
Design of Bio-Conjugated Hydrogels for Regenerative Medicine Applications: From Polymer Scaffold to Biomolecule Choice



Abstract
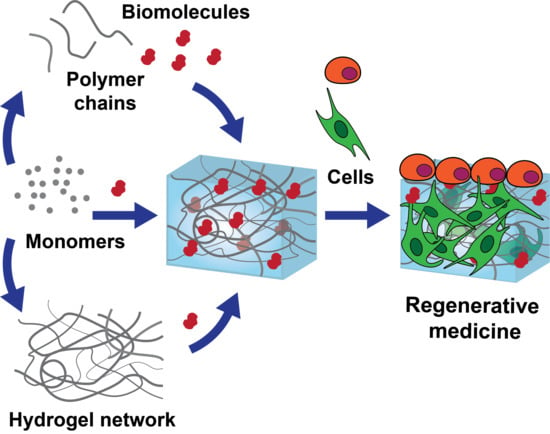
:
1. Introduction
2. General Factors Influencing The Design of Hydrogels for Regenerative Medicine
2.1. Biocompatibility
2.2. Gelling and Mechanical Properties
2.3. Architecture and Shape
2.4. Porosity and Network Density
2.5. Degradability
3. Synthetic Criteria for Hydrogel Production
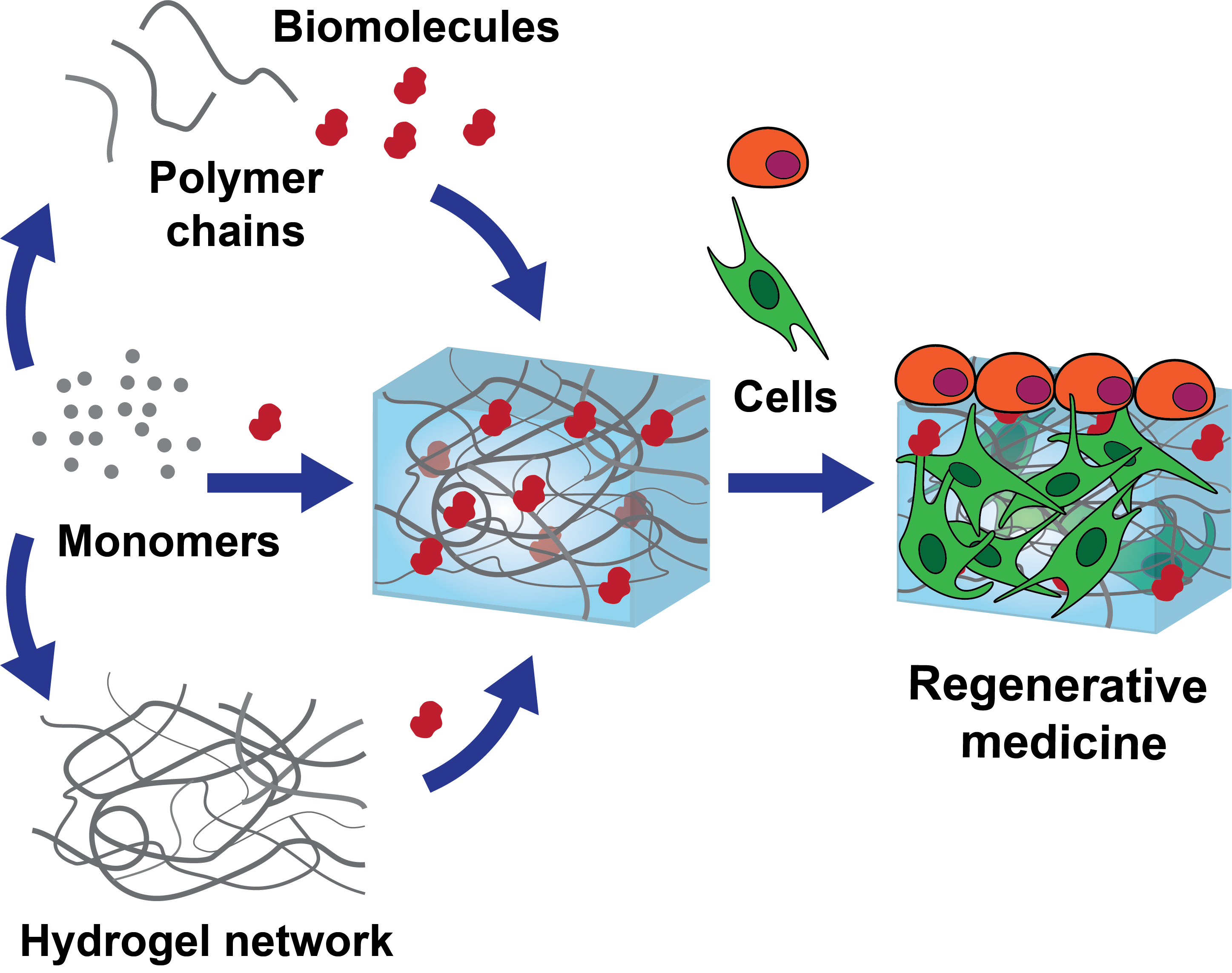
3.1. Polymer Choice
3.2. Polymerization Conditions
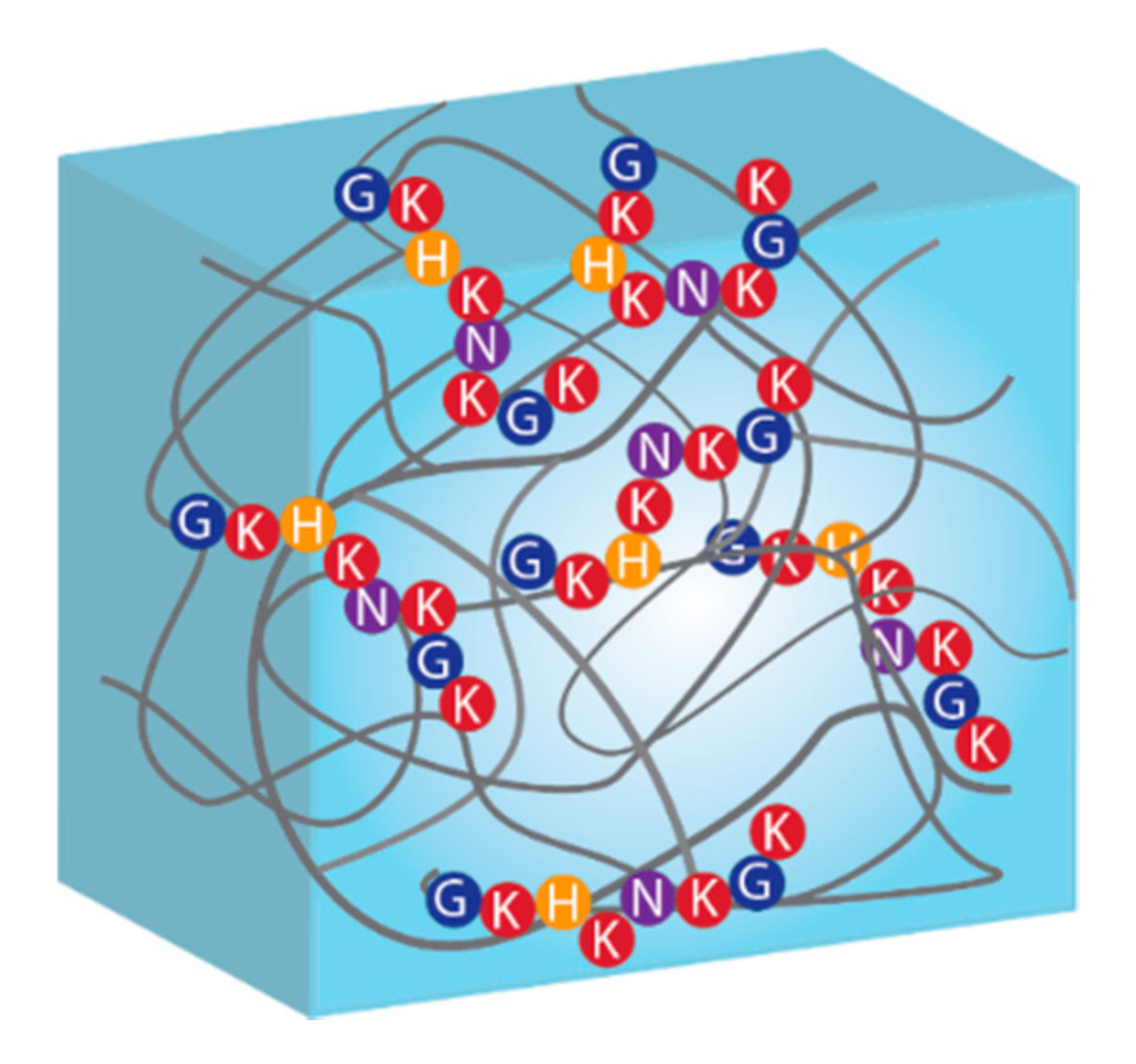
4. Introduction of Biomolecules within the Hydrogel Network
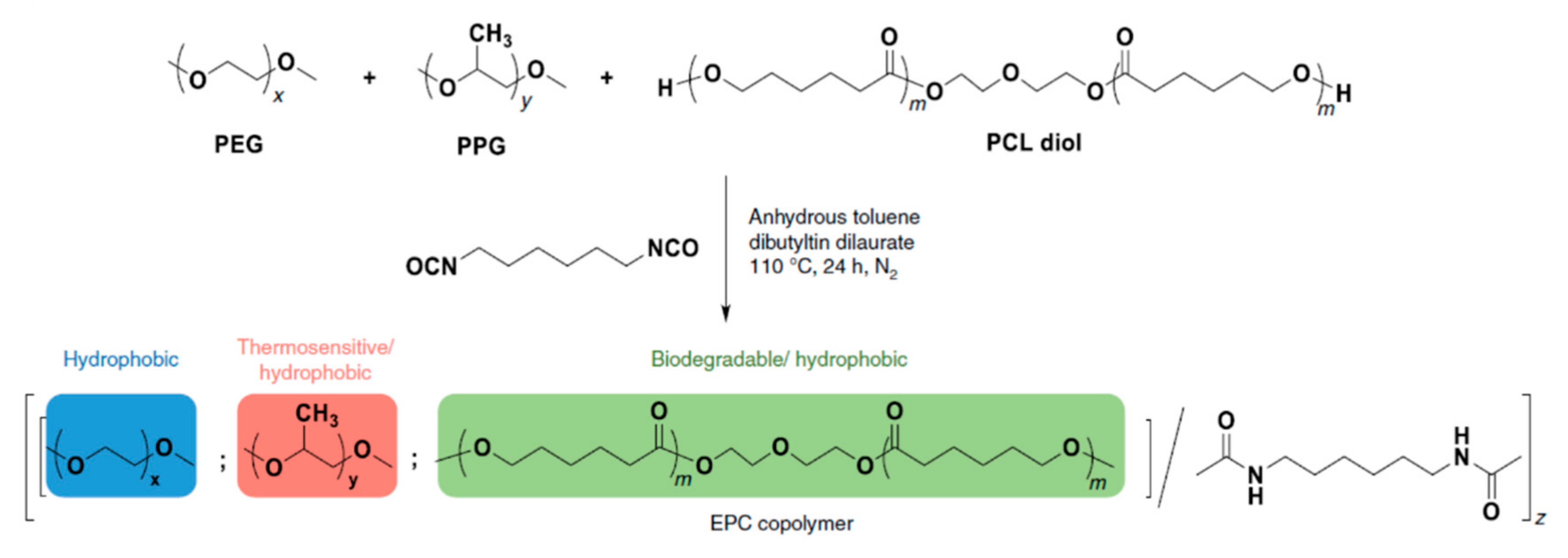
4.1. Covalent Tethering of Biomolecules
4.2. Supramolecular Tethering of Biomolecules
4.3. Uncontrolled Release of Biomolecules from Hydrogel Networks
4.4. ‘On Demand’ Release of Biomolecules from Hydrogels Networks
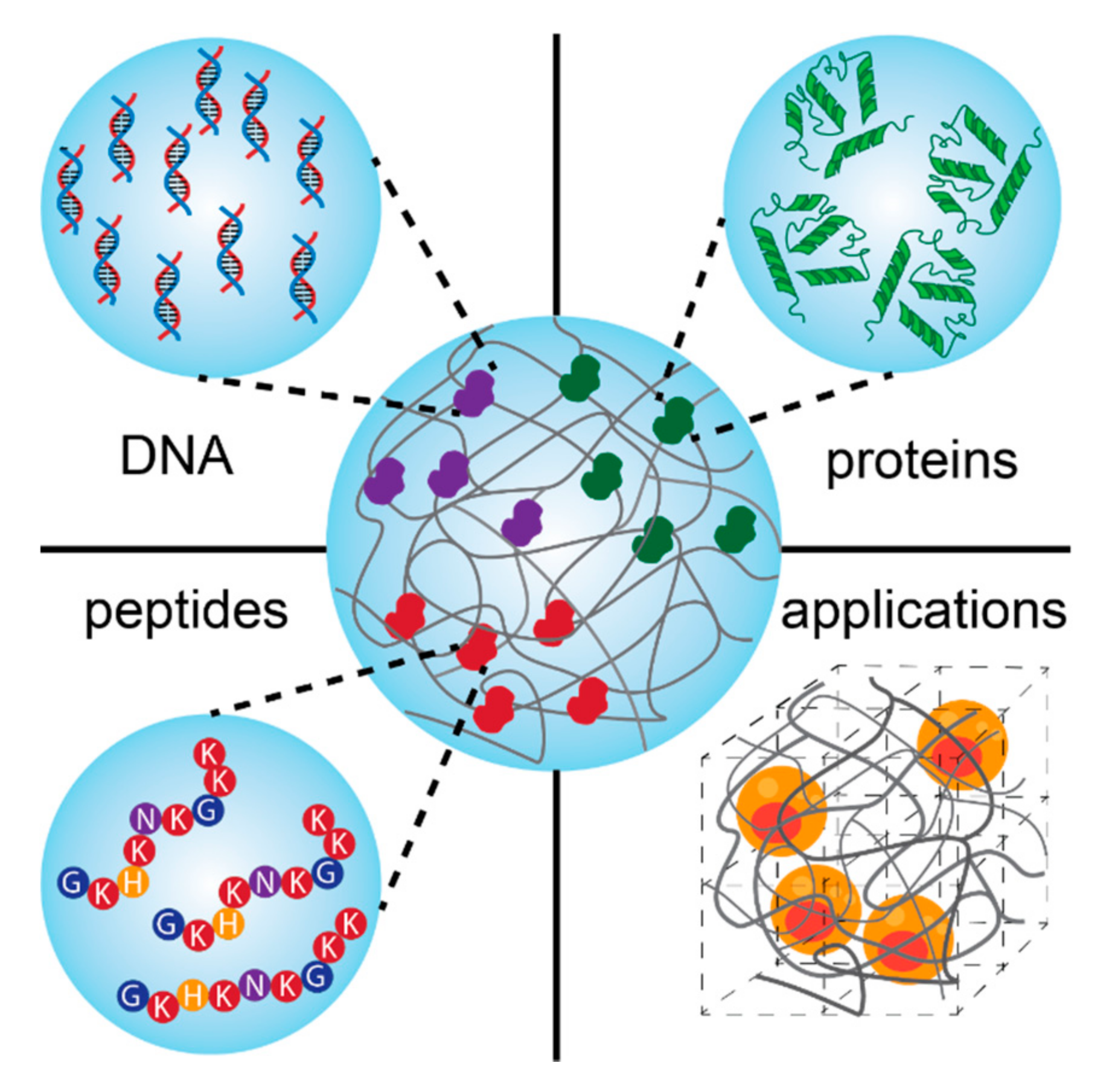
5. Biomolecules for Tissue Regeneration
5.1. Nucleics Acids Conjugated to Synthetic Hydrogels
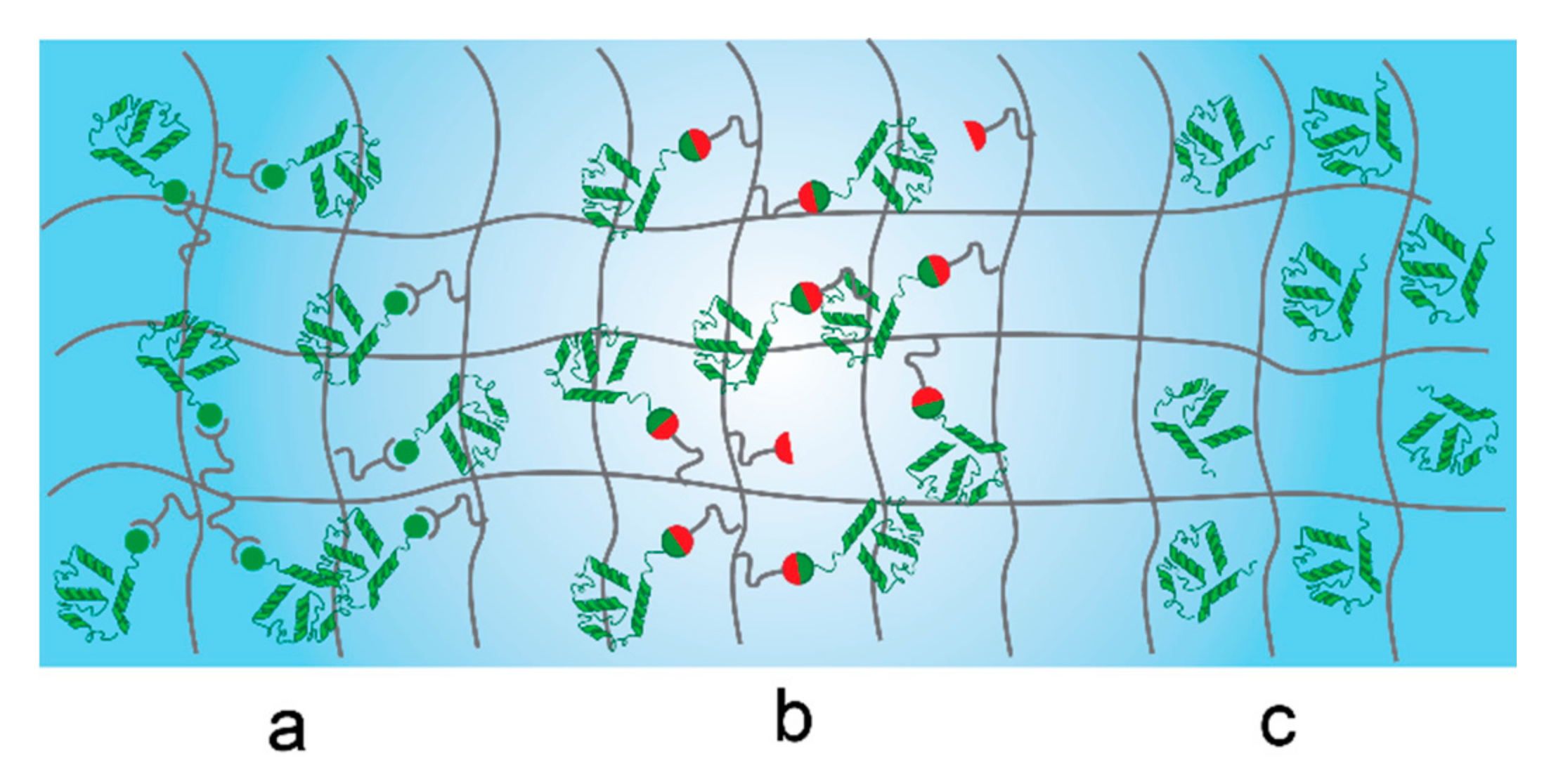
5.2. Proteins Conjugated to Synthetic Networks
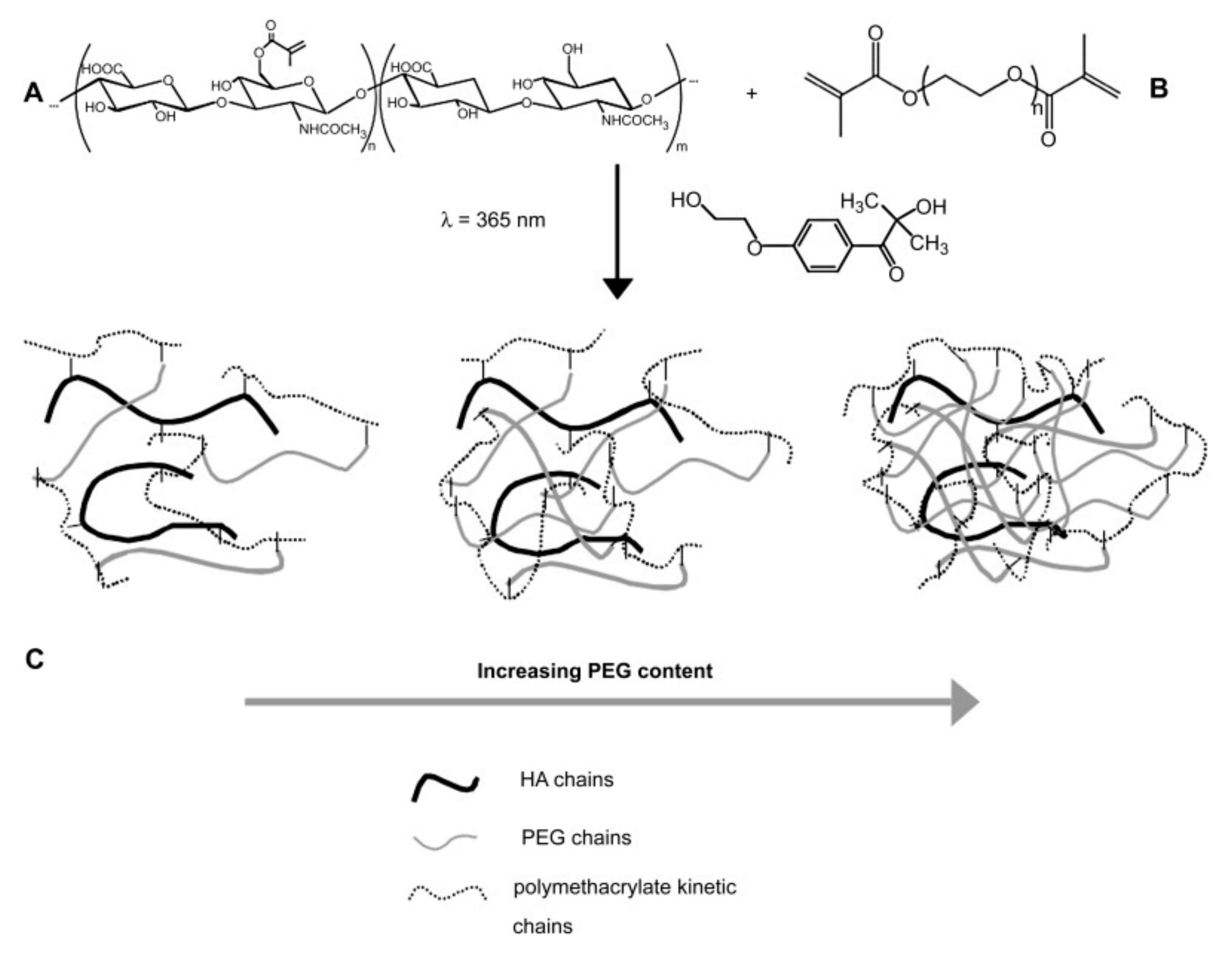
5.3. Carbohydrates Conjugated to Synthetic Hydrogels
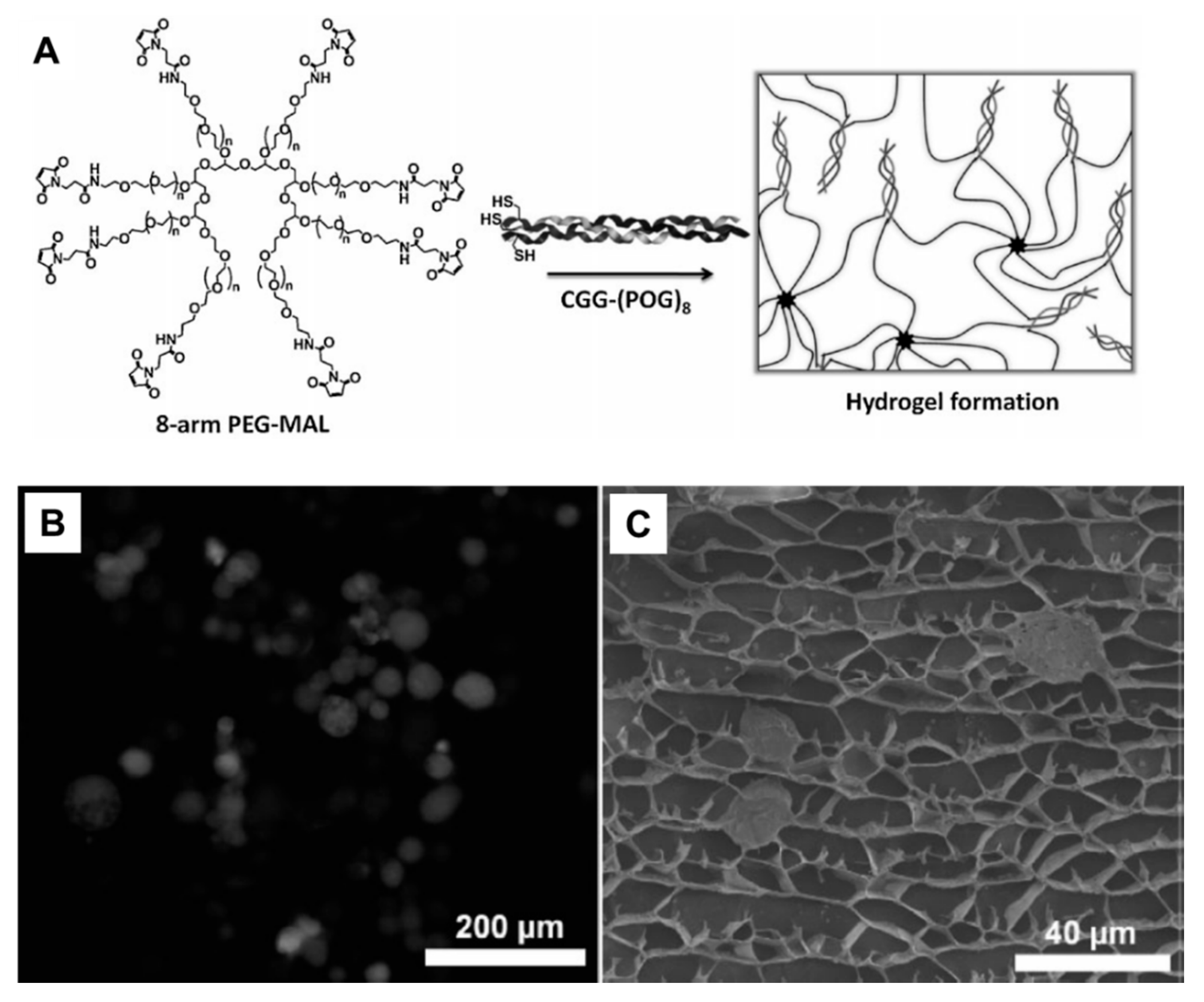
5.4. Peptides Conjugated to Synthetic Hydrogels
6. Conclusions and Future Perspectives
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Abbreviation | Full Name |
| Acs | Acetyl CoA Synthease |
| ATRP | Atom Transfer Radical Polymerization |
| °C | Degree Celsius |
| CGC | Critical Gelation Concentration |
| CL | Cross-linker |
| CMP | Collagen Mimetic Peptide |
| Cryo-SEM | Cryo-Scanning Electron Microscope |
| DAMA | N-(N′,N′-dicarboxymethyl aminopropyl) methacrylamide |
| DMSO | Dimethyl sulfoxide |
| DNA | Deoxyribonucleic acid |
| DTT | Dithiothreitol |
| EtOH | Ethanol |
| Fmoc | Fluorenylmethyloxycarbonyl chloride |
| G’ | Storage Modulus |
| GF | Growth Factor |
| H2O | Water |
| hMSCs | Human Mesenchymal Stem Cells |
| HPMA | Hydroxypropyl methacrylate |
| IA | Itaconic acid |
| J | Joule |
| kPa | KiloPascal |
| LBM | Load Bearing Molecule |
| LCST | Lower Critical Solution Temperature |
| mAB | Monoclonal Anti Bodies |
| MADIX | Macromolecular Design by Interchange of Xantates |
| MeOH | Methanol |
| mm | Millimeter |
| NHS | N-Hydroxysuccinimide |
| nm | Nanometer |
| PAAc | Poly(acrylic acid) |
| PAAm | Poly(acryl amide) |
| PCL | Poly(ε-caprolactone) |
| PDMAEMA | Poly(2-(dimethylamino)ethyl methacrylate) |
| PHEMA | Poly(2-hydroxyethyl methacrylate) |
| PHPMA | Poly(2- hydroxypropyl methacrylate) |
| PEG | Poly(ethylene glycol) |
| PEI | Poly(ethylene imine) |
| PEODA | Poly(ethyleneoxide) diacrylate |
| PGA | Poly(glycolic acid) |
| PLA | Poly(lactic acid) |
| PLGA | Poly(lactic-glycolic acid) |
| PMOXA | Poly(2-methyl-2-oxazoline) |
| PNIPAM | Poly(N-ispropylacrylamide) |
| PNIPMAM | Poly(N-isopropylmethacrylamide) |
| PNVP | Poly(N-vinylpyrrolidone) |
| PPO | Poly(propylene oxide) |
| PU | Poly(urethane) |
| PVA | Poly(vinyl alcohol) |
| PVCL | Poly(vinyl caprolactam) |
| RAFT | Reversible Addition Fragmentation Chain Transfer |
| RGD | Arginylglycylaspartic acid |
| ROMP | Ring-Opening Metathesis Polymerization |
| T | Temperature |
| UCST | Upper Critical Solution Temperature |
| UV | Ultra Violet |
| VPTT | Volume Phase Transition Temperature |
| Wt% | Weight % |
References
- Catoira, M.C.; Fusaro, L.; Di Francesco, D.; Ramella, M.; Boccafoschi, F. Overview of natural hydrogels for regenerative medicine applications. J. Mater. Sci. Mater. Med. 2019, 30, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Annabi, N.; Tamayol, A.; Uquillas, J.A.; Akbari, M.; Bertassoni, L.E.; Cha, C.; Camci-Unal, G.; Dokmeci, M.R.; Peppas, N.A.; Khademhosseini, A. 25th anniversary article: Rational design and applications of hydrogels in regenerative medicine. Adv. Mater. 2014, 26, 85–124. [Google Scholar] [CrossRef]
- Drury, J.L.; Mooney, D.J. Hydrogels for tissue engineering: Scaffold design variables and applications. Biomaterials 2003, 24, 4337–4351. [Google Scholar] [CrossRef]
- Kesireddy, V.; Kasper, F.K. Approaches for building bioactive elements into synthetic scaffolds for bone tissue engineering. J. Mater. Chem. B 2016, 4, 6773–6786. [Google Scholar] [CrossRef] [PubMed]
- Raab, M.; Discher, D.E. Matrix rigidity regulates microtubule network polarization in migration. Cytoskeleton 2017, 74, 114–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantha, S.; Pillai, S.; Khayambashi, P.; Upadhyay, A.; Zhang, Y.; Tao, O.; Pham, H.M.; Tran, S.D. Smart hydrogels in tissue engineering and regenerative medicine. Materials 2019, 12, 3323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Upadhyay, R. Role of biological scaffolds, hydro gels and stem cells in tissue regeneration therapy. Adv. Tissue Eng. Regen. Med. Open Access 2017, 2, 121–135. [Google Scholar] [CrossRef] [Green Version]
- Innocenzi, P. A sol and a gel, what are they? In The Sol-to-Gel Transition; Springer: Berlin/Heidelberg, Germany, 2019; pp. 1–6. [Google Scholar]
- Caló, E.; Khutoryanskiy, V.V. Biomedical applications of hydrogels: A review of patents and commercial products. Eur. Polym. J. 2015, 65, 252–267. [Google Scholar] [CrossRef] [Green Version]
- Seliktar, D. Designing cell-compatible hydrogels for biomedical applications. Science 2012, 336, 1124–1128. [Google Scholar] [CrossRef]
- Chen, J.; Peng, Q.; Peng, X.; Han, L.; Wang, X.; Wang, J.; Zeng, H. Recent advances in mechano-responsive hydrogels for biomedical applications. ACS Appl. Polym. Mater. 2020, 2, 1092–1107. [Google Scholar] [CrossRef]
