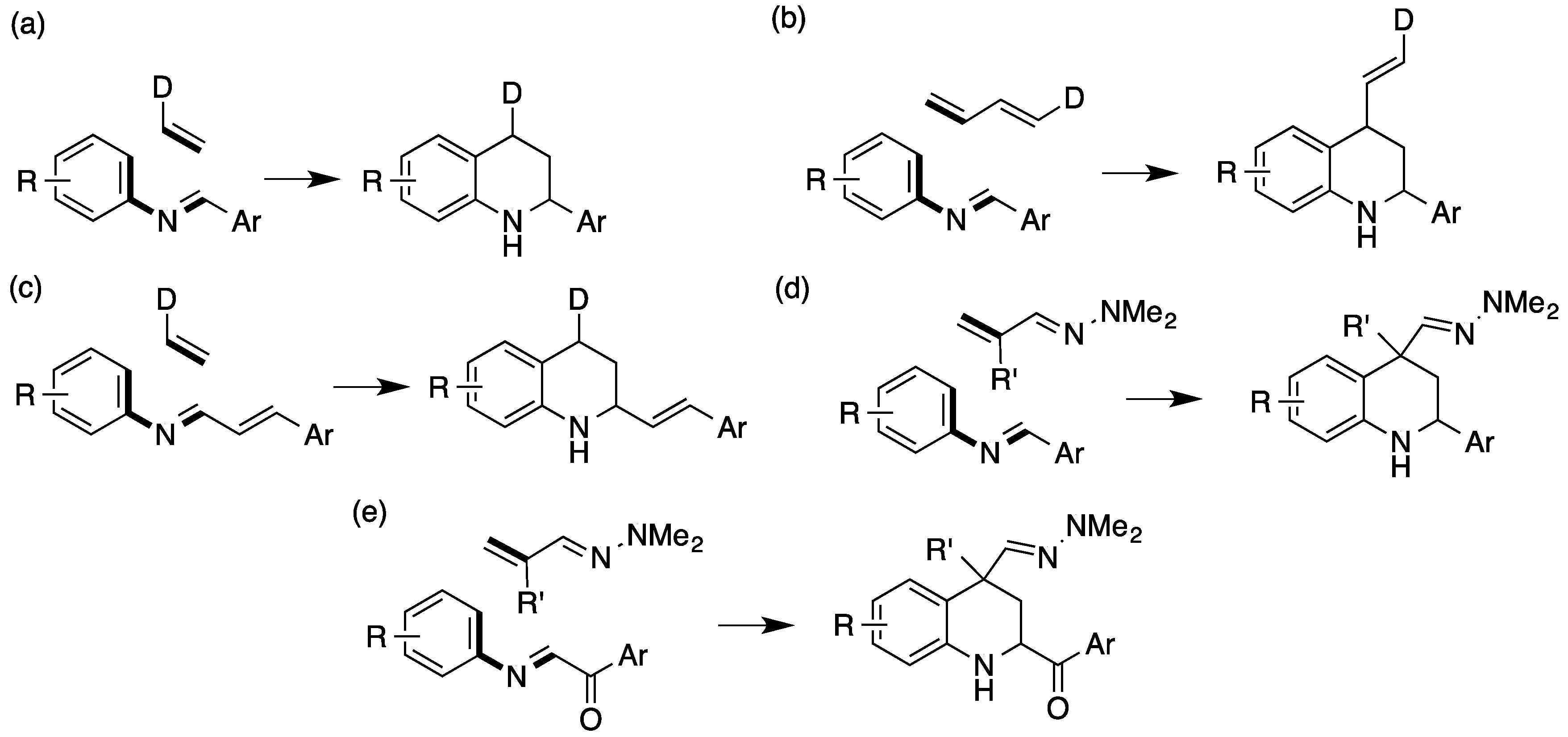
1. Introduction
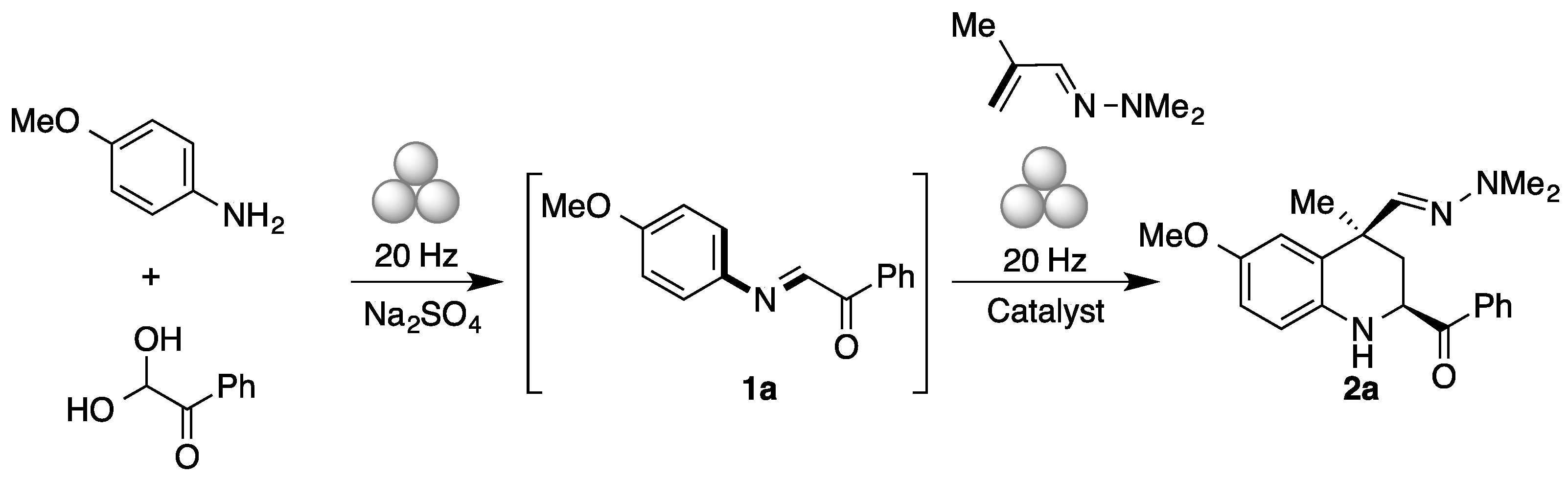
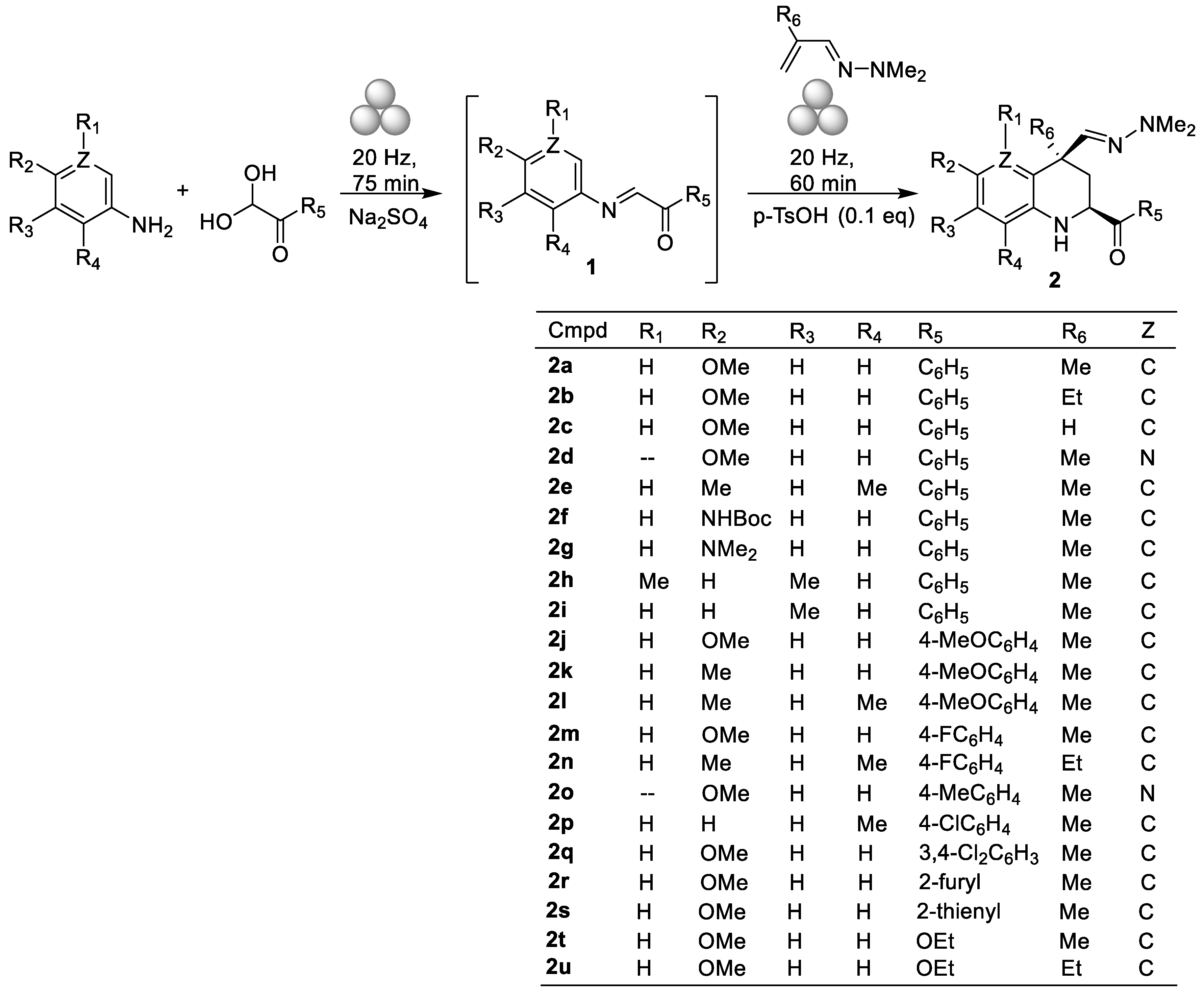
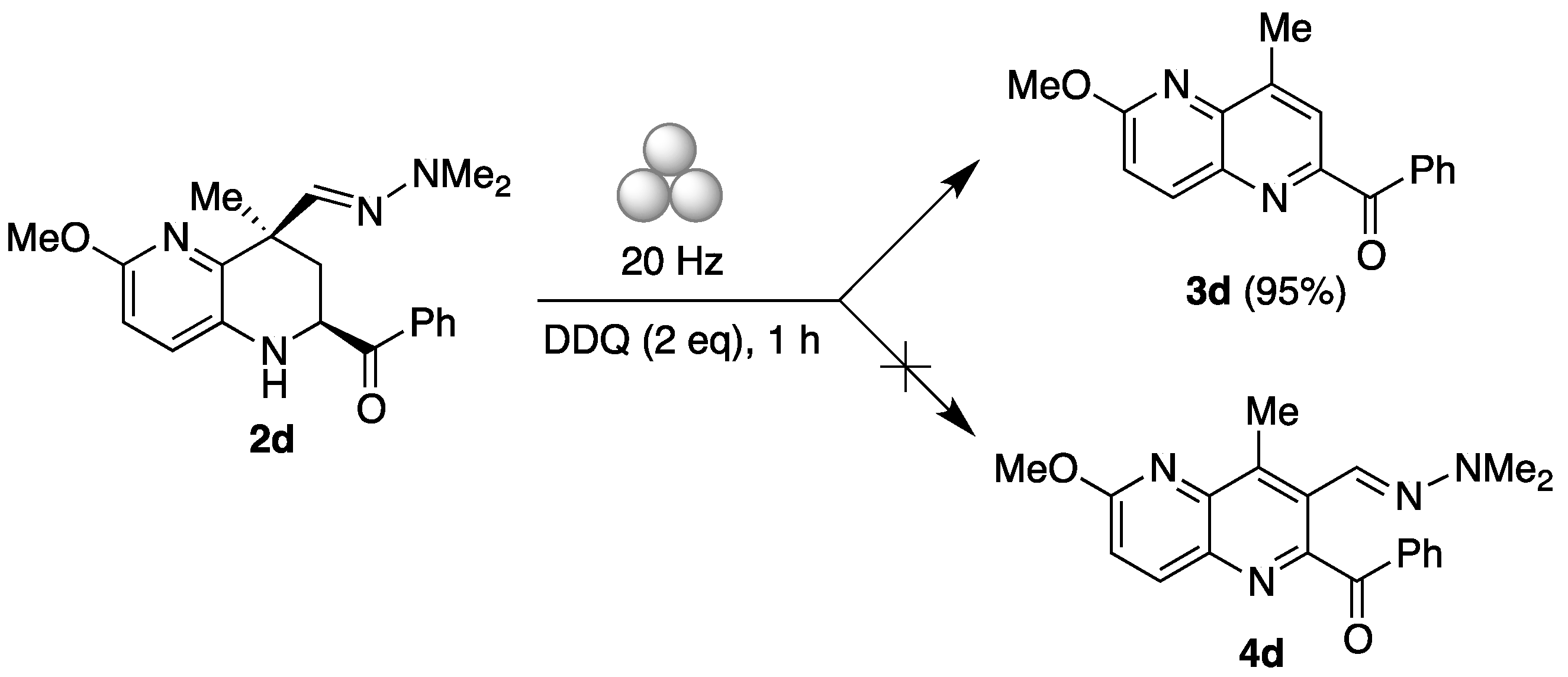
2. Results and Discussion
3. Materials and Methods
3.1. General Experimental Information
3.2. General Procedure for the Synthesis of 2-acyl-1,2,3,4-tetrahydroquinolines and 2-acyl-1,2,3,4-tetrahydro-1,5-naphthyridines (Compounds 2)
3.2.1. (±)-(2. R,4R)-2-Benzoyl-4-((2,2-dimethylhydrazono)methyl)-6-methoxy-4- methyl-1,2,3,4-tetrahydroquinoline (2a)
3.2.2. (±)-(2. R,4R)-2-Benzoyl-4-((2,2-dimethylhydrazono)methyl)-4-ethyl-6-meth- oxy-1,2,3,4-tetrahydroquinoline (2b)
3.2.3. (±)-(2. R,4R)-2-Benzoyl-4-((2,2-dimethylhydrazono)methyl)-6-methoxy-1,2,3,4- tetrahydroquinoline (2c)
3.2.4. (±)-(2. R,4R)-2-Benzoyl-4-((2,2-dimethylhydrazono)methyl)-6-methoxy-4- methyl-1,2,3,4-tetrahydro-1,5-naphthyridine (2d)
3.2.5. (±)-(2. R,4R)-2-Benzoyl-4-((2,2-dimethylhydrazono)methyl)-4,6,8-trimethyl- 1,2,3,4-tetrahydroquinoline (2e)
3.2.6. (±)-(2. R,4R)-tert-Butyl 2-benzoyl-4-((2,2-dimethylhydrazono)methyl)- 4-methyl-1,2,3,4-tetrahydroquinolin-6-yl)carbamate (2f)
3.2.7. (±)-(2. R,4R)-2-Benzoyl-6-(dimethylamino)-4-((2,2-dimethylhydrazono) methyl)-4-methyl-1,2,3,4-tetrahydroquinoline (2g)
3.2.8. (±)-(2. R,4R)-2-Benzoyl-4-((2,2-dimethylhydrazono)methyl)-4,5,7-trimethyl- 1,2,3,4-tetrahydroquinoline (2h)
3.2.9. (±)-(2. R,4R)-2-Benzoyl-4-((2,2-dimethylhydrazono)methyl)-4,7-dimethyl- 1,2,3,4-tetrahydroquinoline (2i)
3.2.10. (±)-(2. R,4R)-2-Benzoyl-4-((2,2-dimethylhydrazono)methyl)-6-methoxy-4- methyl-1,2,3,4-tetrahydroquinoline (2j)
3.2.11. (±)-(2. R,4R)-4-((2,2-Dimethylhydrazono)methyl)-4,6-dimethyl-2-(4-methoxy- benzoyl)-1,2,3,4-tetrahydroquinoline (2k)
3.2.12. (±)-(2. R,4R)-4-((2,2-Dimethylhydrazineylidene)methyl)-2-(4- methoxybenzoyl)-4,6,8-trimethyl-1,2,3,4-tetrahydroquinoline (2l)