- Xu, X.; Liu, Y.; Fu, W.; Yao, M.; Ding, Z.; Xuan, J.; Li, D.; Wang, S.; Xia, Y.; Cao, M. Poly (N-isopropylacrylamide)-based thermoresponsive composite hydrogels for biomedical applications. Polymers 2020, 12, 580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uman, S.; Dhand, A.; Burdick, J.A. Recent advances in shear-thinning and self-healing hydrogels for biomedical applications. J. Appl. Polym. Sci. 2020, 137, 48668. [Google Scholar] [CrossRef] [Green Version]
- Aswathy, S.; Narendrakumar, U.; Manjubala, I. Commercial hydrogels for biomedical applications. Heliyon 2020, 6, e03719. [Google Scholar] [CrossRef] [PubMed]
- Mathur, A.M.; Moorjani, S.K.; Scranton, A.B. Methods for synthesis of hydrogel networks: A review. J. Macromol. Sci. Part C Polym. Rev. 1996, 36, 405–430. [Google Scholar] [CrossRef]
- Shang, J.; Le, X.; Zhang, J.; Chen, T.; Theato, P. Trends in polymeric shape memory hydrogels and hydrogel actuators. Polym. Chem. 2019, 10, 1036–1055. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.-L.; Stoddart, J.F. Azobenzene-based light-responsive hydrogel system. Langmuir 2009, 25, 8442–8446. [Google Scholar] [CrossRef]
- Hu, J.; Zheng, Z.; Liu, C.; Hu, Q.; Cai, X.; Xiao, J.; Cheng, Y. A pH-responsive hydrogel with potent antibacterial activity against both aerobic and anaerobic pathogens. Biomater. Sci. 2019, 7, 581–584. [Google Scholar] [CrossRef]
- Kuckling, D.; Harmon, M.E.; Frank, C.W. Photo-cross-linkable PNIPAAm copolymers. 1. Synthesis and characterization of constrained temperature-responsive hydrogel layers. Macromolecules 2002, 35, 6377–6383. [Google Scholar] [CrossRef]
- Peng, L.; Zhang, H.; Feng, A.; Huo, M.; Wang, Z.; Hu, J.; Gao, W.; Yuan, J. Electrochemical redox responsive supramolecular self-healing hydrogels based on host-guest interaction. Polym. Chem. 2015, 6, 3652–3659. [Google Scholar] [CrossRef]
- Nakamura, T.; Takashima, Y.; Hashidzume, A.; Yamaguchi, H.; Harada, A. A metal–ion-responsive adhesive material via switching of molecular recognition properties. Nat. Commun. 2014, 5, 4622. [Google Scholar] [CrossRef]
- Li, H.; Go, G.; Ko, S.Y.; Park, J.-O.; Park, S. Magnetic actuated pH-responsive hydrogel-based soft micro-robot for targeted drug delivery. Smart Mater. Struct. 2016, 25, 027001. [Google Scholar] [CrossRef]
- Knipe, J.M.; Peppas, N.A. Multi-responsive hydrogels for drug delivery and tissue engineering applications. Regen. Biomater. 2014, 1, 57–65. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Liu, S.; Du, G.; Gao, G.; Fu, J. Multi-responsive and tough hydrogels based on triblock copolymer micelles as multi-functional macro-crosslinkers. Chem. Commun. 2015, 51, 8512–8515. [Google Scholar] [CrossRef] [PubMed]
- Xinming, L.; Yingde, C.; Lloyd, A.W.; Mikhalovsky, S.V.; Sandeman, S.R.; Howel, C.A.; Liewen, L. Polymeric hydrogels for novel contact lens-based ophthalmic drug delivery systems: A review. Contact Lens Anterior Eye 2008, 31, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Mooney, D.J. Designing hydrogels for controlled drug delivery. Nat. Rev. Mater. 2016, 1, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Mondal, S.; Das, S.; Nandi, A.K. A review on recent advances in polymer and peptide hydrogels. Soft Matter 2020, 16, 1404–1454. [Google Scholar] [CrossRef]
- Radvar, E.; Azevedo, H.S. Supramolecular peptide/polymer hybrid hydrogels for biomedical applications. Macromol. Biosci. 2019, 19, 1800221. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Xue, B.; Cao, Y. 100th anniversary of macromolecular science viewpoint: Synthetic protein hydrogels. ACS Macro Lett. 2020, 9, 512–524. [Google Scholar] [CrossRef]
- Chen, W.; Tabata, Y.; Wah Tong, Y. Fabricating tissue engineering scaffolds for simultaneous cell growth and drug delivery. Curr. Pharm. Des. 2010, 16, 2388–2394. [Google Scholar] [CrossRef] [PubMed]
- Vasile, C.; Pamfil, D.; Stoleru, E.; Baican, M. New developments in medical applications of hybrid hydrogels containing natural polymers. Molecules 2020, 25, 1539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olson, J.L.; Atala, A.; Yoo, J.J. Tissue engineering: Current strategies and future directions. Chonnam Med. J. 2011, 47, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, N.K.; Allen, P.; Song, Y.H.; Wachs, R.A.; Du, Y.; Ellington, A.D.; Schmidt, C.E. Oligonucleotide-functionalized hydrogels for sustained release of small molecule (aptamer) therapeutics. Acta Biomater. 2020, 102, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Ramasubramanian, L.; Kumar, P.; Wang, A. Engineering extracellular vesicles as nanotherapeutics for regenerative medicine. Biomolecules 2020, 10, 48. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.-H.; Kang, L.; Feng, W.-H.; Cheng, T.-L.; Tsai, W.-C.; Huang, H.-T.; Lee, H.-C.; Chen, C.-H. Effects of lipids and lipoproteins on mesenchymal stem cells used in cardiac tissue regeneration. Int. J. Mol. Sci. 2020, 21, 4770. [Google Scholar] [CrossRef]
- Spicer, C.D. Hydrogel scaffolds for tissue engineering: The importance of polymer choice. Polym. Chem. 2020, 11, 184–219. [Google Scholar] [CrossRef]
- Goor, O.J.; Hendrikse, S.I.; Dankers, P.Y.; Meijer, E. From supramolecular polymers to multi-component biomaterials. Chem. Soc. Rev. 2017, 46, 6621–6637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernard, M.; Jubeli, E.; Pungente, M.D.; Yagoubi, N. Biocompatibility of polymer-based biomaterials and medical devices-regulations, in vitro screening and risk-management. Biomater. Sci. 2018, 6, 2025–2053. [Google Scholar] [CrossRef]
- Sionkowska, A. Current research on the blends of natural and synthetic polymers as new biomaterials. Prog. Polym. Sci. 2011, 36, 1254–1276. [Google Scholar] [CrossRef]
- Lee, K.Y.; Mooney, D.J. Hydrogels for tissue engineering. Chem. Rev. 2001, 101, 1869–1880. [Google Scholar] [CrossRef]
- Andrews, L.; Clary, J.J. Review of the toxicity of multifunctional acrylates. J. Toxicol. Environ. Health Part A Curr. Issues 1986, 19, 149–164. [Google Scholar] [CrossRef]
- King, D.J.; Noss, R.R. Toxicity of polyacrylamide and acrylamide monome. Rev. Environ. Health 1989, 8, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Loos, A.; Rohde, R.; Haverich, A.; Barlach, S. In vitro and in vivo biocompatibility testing of absorbable metal stents. Macromol. Symp. 2007, 253, 103–108. [Google Scholar] [CrossRef]
- Zhang, P.; Sun, F.; Liu, S.; Jiang, S. Anti-PEG antibodies in the clinic: Current issues and beyond PEGylation. J. Control. Release 2016, 244, 184–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.-A.; Yeom, J.; Hwang, B.W.; Hoffman, A.S.; Hahn, S.K. In situ-forming injectable hydrogels for regenerative medicine. Prog. Polym. Sci. 2014, 39, 1973–1986. [Google Scholar] [CrossRef]
- Hennink, W.E.; Van Nostrum, C.F. Novel crosslinking methods to design hydrogels. Adv. Drug Deliv. Rev. 2012, 64, 223–236. [Google Scholar] [CrossRef]
- Plamper, F.A.; Richtering, W. Functional microgels and microgel systems. Acc. Chem. Res. 2017, 50, 131–140. [Google Scholar] [CrossRef]
- Chen, Q.; Chen, H.; Zhu, L.; Zheng, J. Fundamentals of double network hydrogels. J. Mater. Chem. B 2015, 3, 3654–3676. [Google Scholar] [CrossRef]
- Lake, G.; Thomas, A. The strength of highly elastic materials. Math. Phys. Sci. 1967, 300, 108–119. [Google Scholar]
- Sun, T.L.; Luo, F.; Hong, W.; Cui, K.; Huang, Y.; Zhang, H.J.; King, D.R.; Kurokawa, T.; Nakajima, T.; Gong, J.P. Bulk energy dissipation mechanism for the fracture of tough and self-healing hydrogels. Macromolecules 2017, 50, 2923–2931. [Google Scholar] [CrossRef]
- Zhang, Y.; Yu, J.; Ren, K.; Zuo, J.; Ding, J.; Chen, X. Thermosensitive hydrogels as scaffolds for cartilage tissue engineering. Biomacromolecules 2019, 20, 1478–1492. [Google Scholar] [CrossRef]
- Thakar, H.; Sebastian, S.M.; Mandal, S.; Pople, A.; Agarwal, G.; Srivastava, A. Biomolecule-conjugated macroporous hydrogels for biomedical applications. ACS Biomater. Sci. Eng. 2019, 5, 6320–6341. [Google Scholar] [CrossRef]
- Chen, M.H.; Chung, J.J.; Mealy, J.E.; Zaman, S.; Li, E.C.; Arisi, M.F.; Atluri, P.; Burdick, J.A. Injectable supramolecular hydrogel/microgel composites for therapeutic delivery. Macromol. Biosci. 2019, 19, 1800248. [Google Scholar] [CrossRef] [Green Version]
- Bencherif, S.A.; Sands, R.W.; Bhatta, D.; Arany, P.; Verbeke, C.S.; Edwards, D.A.; Mooney, D.J. Injectable preformed scaffolds with shape-memory properties. Proc. Natl. Acad. Sci. USA 2012, 109, 19590–19595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De France, K.J.; Xu, F.; Hoare, T. Structured macroporous hydrogels: Progress, challenges, and opportunities. Adv. Healthc. Mater. 2018, 7, 1700927. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.H.; Herman, E.S.; Lyon, L.A. Network deconstruction reveals network structure in responsive microgels. J. Phys. Chem. B 2011, 115, 3761–3764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izak-Nau, E.; Demco, D.E.; Braun, S.; Baumann, C.; Pich, A.; Göstl, R. Shear-induced structural and functional transformations of poly (N-vinylcaprolactam) microgels. ACS Appl. Polym. Mater. 2020, 2, 1682–1691. [Google Scholar] [CrossRef] [Green Version]
- Yount, W.C.; Loveless, D.M.; Craig, S.L. Strong means slow: Dynamic contributions to the bulk mechanical properties of supramolecular networks. Angew. Chem. Int. Ed. 2005, 44, 2746–2748. [Google Scholar] [CrossRef]
- Chivers, P.R.; Smith, D.K. Shaping and structuring supramolecular gels. Nat. Rev. Mater. 2019, 4, 463–478. [Google Scholar] [CrossRef] [Green Version]
- Rodell, C.B.; MacArthur, J.W., Jr.; Dorsey, S.M.; Wade, R.J.; Wang, L.L.; Woo, Y.J.; Burdick, J.A. Shear-thinning supramolecular hydrogels with secondary autonomous covalent crosslinking to modulate viscoelastic properties in vivo. Adv. Funct. Mater. 2015, 25, 636–644. [Google Scholar] [CrossRef]
- Patenaude, M.; Campbell, S.; Kinio, D.; Hoare, T. Tuning gelation time and morphology of injectable hydrogels using ketone-hydrazide cross-linking. Biomacromolecules 2014, 15, 781–790. [Google Scholar] [CrossRef]
- Kalaf, E.A.G.; Flores, R.; Bledsoe, J.G.; Sell, S.A. Characterization of slow-gelling alginate hydrogels for intervertebral disc tissue-engineering applications. Mater. Sci. Eng. C 2016, 63, 198–210. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhao, C.; Cao, H.; Wang, G.; Song, L.; Niu, G.; Yang, H.; Ma, J.; Zhu, S. Hyperbranched poly(amine-ester) based hydrogels for controlled multi-drug release in combination chemotherapy. Biomaterials 2010, 31, 5445–5454. [Google Scholar] [CrossRef] [PubMed]
- Saunders, L.; Ma, P.X. Self-healing supramolecular hydrogels for tissue engineering applications. Macromol. Biosci. 2019, 19, 1800313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Zhang, J.; Li, Y.; Wang, N.; Liu, W. A nucleoside responsive diaminotriazine-based hydrogen bonding strengthened hydrogel. Mater. Lett. 2015, 142, 71–74. [Google Scholar] [CrossRef]
- Ma, M.; Kuang, Y.; Gao, Y.; Zhang, Y.; Gao, P.; Xu, B. Aromatic−aromatic interactions induce the self-assembly of pentapeptidic derivatives in water to form nanofibers and supramolecular hydrogels. J. Am. Chem. Soc. 2010, 132, 2719–2728. [Google Scholar] [CrossRef]
- Vinner, G.K.; Vladisavljević, G.T.; Clokie, M.R.; Malik, D.J. Microencapsulation of clostridium difficile specific bacteriophages using microfluidic glass capillary devices for colon delivery using pH triggered release. PLoS ONE 2017, 12, e0186239. [Google Scholar] [CrossRef] [Green Version]
- Menyo, M.S.; Hawker, C.J.; Waite, J.H. Versatile tuning of supramolecular hydrogels through metal complexation of oxidation-resistant catechol-inspired ligands. Soft Matter 2013, 9, 10314–10323. [Google Scholar] [CrossRef]