3.2.13. (±)-(2. R,4R)-4-((2,2-Dimethylhydrazono)methyl)-2-(4-fluorobenzoyl)-6- methoxy-4-methyl-1,2,3,4-tetrahydroquinoline (2m)
3.2.14. (±)-(2. R,4R)-4-((2,2-Dimethylhydrazono)methyl)-4-ethyl-2- (4-fluorobenzoyl)6,8-dimethyl-1,2,3,4-tetrahydroquinoline (2n)
3.2.15. (±)-(2. R,4R)-4-((2,2-Dimethylhydrazono)methyl)-6-methoxy-4-methyl-2- (4-methylbenzoyl)-1,2,3,4-tetrahydro-1,5-naphthyridine (2o)
3.2.16. (±)-(2. R,4R)-2-(4-Chlorobenzoyl)-4-((2,2-dimethylhydrazono)methyl)- 4,8-dimethyl-1,2,3,4-tetrahydroquinoline (2p)
3.2.17. (±)-(2. R,4R)-2-(3,4-Dichlorobenzoyl)-4-((2,2-dimethylhydrazono)methyl)- 6-methoxy-4-methyl-1,2,3,4-tetrahydroquinoline (2q)
3.2.18. (±)-(2. R,4R)-4-((2,2-Dimethylhydrazono)methyl)-2-(2-furylcarbonyl)- 6-methoxy-4-methyl-1,2,3,4-tetrahydroquinoline (2r)
3.2.19. (±)-(2. R,4R)-4-((2,2-Dimethylhydrazono)methyl)-6-methoxy-4-methyl-2- (thiophen-2-ylcarbonyl)-1,2,3,4-tetrahydroquinoline (2s)
3.2.20. (±)-(2. R,4R)-Ethyl 4-((E)-(2,2-dimethylhydrazono)methyl)-6-methoxy- 4-methyl-1,2,3,4-tetrahydroquinoline-2-carboxylate (2t)
3.2.21. (±)-(2. R,4R)-Ethyl 4-((2,2-dimethylhydrazono)methyl)-4-ethyl-6-methoxy- 1,2,3,4-tetrahydroquinoline-2-carboxylate (2u)
3.3. 6-Methoxy-4-methyl-1,5-naphthyridin-2-yl)(phenyl)methanone (3d)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Kim, W.G.; Kim, J.P.; Kim, C.J.; Lee, K.H.; Yoo, I.D. Benzastatins A, B, C, and D: New free radical scavengers from Streptomyces nitrosporeus 30643. I. Taxonomy, fermentation, isolation, physico-chemical properties and biological activities. J. Antibiot. 1996, 49, 20–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, S.G.; Fletcher, A.M.; Houlsby, I.T.T.; Roberts, P.M.; Thomson, J.E. Structural revision of the hancock alkaloid (−)-galipeine. J. Org. Chem. 2017, 82, 10673–10679. [Google Scholar] [CrossRef] [PubMed]