- Jensen, B.E.; Dávila, I.; Zelikin, A.N. Poly (vinyl alcohol) physical hydrogels: Matrix-mediated drug delivery using spontaneously eroding substrate. J. Phys. Chem. B 2016, 120, 5916–5926. [Google Scholar] [CrossRef]
- Kuang, H.; He, H.; Zhang, Z.; Qi, Y.; Xie, Z.; Jing, X.; Huang, Y. Injectable and biodegradable supramolecular hydrogels formed by nucleobase-terminated poly (ethylene oxide) s and α-cyclodextrin. J. Mater. Chem. B 2014, 2, 659–667. [Google Scholar] [CrossRef]
- Takigawa, T.; Yamawaki, T.; Takahashi, K.; Masuda, T. Change in Young’s modulus of poly (N-isopropylacrylamide) gels by volume phase transition. Polym. Gels Netw. 1998, 5, 585–589. [Google Scholar] [CrossRef]
- Meier-Koll, A.; Pipich, V.; Busch, P.; Papadakis, C.M.; Müller-Buschbaum, P. Phase separation in semidilute aqueous poly(N-isopropylacrylamide) solutions. Langmuir 2012, 28, 8791–8798. [Google Scholar] [CrossRef] [PubMed]
- Hou, L.; Wu, P. LCST transition of PNIPAM-b-PVCL in water: Cooperative aggregation of two distinct thermally responsive segments. Soft Matter 2014, 10, 3578–3586. [Google Scholar] [CrossRef] [PubMed]
- Vishnevetskaya, N.S.; Hildebrand, V.; Niebuur, B.-J.; Grillo, I.; Filippov, S.K.; Laschewsky, A.; Müller-Buschbaum, P.; Papadakis, C.M. “Schizophrenic” micelles from doubly thermoresponsive polysulfobetaine-b-poly(N-isopropylmethacrylamide) diblock copolymers. Macromolecules 2017, 50, 3985–3999. [Google Scholar] [CrossRef]
- Idziak, I.; Avoce, D.; Lessard, D.; Gravel, D.; Zhu, X. Thermosensitivity of aqueous solutions of poly (N, N-diethylacrylamide). Macromolecules 1999, 32, 1260–1263. [Google Scholar] [CrossRef]
- Verdonck, B.; Goethals, E.J.; Du Prez, F.E. Block copolymers of methyl vinyl ether and isobutyl vinyl ether with thermo-adjustable amphiphilic properties. Macromol. Chem. Phys. 2003, 204, 2090–2098. [Google Scholar] [CrossRef]
- Haq, M.A.; Su, Y.; Wang, D. Mechanical properties of PNIPAM based hydrogels: A review. Mater. Sci. Eng. C 2017, 70, 842–855. [Google Scholar] [CrossRef]
- Liu, W.; Gong, X.; Zhu, Y.; Wang, J.; Ngai, T.; Wu, C. Probing sol–gel matrices and dynamics of star PEG hydrogels near overlap concentration. Macromolecules 2019, 52, 8956–8966. [Google Scholar] [CrossRef]
- Zhu, D.Y.; Hong, Z.P.; Xue, Y.M.; Chen, X.J.; Zhang, L.Y.; Gao, L.; Wang, Y.X.; Yang, C.F.; Guo, J.W. Injectable, remoldable hydrogels with thermoresponsiveness, self-healing and cytocompatibility constructed via orthogonal assembly of well-defined star and linear polymers. J. Mater. Chem. B 2019, 7, 3232–3242. [Google Scholar] [CrossRef]
- Creusen, G.; Roshanasan, A.; Lopez, J.G.; Peneva, K.; Walther, A. Bottom-up design of model network elastomers and hydrogels from precise star polymers. Polym. Chem. 2019, 10, 3740–3750. [Google Scholar] [CrossRef]
- Lyu, Z.; Ding, L.; Huang, A.Y.T.; Kao, C.L.; Peng, L. Poly(amidoamine) dendrimers: Covalent and supramolecular synthesis. Mater. Today Chem. 2019, 13, 34–48. [Google Scholar] [CrossRef]
- Xu, Q.; Guo, L.; Sigen, A.; Gao, Y.; Zhou, D.; Greiser, U.; Creagh-Flynn, J.; Zhang, H.; Dong, Y.; Cutlar, L. Injectable hyperbranched poly (β-amino ester) hydrogels with on-demand degradation profiles to match wound healing processes. Chem. Sci. 2018, 9, 2179–2187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurzbach, D.; Junk, M.J.N.; Hinderberger, D. Nanoscale inhomogeneities in thermoresponsive polymers. Macromol. Rapid Commun. 2013, 34, 119–134. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Zhang, S.; Wan, Y.; Fu, W.; Li, Z. Hydrogels assembled from star-shaped polypeptides with a dendrimer as the core. Soft Matter 2015, 11, 2945–2951. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.L.; Samanta, S.; Sarkar, S.; Singha, N.K. A self-healable and antifouling hydrogel based on PDMS centered ABA tri-block copolymer polymersomes: A potential material for therapeutic contact lenses. J. Mater. Chem. B 2020, 8, 226–243. [Google Scholar] [CrossRef] [PubMed]
- Fu, J. Triblock Copolymer Micelle-Crosslinked Hydrogels. In Advances in Polymer Science; Springer: Berlin/Heidelberg, Germany, 2020. [Google Scholar]
- Guermani, E.; Shaki, H.; Mohanty, S.; Mehrali, M.; Arpanaei, A.; Gaharwar, A.K.; Dolatshahi-Pirouz, A. Engineering complex tissue-like microgel arrays for evaluating stem cell differentiation. Sci. Rep. 2016, 6, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Merkel, T.J.; Pandya, A.; Napier, M.E.; Luft, J.C.; Daniel, W.; Sheiko, S.; DeSimone, J.M. Low modulus biomimetic microgel particles with high loading of hemoglobin. Biomacromolecules 2012, 13, 2748–2759. [Google Scholar] [CrossRef]
- Pich, A.; Richtering, W. Chemical Design of Responsive Microgels; Springer: Berlin/Heidelberg, Germany, 2010; Volume 234. [Google Scholar]
- Wachiralarpphaithoon, C.; Iwasaki, Y.; Akiyoshi, K. Enzyme-degradable phosphorylcholine porous hydrogels cross-linked with polyphosphoesters for cell matrices. Biomaterials 2007, 28, 984–993. [Google Scholar] [CrossRef]
- Lee, P.I. Kinetics of drug release from hydrogel matrices. J. Control. Release 1985, 2, 277–288. [Google Scholar] [CrossRef]
- Annabi, N.; Nichol, J.W.; Zhong, X.; Ji, C.; Koshy, S.; Khademhosseini, A.; Dehghani, F. Controlling the porosity and microarchitecture of hydrogels for tissue engineering. Tissue Eng. Part B Rev. 2010, 16, 371–383. [Google Scholar] [CrossRef]
- Li, H.; Wu, C.-W.; Wang, S.; Zhang, W. Mechanically strong poly (vinyl alcohol) hydrogel with macropores and high porosity. Mater. Lett. 2020, 266, 127504. [Google Scholar] [CrossRef]
- Appel, E.A.; Forster, R.A.; Rowland, M.J.; Scherman, O.A. The control of cargo release from physically crosslinked hydrogels by crosslink dynamics. Biomaterials 2014, 35, 9897–9903. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Rudov, A.A.; Schroeder, R.; Portnov, I.V.; Richtering, W.; Potemkin, I.I.; Pich, A. Distribution of ionizable groups in polyampholyte microgels controls interactions with captured proteins: From blockade and “levitation” to accelerated release. Biomacromolecules 2019, 20, 1578–1591. [Google Scholar] [CrossRef] [PubMed]
- Patenaude, M.; Hoare, T. Injectable, degradable thermoresponsive poly (N-isopropylacrylamide) hydrogels. ACS Macro Lett. 2012, 1, 409–413. [Google Scholar] [CrossRef]
- Ramot, Y.; Haim-Zada, M.; Domb, A.J.; Nyska, A. Biocompatibility and safety of PLA and its copolymers. Adv. Drug Deliv. Rev. 2016, 107, 153–162. [Google Scholar] [CrossRef]
- Ceonzo, K.; Gaynor, A.; Shaffer, L.; Kojima, K.; Vacanti, C.A.; Stahl, G.L. Polyglycolic acid-induced inflammation: Role of hydrolysis and resulting complement activation. Tissue Eng. 2006, 12, 301–308. [Google Scholar] [CrossRef]
- Langer, R.; Folkman, J. Polymers for the sustained release of proteins and other macromolecules. Nature 1976, 263, 797–800. [Google Scholar] [CrossRef]
- Elsawy, M.A.; Kim, K.-H.; Park, J.-W.; Deep, A. Hydrolytic degradation of polylactic acid (PLA) and its composites. Renew. Sustain. Energy Rev. 2017, 79, 1346–1352. [Google Scholar] [CrossRef]
- Hu, W.; Wang, Z.; Xiao, Y.; Zhang, S.; Wang, J. Advances in crosslinking strategies of biomedical hydrogels. Biomater. Sci. 2019, 7, 843–855. [Google Scholar] [CrossRef]
- Zhang, J.; Sun, R.; DeSouza-Edwards, A.O.; Frueh, J.; Sukhorukov, G.B. Microchamber arrays made of biodegradable polymers for enzymatic release of small hydrophilic cargos. Soft Matter 2020, 16, 2266–2275. [Google Scholar] [CrossRef]
- Cai, Q.; Li, X.; Zhu, W. High molecular weight biodegradable poly(ethylene glycol) via carboxyl-ester transesterification. Macromolecules 2020, 53, 2177–2186. [Google Scholar] [CrossRef]
- Dechy-Cabaret, O.; Martin-Vaca, B.; Bourissou, D. Controlled ring-opening polymerization of lactide and glycolide. Chem. Rev. 2004, 104, 6147–6176. [Google Scholar] [CrossRef] [PubMed]
- Kawai, F.; Nakadai, K.; Nishioka, E.; Nakajima, H.; Ohara, H.; Masaki, K.; Iefuji, H. Different enantioselectivity of two types of poly (lactic acid) depolymerases toward poly (l-lactic acid) and poly (d-lactic acid). Polym. Degrad. Stab. 2011, 96, 1342–1348. [Google Scholar] [CrossRef]
- Grizzi, I.; Garreau, H.; Li, S.; Vert, M. Hydrolytic degradation of devices based on poly (DL-lactic acid) size-dependence. Biomaterials 1995, 16, 305–311. [Google Scholar] [CrossRef]
- Park, T.G. Degradation of poly (D, L-lactic acid) microspheres: Effect of molecular weight. J. Control. Release 1994, 30, 161–173. [Google Scholar] [CrossRef]
- Wang, Y.; Tashiro, Y.; Sonomoto, K. Fermentative production of lactic acid from renewable materials: Recent achievements, prospects, and limits. J. Biosci. Bioeng. 2015, 119, 10–18. [Google Scholar] [CrossRef]
- Sun, H.; Mei, L.; Song, C.; Cui, X.; Wang, P. The in vivo degradation, absorption and excretion of PCL-based implant. Biomaterials 2006, 27, 1735–1740. [Google Scholar] [CrossRef]
- Yuan, J.; Xiong, W.; Zhou, X.; Zhang, Y.; Shi, D.; Li, Z.; Lu, H. 4-Hydroxyproline-derived sustainable polythioesters: Controlled ring-opening polymerization, complete recyclability, and facile functionalization. J. Am. Chem. Soc. 2019, 141, 4928–4935. [Google Scholar] [CrossRef] [Green Version]
- Urbánek, T.; Jäger, E.; Jäger, A.; Hrubý, M. Selectively biodegradable polyesters: Nature-inspired construction materials for future biomedical applications. Polymers 2019, 11, 1061. [Google Scholar] [CrossRef] [Green Version]
- Ye, H.; Zhang, K.; Kai, D.; Li, Z.; Loh, X.J. Polyester elastomers for soft tissue engineering. Chem. Soc. Rev. 2018, 47, 4545–4580. [Google Scholar] [CrossRef]
- Gonçalves, F.; Fonseca, A.; Domingos, M.; Gloria, A.; Serra, A.; Coelho, J. The potential of unsaturated polyesters in biomedicine and tissue engineering: Synthesis, structure-properties relationships and additive manufacturing. Prog. Polym. Sci. 2017, 68, 1–34. [Google Scholar] [CrossRef] [Green Version]
- Díaz, A.; Katsarava, R.; Puiggalí, J. Synthesis, properties and applications of biodegradable polymers derived from diols and dicarboxylic acids: From polyesters to poly(ester amide)s. Int. J. Mol. Sci. 2014, 15, 7064–7123. [Google Scholar]
- Sartori, S.; Chiono, V.; Tonda-Turo, C.; Mattu, C.; Gianluca, C. Biomimetic polyurethanes in nano and regenerative medicine. J. Mater. Chem. B 2014, 2, 5128–5144. [Google Scholar] [CrossRef]
- Kamaci, M. Polyurethane-based hydrogels for controlled drug delivery applications. Eur. Polym. J. 2020, 123, 109444. [Google Scholar] [CrossRef]
- Lundin, J.G.; Daniels, G.C.; McGann, C.L.; Stanbro, J.; Watters, C.; Stockelman, M.; Wynne, J.H. Multi-functional polyurethane hydrogel foams with tunable mechanical properties for wound dressing applications. Macromol. Mater. Eng. 2017, 302, 1600375. [Google Scholar] [CrossRef]
- Bhattacharyya, A.; Mukhopadhyay, P.; Pramanik, N.; Kundu, P.P. Effect of polyethylene glycol on bis (2-hydroxyethyl) terephthalate-based polyurethane/alginate ph-sensitive blend for oral protein delivery. Adv. Polym. Technol. 2016, 35. [Google Scholar] [CrossRef]
- Liu, Z.; Liow, S.S.; Lai, S.L.; Alli-Shaik, A.; Holder, G.E.; Parikh, B.H.; Krishnakumar, S.; Li, Z.; Tan, M.J.; Gunaratne, J. Retinal-detachment repair and vitreous-like-body reformation via a thermogelling polymer endotamponade. Nat. Biomed. Eng. 2019, 3, 598–610. [Google Scholar] [CrossRef]
- Xue, K.; Liu, Z.; Jiang, L.; Kai, D.; Li, Z.; Su, X.; Loh, X.J. A new highly transparent injectable PHA-based thermogelling vitreous substitute. Biomater. Sci. 2020, 8, 926–936. [Google Scholar] [CrossRef]