- Davies, S.G.; Fletcher, A.M.; Houlsby, I.T.T.; Roberts, P.M.; Thomson, J.E.; Zimmer, D. The hancock alkaloids (−)-cuspareine, (−)-galipinine, (−)-galipeine, and (−)-angustureine: Asymmetric syntheses and corrected 1H and 13C NMR data. J. Nat. Prod. 2018, 81, 2731–2742. [Google Scholar] [CrossRef]
- Neff, S.A.; Lee, S.U.; Asami, Y.; Ahn, J.S.; Oh, H.; Baltrusaitis, H.; Gloer, J.B.; Wicklow, D.T. Aflaquinolones A−G: Secondary metabolites from marine and fungicolous isolates of Aspergillus spp. J. Nat. Prod. 2012, 75, 464–472. [Google Scholar] [CrossRef] [Green Version]
- Pappoppula, M.; Aponick, A. Enantioselective total synthesis of (−)-martinellic acid. Angew. Chem. Int. Ed. 2015, 54, 15827–15830. [Google Scholar] [CrossRef]
- Sridharan, V.; Suryavanshi, P.A.; Menéndez, J.C. Advances in the chemistry of tetrahydroquinolines. Chem. Rev. 2011, 111, 7157–7259. [Google Scholar] [CrossRef]
- Muthukrishnan, I.; Sridharan, V.; Menéndez, J.C. Progress in the chemistry of tetrahydroquinolines. Chem. Rev. 2019, 119, 5057–5191. [Google Scholar] [CrossRef] [PubMed]
- Kouznetsov, V.V. Recent synthetic developments in a powerful imino Diels–Alder reaction (Povarov reaction): Application to the synthesis of N-polyheterocycles and related alkaloids. Tetrahedron 2009, 65, 2721–2750. [Google Scholar] [CrossRef]
- Ghashghaei, O.; Masdeu, C.; Alonso, C.; Palacios, F.; Lavilla, R. Recent advances of the Povarov reaction in medicinal chemistry. Drug Discov. Today Technol. 2018, 29, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Sridharan, V.; Avendaño, C.; Menéndez, J.C. CAN-catalyzed vinylogous Povarov reactions: The first three-component synthesis of 2-functionalized tetrahydroquinolines from anilines, cinnamaldehyde and vinyl ethers. Synlett 2007, 18, 1079–1082. [Google Scholar] [CrossRef]
- Sridharan, V.; Perumal, P.T.; Avendaño, C.; Menéndez, J.C. The first aza Diels–Alder reaction involving an α,β-unsaturated hydrazone as the dienophile: Stereoselective synthesis of C-4 functionalized 1,2,3,4-tetrahydroquinolines containing a quaternary stereocenter. Org. Biomol. Chem. 2007, 5, 1351–1353. [Google Scholar] [CrossRef]
- Sridharan, V.; Ribelles, P.; Estévez, V.; Villacampa, M.; Ramos, M.T.; Perumal, P.T.; Menéndez, J.C. New types of reactivity of α,β-unsaturated N,N-dimethylhydrazones: Chemodivergent, diastereoselective synthesis of functionalized tetrahydroquinolines and hexahydropyrrolo[3,2-b]indoles. Chem. Eur. J. 2012, 18, 5056–5063. [Google Scholar] [CrossRef]
- Bianchini, G.; Ribelles, P.; Becerra, D.; Ramos, M.T.; Menéndez, J.C. Efficient synthesis of 2-acylquinolines based on an aza-vinylogous Povarov reaction. Org. Chem. Front. 2016, 3, 412–422. [Google Scholar] [CrossRef]
- Woo, G.C.; Beeler, A.B.; Snyder, J.K. 1,2,3,4-Tetrahydro-1,5-naphthyridines and related heterocyclic scaffolds: Exploration of suitable chemistry for library development. Tetrahedron 2007, 63, 5649–5655. [Google Scholar] [CrossRef]
- Tanaka, K.; Toda, F. Solvent-free Organic Synthesis. Chem. Rev. 2000, 100, 1025–1074. [Google Scholar] [CrossRef]
- Porcheddu, A.; Delogu, F.; De Luca, L.; Colacino, E. From Lossen transposition to solventless “Medicinal Mechanochemistry”. ACS Sustain. Chem. Eng. 2019, 7, 12044–12051. [Google Scholar] [CrossRef]
- Clarke, C.J.; Tu, W.-C.; Levers, O.; Bröhl, A.; Hallett, J.P. Green and sustainable solvents in chemical processes. Chem. Rev. 2018, 118, 747–800. [Google Scholar] [CrossRef]
- Byrne, F.P.; Jin, S.; Paggiola, G.; Petchey, T.H.M.; Clark, J.H.; Farmer, T.J.; Hunt, A.J.; McElroy, C.R.; Sherwood, J. Tools and techniques for solvent selection: Green solvent selection guides. Sustain. Chem. Process. 2016, 4, 7. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, K.; Kaupp, G. Solvent-Free Organic Synthesis, 2nd ed.; Wiley: Hoboken, NJ, USA, 2008; ISBN 978-3527322640. [Google Scholar]