- Mi, H.-Y.; Jing, X.; Yilmaz, G.; Hagerty, B.S.; Enriquez, E.; Turng, L.-S. In situ synthesis of polyurethane scaffolds with tunable properties by controlled crosslinking of tri-block copolymer and polycaprolactone triol for tissue regeneration. Chem. Eng. J. 2018, 348, 786–798. [Google Scholar] [CrossRef]
- Chimisso, V.; Fodor, C.; Meier, W. Effect of divalent cation on swelling behavior of anionic microgels: Quantification and dynamics of ion uptake and release. Langmuir 2019, 35, 13413–13420. [Google Scholar] [CrossRef]
- Ter Schiphorst, J.; Coleman, S.; Stumpel, J.E.; Ben Azouz, A.; Diamond, D.; Schenning, A.P.H.J. Molecular design of light-responsive hydrogels, for in situ generation of fast and reversible valves for microfluidic applications. Chem. Mater. 2015, 27, 5925–5931. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Chen, J.; Deng, C.; Suuronen, E.J.; Zhong, Z. Click hydrogels, microgels and nanogels: Emerging platforms for drug delivery and tissue engineering. Biomaterials 2014, 35, 4969–4985. [Google Scholar] [CrossRef] [PubMed]
- Pertici, V.; Trimaille, T.; Gigmes, D. Inputs of macromolecular engineering in the design of injectable hydrogels based on synthetic thermoresponsive polymers. Macromolecules 2020, 53, 682–692. [Google Scholar] [CrossRef] [Green Version]
- Akhtar, M.F.; Hanif, M.; Ranjha, N.M. Methods of synthesis of hydrogels: A review. Saudi Pharm. J. 2016, 24, 554–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nezhad-Mokhtari, P.; Ghorbani, M.; Roshangar, L.; Rad, J.S. A review on the construction of hydrogel scaffolds by various chemically techniques for tissue engineering. Eur. Polym. J. 2019, 117, 64–76. [Google Scholar] [CrossRef]
- Huang, Q.; Zou, Y.; Arno, M.C.; Chen, S.; Wang, T.; Gao, J.; Dove, A.P.; Du, J. Hydrogel scaffolds for differentiation of adipose-derived stem cells. Chem. Soc. Rev. 2017, 46, 6255–6275. [Google Scholar] [CrossRef] [PubMed]
- Mol, E.A.; Lei, Z.; Roefs, M.T.; Bakker, M.H.; Goumans, M.J.; Doevendans, P.A.; Dankers, P.Y.; Vader, P.; Sluijter, J.P. Injectable supramolecular ureidopyrimidinone hydrogels provide sustained release of extracellular vesicle therapeutics. Adv. Healthc. Mater. 2019, 8, 1900847. [Google Scholar] [CrossRef] [Green Version]
- Heo, D.N.; Lee, S.-J.; Timsina, R.; Qiu, X.; Castro, N.J.; Zhang, L.G. Development of 3D printable conductive hydrogel with crystallized PEDOT:PSS for neural tissue engineering. Mater. Sci. Eng. C 2019, 99, 582–590. [Google Scholar] [CrossRef]
- Alvarez-Lorenzo, C.; García-González, C.A.; Concheiro, A. Cyclodextrins as versatile building blocks for regenerative medicine. J. Control. Release 2017, 268, 269–281. [Google Scholar] [CrossRef]
- Rowland, M.J.; Parkins, C.C.; McAbee, J.H.; Kolb, A.K.; Hein, R.; Loh, X.J.; Watts, C.; Scherman, O.A. An adherent tissue-inspired hydrogel delivery vehicle utilised in primary human glioma models. Biomaterials 2018, 179, 199–208. [Google Scholar] [CrossRef]
- Hughes, A.; Tai, H.; Tochwin, A.; Wang, W. Biodegradable and biocompatible PDLLA-PEG1k-PDLLA diacrylate macromers: Synthesis, characterisation and preparation of soluble hyperbranched polymers and crosslinked hydrogels. Processes 2017, 5, 18. [Google Scholar] [CrossRef]
- Badea, A.; McCracken, J.M.; Tillmaand, E.G.; Kandel, M.E.; Oraham, A.W.; Mevis, M.B.; Rubakhin, S.S.; Popescu, G.; Sweedler, J.V.; Nuzzo, R.G. 3D-printed pHEMA materials for topographical and biochemical modulation of dorsal root ganglion cell response. ACS Appl. Mater. Interfaces 2017, 9, 30318–30328. [Google Scholar] [CrossRef]
- Gu, J.; Li, X.; Ma, H.; Guan, Y.; Zhang, Y. One-step synthesis of PHEMA hydrogel films capable of generating highly ordered wrinkling patterns. Polymer 2017, 110, 114–123. [Google Scholar] [CrossRef]
- McCracken, J.M.; Badea, A.; Kandel, M.E.; Gladman, A.S.; Wetzel, D.J.; Popescu, G.; Lewis, J.A.; Nuzzo, R.G. Programming Mechanical and Physicochemical Properties of 3D hydrogel cellular microcultures via direct ink writing. Adv. Healthc. Mater. 2016, 5, 1025–1039. [Google Scholar] [CrossRef] [PubMed]
- Lienemann, P.S.; Karlsson, M.; Sala, A.; Wischhusen, H.M.; Weber, F.E.; Zimmermann, R.; Weber, W.; Lutolf, M.P.; Ehrbar, M. A versatile approach to engineering biomolecule-presenting cellular microenvironments. Adv. Healthc. Mater. 2013, 2, 292–296. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Xu, J.; Wei, K.; Xu, Y.J.; Choi, C.K.K.; Zhu, M.; Bian, L. Bioactive nanocomposite poly (ethylene glycol) hydrogels crosslinked by multifunctional layered double hydroxides nanocrosslinkers. Macromol. Biosci. 2016, 16, 1019–1026. [Google Scholar] [CrossRef] [PubMed]
- Berberich, O.; Blöhbaum, J.; Hölscher-Doht, S.; Meffert, R.H.; Teßmar, J.; Blunk, T.; Groll, J. Catechol-modified poly(oxazoline)s with tunable degradability facilitate cell invasion and lateral cartilage integration. J. Ind. Eng. Chem. 2019, 80, 757–769. [Google Scholar] [CrossRef]
- Dargaville, T.R.; Park, J.-R.; Hoogenboom, R. Poly(2-oxazoline) hydrogels: State-of-the-art and emerging applications. Macromol. Biosci. 2018, 18, 1800070. [Google Scholar] [CrossRef]
- Kumar, A.; Han, S.S. PVA-based hydrogels for tissue engineering: A review. Int. J. Polym. Mater. Polym. Biomater. 2017, 66, 159–182. [Google Scholar] [CrossRef]
- Ben Halima, N. Poly(vinyl alcohol): Review of its promising applications and insights into biodegradation. RSC Adv. 2016, 6, 39823–39832. [Google Scholar] [CrossRef]
- Anderson, D.E.J.; Truong, K.P.; Hagen, M.W.; Yim, E.K.F.; Hinds, M.T. Biomimetic modification of poly(vinyl alcohol): Encouraging endothelialization and preventing thrombosis with antiplatelet monotherapy. Acta Biomater. 2019, 86, 291–299. [Google Scholar] [CrossRef]