- Zangade, S.; Patil, P. A review on solvent-free methods in organic synthesis. Curr. Org. Chem. 2019, 23, 2295–2318. [Google Scholar] [CrossRef]
- Obst, M.; König, B. Organic synthesis without conventional solvents. Eur. J. Org. Chem. 2018, 4213–4232. [Google Scholar] [CrossRef]
- Erythropel, H.C.; Zimmerman, J.B.; de Winter, T.M.; Petitjean, L.; Melnikov, F.; Ho Lam, C.; Lounsbury, A.W.; Mellor, K.E.; Janković, N.Z.; Tu, Q.; et al. The Green ChemisTREE: 20 years after taking root with the 12 principles. Green Chem. 2018, 20, 1929–1961. [Google Scholar] [CrossRef]
- Hernández, J.G.; Vila-Ortiz, C.G.; Juaristi, E. Useful chemical activation alternatives in solvent-free organic reactions. In Comprehensive Organic Synthesis; Molander, G.A., Knochel, P., Eds.; Elsevier: Oxford, UK, 2014; Volume 9, pp. 287–314. [Google Scholar]
- Claramunt, R.M.; López, C.; Sanz, D.; Elguero, J. Mechano heterocyclic chemistry: Grinding and ball mills. Adv. Heterocycl. Chem. 2014, 112, 117–143. [Google Scholar]
- Margetic, D.; Strukil, V. Mechanochemical Organic Synthesis; Elsevier: Amsterdam, The Netherlands, 2016; ISBN 9780128021842. [Google Scholar]
- Achar, T.K.; Bose, A.; Mal, P. Mechanochemical synthesis of small organic molecules. Beilstein J. Org. Chem. 2017, 13, 1907–1931. [Google Scholar] [CrossRef] [Green Version]
- Leonardi, M.; Villacampa, M.; Menéndez, J.C. Multicomponent mechanochemical synthesis. Chem. Sci. 2018, 9, 2042–2064. [Google Scholar] [CrossRef] [Green Version]
- Howard, J.L.; Cao, C.; Browne, D.L. Mechanochemistry as an emerging tool for molecular synthesis: What can it offer? Chem. Sci. 2018, 9, 3080–3094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, D.; Friščić, T. Mechanochemistry for organic chemists: An update. Eur. J. Org. Chem. 2018, 2018, 18–33. [Google Scholar] [CrossRef]
- Leonardi, M.; Villacampa, M.; Menéndez, J.C. Mechanochemical synthesis of biologically relevant heterocycles. In Green Synthetic Processes and Procedures, Ballini, R., Ed.; Royal Society of Chemistry: London, UK, 2019; Chapter 8; ISBN 978-1-78801-512-7. [Google Scholar]
- Friščić, T.; Mottillo, C.; Titi, H.M. Mechanochemistry for synthesis. Angew. Chem. Int. Ed. 2020, 59, 1018–1029. [Google Scholar] [CrossRef]
- Tan, Y.-J.; Zhang, Z.; Wang, F.-J.; Wu, H.-H.; Li, Q.-H. Mechanochemical milling promoted solvent-free imino Diels–Alder reaction catalyzed by FeCl3: Diastereoselective synthesis of cis-2,4-diphenyl-1,2,3,4-tetrahydroquinolines. RSC Adv. 2014, 4, 35635–35638. [Google Scholar] [CrossRef]
- Tan, Y.-J.; Wang, F.-J.; Asirib, A.A.; Marwanib, H.D.; Zhang, Z. FeCl3-mediated one-pot cyclization–aromatization of anilines, benzaldehydes, and phenylacetylenes under ball milling: A new alternative for the synthesis of 2,4-diphenylquinolines. J. Chin. Chem. Soc. 2018, 65, 65–73. [Google Scholar] [CrossRef]
- Kouznetsov, V.V.; Merchán-Arenas, D.R.; Martínez-Bonilla, C.A.; Marcías, M.A.; Roussel, P.; Gauthier, G.H. Grinding and milling: Two efficient methodologies in the solvent-free phosphomolybdic acid-catalyzed and mechanochemical synthesis of cis-4-amido-N-yl-2- methyltetrahydroquinolines. J. Braz. Chem. Soc. 2016, 27, 2246–2255. [Google Scholar] [CrossRef]
- Andersen, J.M.; Mack, J. Decoupling the Arrhenius equation via mechanochemistry. Chem. Sci. 2017, 8, 5447–5453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuertes, M.; Masdeu, C.; Martín-Encinas, E.; Selas, A.; Rubiales, G.; Palacios, F.; Alonso, C. Fused 1,5-Naphthyridines: Synthetic Toolsand Applications. Molecules 2020, 25, 3508. [Google Scholar] [CrossRef]