- Rahimi, N.; Molin, D.G.; Cleij, T.J.; Van Zandvoort, M.A.; Post, M.J. Electrosensitive polyacrylic acid/fibrin hydrogel facilitates cell seeding and alignment. Biomacromolecules 2012, 13, 1448–1457. [Google Scholar] [CrossRef] [PubMed]
- Fliervoet, L.A.L.; Zhang, H.; Van Groesen, E.; Fortuin, K.; Duin, N.J.C.B.; Remaut, K.; Schiffelers, R.M.; Hennink, W.E.; Vermonden, T. Local release of siRNA using polyplex-loaded thermosensitive hydrogels. Nanoscale 2020, 12, 10347–10360. [Google Scholar] [CrossRef] [PubMed]
- Singh Chandel, A.K.; Kannan, D.; Nutan, B.; Singh, S.; Jewrajka, S.K. Dually crosslinked injectable hydrogels of poly(ethylene glycol) and poly((2-dimethylamino)ethyl methacrylate)-b-poly(N-isopropyl acrylamide) as a wound healing promoter. J. Mater. Chem. B 2017, 5, 4955–4965. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.L.; Sloand, J.N.; Gaffey, A.C.; Venkataraman, C.M.; Wang, Z.; Trubelja, A.; Hammer, D.A.; Atluri, P.; Burdick, J.A. Injectable, guest–host assembled polyethylenimine hydrogel for siRNA delivery. Biomacromolecules 2017, 18, 77–86. [Google Scholar] [CrossRef]
- Kim, Y.-M.; Song, S.-C. Targetable micelleplex hydrogel for long-term, effective, and systemic siRNA delivery. Biomaterials 2014, 35, 7970–7977. [Google Scholar] [CrossRef]
- Xue, H.; Hu, L.; Xiong, Y.; Zhu, X.; Wei, C.; Cao, F.; Zhou, W.; Sun, Y.; Endo, Y.; Liu, M.; et al. Quaternized chitosan-Matrigel-polyacrylamide hydrogels as wound dressing for wound repair and regeneration. Carbohydr. Polym. 2019, 226, 115302. [Google Scholar] [CrossRef]
- Steichen, S.; O’Connor, C.; Peppas, N.A. Development of a P((MAA-co-NVP)-g-EG) hydrogel platform for oral protein delivery: Effects of hydrogel composition on environmental response and protein partitioning. Macromol. Biosci. 2017, 17, 1600266. [Google Scholar] [CrossRef]
- Lanzalaco, S.; Armelin, E. Poly(N-isopropylacrylamide) and copolymers: A review on recent progresses in biomedical applications. Gels 2017, 3, 36. [Google Scholar] [CrossRef]
- Lee, H.; Kim, G. Enhanced cellular activities of polycaprolactone/alginate-based cell-laden hierarchical scaffolds for hard tissue engineering applications. J. Colloid Interface Sci. 2014, 430, 315–325. [Google Scholar] [CrossRef]
- Tseng, H.; Puperi, D.S.; Kim, E.J.; Ayoub, S.; Shah, J.V.; Cuchiara, M.L.; West, J.L.; Grande-Allen, K.J. Anisotropic poly(ethylene glycol)/polycaprolactone hydrogel–fiber composites for heart valve tissue engineering. Tissue Eng. Part. A 2014, 20, 2634–2645. [Google Scholar] [CrossRef]
- Basu, A.; Kunduru, K.R.; Doppalapudi, S.; Domb, A.J.; Khan, W. Poly(lactic acid) based hydrogels. Adv. Drug Deliv. Rev. 2016, 107, 192–205. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.L.; Kim, Y.S.; Xie, V.Y.; Smith, B.T.; Watson, E.; Lam, J.; Pearce, H.A.; Engel, P.S.; Mikos, A.G. Modular, tissue-specific, and biodegradable hydrogel cross-linkers for tissue engineering. Sci. Adv. 2019, 5, eaaw7396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karam, J.-P.; Muscari, C.; Sindji, L.; Bastiat, G.; Bonafè, F.; Venier-Julienne, M.-C.; Montero-Menei, N.C. Pharmacologically active microcarriers associated with thermosensitive hydrogel as a growth factor releasing biomimetic 3D scaffold for cardiac tissue-engineering. J. Control. Release 2014, 192, 82–94. [Google Scholar] [CrossRef] [Green Version]
- Fletcher, N.A.; Babcock, L.R.; Murray, E.A.; Krebs, M.D. Controlled delivery of antibodies from injectable hydrogels. Mater. Sci. Eng. C 2016, 59, 801–806. [Google Scholar] [CrossRef] [PubMed]
- Sonzogni, A.S.; Yealland, G.; Kar, M.; Wedepohl, S.; Gugliotta, L.M.; Gonzalez, V.D.G.; Hedtrich, S.; Calderón, M.; Minari, R.J. Effect of delivery platforms structure on the epidermal antigen transport for topical vaccination. Biomacromolecules 2018, 19, 4607–4616. [Google Scholar] [CrossRef]
- Zhang, C.; Gau, E.; Sun, W.; Zhu, J.; Schmidt, B.M.; Pich, A.; Shi, X. Influence of size, crosslinking degree and surface structure of poly(N-vinylcaprolactam)-based microgels on their penetration into multicellular tumor spheroids. Biomater. Sci. 2019, 7, 4738–4747. [Google Scholar] [CrossRef]
- Lynch, B.; Crawford, K.; Baruti, O.; Abdulahad, A.; Webster, M.; Puetzer, J.; Ryu, C.; Bonassar, L.J.; Mendenhall, J. The effect of hypoxia on thermosensitive poly(N-vinylcaprolactam) hydrogels with tunable mechanical integrity for cartilage tissue engineering. J. Biomed. Mater. Res. Part B Appl. Biomater. 2017, 105, 1863–1873. [Google Scholar] [CrossRef]
- Chiu, W.Y.; Carratt, G.M.; Soong, D.S. A computer model for the gel effect in free-radical polymerization. Macromolecules 1983, 16, 348–357. [Google Scholar] [CrossRef]
- Webster, O.W. Living polymerization methods. Science 1991, 251, 887–893. [Google Scholar] [CrossRef]
- Matyjaszewski, K.; Pintauer, T.; Gaynor, S. Removal of copper-based catalyst in atom transfer radical polymerization using ion exchange resins. Macromolecules 2000, 33, 1476–1478. [Google Scholar] [CrossRef]
- Semsarilar, M.; Perrier, S. ‘Green’reversible addition-fragmentation chain-transfer (RAFT) polymerization. Nat. Chem. 2010, 2, 811. [Google Scholar] [CrossRef] [PubMed]
- Perrier, S.; Takolpuckdee, P. Macromolecular design via reversible addition–fragmentation chain transfer (RAFT)/xanthates (MADIX) polymerization. J. Polym. Sci. Part A Polym. Chem. 2005, 43, 5347–5393. [Google Scholar] [CrossRef]
- Culver, H.R.; Clegg, J.R.; Peppas, N.A. Analyte-responsive hydrogels: Intelligent materials for biosensing and drug delivery. Acc. Chem. Res. 2017, 50, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Abdou, P.; Wang, Z.; Chen, Q.; Chan, A.; Zhou, D.R.; Gunadhi, V.; Gu, Z. Advances in engineering local drug delivery systems for cancer immunotherapy. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2020, e1632. [Google Scholar] [CrossRef]