- Clerigué, J.; Bianchini, G.; Ribelles, P.; Tejero, T.; Merino, P.; Ramos, M.T.; Menéndez, J.C. Rearrangement reactions in aza-vinylogous Povarov products: Metal-free synthesis of C3-functionalized quinolines and studies on their synthetic application. Eur. J. Org. Chem. 2019, 38, 6452–6464. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Catalyst (eqs.) | Yield (%) | Dr (cis:trans) | Milling Type (Frequency) | Jar Material a |
|---|---|---|---|---|---|
| 1 | − | 31 | 67:33 | HSVM (20 Hz) | ZrO2 |
| 2 | CAN (0.1) | 54 | 69:31 | HSVM (20 Hz) | ZrO2 |
| 3 | FeCl3 (0.1) | 51 | 70:30 | HSVM (20 Hz) | ZrO2 |
| 4 | AlCl3 (0.1) | 49 | 72:28 | HSVM (20 Hz) | ZrO2 |
| 5 | Sc(TfO)3 (0.1) | 72 | 73:27 | HSVM (20 Hz) | ZrO2 |
| 6 | Yb(TfO)3 (0.1) | 73 | 78:22 | HSVM (20 Hz) | ZrO2 |
| 7 | Eu(hfc)3 (0.05) | 35 | 55:45 | HSVM (20 Hz) | ZrO2 |
| 8 | BF3 ·Et2O (0.1) | 69 | 75:25 | HSVM (20 Hz) | ZrO2 |
| 9 | ZnCl2 (0.1) | 60 | 75:25 | HSVM (20 Hz) | ZrO2 |
| 10 | InCl3 (0.1) | 90 | 71:29 | HSVM (20 Hz) | ZrO2 |
| 11 | InCl3 (0.2) | 78 | 69:31 | HSVM (20 Hz) | ZrO2 |
| 12 | InCl3 (0.1) | 72 | 71:29 | HSVM (20 Hz) | ZrO2 b |
| 13 | InCl3 (0.1) | 67 | 71:29 | HSVM (25 Hz) | ZrO2 |
| 14 | InCl3 (0.1) | 45 | 71:29 | HSVM (10 Hz) | Stainless steel c |
| 15 | InCl3 (0.1) | 30 | 71:29 | HSVM (20 Hz) | Stainless steel d |
| 16 | InCl3 (0.1) | 16 | 71:29 | HSVM (30 Hz) | Stainless steel d |
| 17 | InCl3 (0.1) | 22 | 71:29 | PBM (400 rpm) | Stainless steel e |
| 18 | InCl3 (0.1) | 48 | 71:29 | PBM (600 rpm) | Stainless steel e |
| 19 | (±)-CSA (0.1) | 67 | 70:30 | HSVM (20 Hz) | ZrO2 |
| 20 | p-TsOH (0.1) | 90 | 75:25 | HSVM (20 Hz) | ZrO2 |
| 21 | p-TsOH (0.1) | 67 f | 75:25 | HSVM (20 Hz) | ZrO2 |
| 22 | p-TsOH (0.1) | 54 g | 75:25 | HSVM (20 Hz) | ZrO2 |
| 23 | p-TsOH (1) | 87 | 75:25 | HSVM (20 Hz) | ZrO2 |
| 24 | p-TsOH (0.1) | 81h | 75:25 | HSVM (20 Hz) | ZrO2 |
| 25 | p-TsOH (0.1) | 56 i | 75:25 | HSVM (20 Hz) | ZrO2 |
| Cmpd | Mechanochemical Synthesis | Solution Synthesis a | ||||
|---|---|---|---|---|---|---|
| Time, h | Yield, % | Dr, cis:trans | Time, h | Yield, % | Dr, cis:trans | |
| 2a | 1 | 90 | 75:25 | 3 | 72 | 82:18 |
| 2b | 1 | 62 | 75:25 | 3 | 63 | 91:09 |
| 2c | 1 | 67 | 86:14 | 1 | 72 | 100:0 |
| 2d | 1 | 98 | 86:14 | − | − | − |
| 2e | 1 | 76 | 69:31 | 4 | 95 | 98:02 |
| 2f | 1 | 72 | 74:26 | − | − | − |
| 2g | 1 | 59 | 76:24 | − | − | − |
| 2h | 1 | 76 | 55:45 | 5 | 50 | 50:50 |
| 2i | 1 | 41 | 72:28 | − | − | − |
| 2j | 1 | 61 | 73:27 | 3 | 70 | 83:17 |
| 2k | 1 | 71 | 74:26 | − | − | − |
| 2l | 1 | 99 | 75:25 | − | − | − |
| 2m | 1 | 93 | 72:28 | 3 | 75 | 84:16 |
| 2n | 1 | 99 | 75:25 | − | − | − |
| 2o | 1 | 85 | 71:29 | − | − | − |
| 2p | 1 | 60 | 75:25 | − | − | − |
| 2q | 1 | 81 | 82:18 | − | − | − |
| 2r | 1 | 85 | 73:27 | 2 | 88 | 100:0 |
| 2s | 1 | 72 | 70:30 | − | − | − |
| 2t | 1 | 70 | 66:34 | − | − | − |
| 2u | 1 | 64 | 75:25 | 2 | 74 | 87:13 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Clerigué, J.; Ramos, M.T.; Menéndez, J.C. Mechanochemical Aza-Vinylogous Povarov Reactions for the Synthesis of Highly Functionalized 1,2,3,4-Tetrahydroquinolines and 1,2,3,4-Tetrahydro-1,5-Naphthyridines. Molecules 2021, 26, 1330. https://doi.org/10.3390/molecules26051330
Clerigué J, Ramos MT, Menéndez JC. Mechanochemical Aza-Vinylogous Povarov Reactions for the Synthesis of Highly Functionalized 1,2,3,4-Tetrahydroquinolines and 1,2,3,4-Tetrahydro-1,5-Naphthyridines. Molecules. 2021; 26(5):1330. https://doi.org/10.3390/molecules26051330
Chicago/Turabian StyleClerigué, José, M. Teresa Ramos, and J. Carlos Menéndez. 2021. "Mechanochemical Aza-Vinylogous Povarov Reactions for the Synthesis of Highly Functionalized 1,2,3,4-Tetrahydroquinolines and 1,2,3,4-Tetrahydro-1,5-Naphthyridines" Molecules 26, no. 5: 1330. https://doi.org/10.3390/molecules26051330