- Oliva, N.; Conde, J.O.; Wang, K.; Artzi, N. Designing hydrogels for on-demand therapy. Acc. Chem. Res. 2017, 50, 669–679. [Google Scholar] [CrossRef] [PubMed]
- Chao, Y.; Chen, Q.; Liu, Z. Smart injectable hydrogels for cancer immunotherapy. Adv. Funct. Mater. 2020, 30, 1902785. [Google Scholar] [CrossRef]
- Thi, T.T.H.; Lee, Y.; Le Thi, P.; Park, K.D. Engineered horseradish peroxidase-catalyzed hydrogels with high tissue adhesiveness for biomedical applications. J. Ind. Eng. Chem. 2019, 78, 34–52. [Google Scholar]
- Wang, F.; Lu, C.-H.; Willner, I. From cascaded catalytic nucleic acids to enzyme-DNA nanostructures: Controlling reactivity, sensing, logic operations, and assembly of complex structures. Chem. Rev. 2014, 114, 2881–2941. [Google Scholar] [CrossRef]
- Li, F.; Lyu, D.; Liu, S.; Guo, W. DNA hydrogels and microgels for biosensing and biomedical applications. Adv. Mater. 2020, 32, 1806538. [Google Scholar] [CrossRef]
- Li, C.; Faulkner-Jones, A.; Dun, A.R.; Jin, J.; Chen, P.; Xing, Y.; Yang, Z.; Li, Z.; Shu, W.; Liu, D. Rapid formation of a supramolecular polypeptide–DNA hydrogel for in situ three-dimensional multilayer bioprinting. Angew. Chem. 2015, 127, 4029–4033. [Google Scholar] [CrossRef] [Green Version]
- Basu, S.; Pacelli, S.; Feng, Y.; Lu, Q.; Wang, J.; Paul, A. Harnessing the noncovalent interactions of DNA backbone with 2D silicate nanodisks to fabricate injectable therapeutic hydrogels. ACS Nano 2018, 12, 9866–9880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daamen, W.F.; Veerkamp, J.; Van Hest, J.; Van Kuppevelt, T. Elastin as a biomaterial for tissue engineering. Biomaterials 2007, 28, 4378–4398. [Google Scholar] [CrossRef] [PubMed]
- Antoine, E.E.; Vlachos, P.P.; Rylander, M.N. Review of collagen I hydrogels for bioengineered tissue microenvironments: Characterization of mechanics, structure, and transport. Tissue Eng. Part. B Rev. 2014, 20, 683–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, J.; Zhang, X.; Cai, Y.; Wei, Y. Small globular protein motif forms particulate hydrogel under various pH conditions. Biomacromolecules 2011, 12, 1578–1584. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Li, P.; Dong, C.; Jiang, H.; Xue, B.; Gao, X.; Qin, M.; Wang, W.; Chen, B.; Cao, Y. Rationally designed synthetic protein hydrogels with predictable mechanical properties. Nat. Commun. 2018, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Balazs, E.A.; Laurent, T. The Chemistry, Biology and Medical Applications of Hyaluronan and its Derivatives; Portland Press: London, UK, 1998. [Google Scholar]
- Shah, D.N.; Recktenwall-Work, S.M.; Anseth, K.S. The effect of bioactive hydrogels on the secretion of extracellular matrix molecules by valvular interstitial cells. Biomaterials 2008, 29, 2060–2072. [Google Scholar] [CrossRef] [Green Version]
- Van Dijk, M.; Van Nostrum, C.F.; Hennink, W.E.; Rijkers, D.T.; Liskamp, R.M. Synthesis and characterization of enzymatically biodegradable PEG and peptide-based hydrogels prepared by click chemistry. Biomacromolecules 2010, 11, 1608–1614. [Google Scholar] [CrossRef]
- Rubert Pérez, C.M.; Panitch, A.; Chmielewski, J. A collagen peptide-based physical hydrogel for cell encapsulation. Macromol. Biosci. 2011, 11, 1426–1431. [Google Scholar] [CrossRef]
- Grieshaber, S.E.; Farran, A.J.; Lin-Gibson, S.; Kiick, K.L.; Jia, X. Synthesis and characterization of elastin—mimetic hybrid polymers with multiblock, alternating molecular architecture and elastomeric properties. Macromolecules 2009, 42, 2532–2541. [Google Scholar] [CrossRef] [Green Version]
- Hamley, I.W.; Cheng, G.; Castelletto, V. A Thermoresponsive Hydrogel Based on Telechelic PEG end-capped with hydrophobic dipeptides. Macromol. Biosci. 2011, 11, 1068–1078. [Google Scholar] [CrossRef]
- Anderson, S.B.; Lin, C.-C.; Kuntzler, D.V.; Anseth, K.S. The performance of human mesenchymal stem cells encapsulated in cell-degradable polymer-peptide hydrogels. Biomaterials 2011, 32, 3564–3574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.H.; Liao, W.C.; Sohn, Y.S.; Fadeev, M.; Cecconello, A.; Nechushtai, R.; Willner, I. Stimuli-responsive nucleic acid-based polyacrylamide hydrogel-coated metal–organic framework nanoparticles for controlled drug release. Adv. Funct. Mater. 2018, 28, 1705137. [Google Scholar] [CrossRef]
- Lu, H.; Yuan, L.; Yu, X.; Wu, C.; He, D.; Deng, J. Recent advances of on-demand dissolution of hydrogel dressings. Burns Trauma 2018, 6, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Wang, Y.; Yang, H.; Peng, K.; Zhang, X. Injectable self-healing hydrogels formed via thiol/disulfide exchange of thiol functionalized F127 and dithiolane modified PEG. J. Mater. Chem. B 2017, 5, 4121–4127. [Google Scholar] [CrossRef] [PubMed]
- Stahl, P.J.; Romano, N.H.; Wirtz, D.; Yu, S.M. PEG-based hydrogels with collagen mimetic peptide-mediated and tunable physical cross-links. Biomacromolecules 2010, 11, 2336–2344. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhu, C.N.; Zeng, H.; Ji, X.; Xie, T.; Yan, X.; Wu, Z.L.; Huang, F. Reversible ion-conducting switch in a novel single-ion supramolecular hydrogel enabled by photoresponsive host–guest molecular recognition. Adv. Mater. 2019, 31, 1807328. [Google Scholar] [CrossRef] [PubMed]
- Xiao, T.; Xu, L.; Zhou, L.; Sun, X.-Q.; Lin, C.; Wang, L. Dynamic hydrogels mediated by macrocyclic host–guest interactions. J. Mater. Chem. B 2019, 7, 1526–1540. [Google Scholar] [CrossRef]
- Domiński, A.; Konieczny, T.; Kurcok, P. α-cyclodextrin-based polypseudorotaxane hydrogels. Materials 2020, 13, 133. [Google Scholar] [CrossRef] [Green Version]
- Huynh, V.; Wylie, R.G. Competitive affinity release for long-term delivery of antibodies from hydrogels. Angew. Chem. Int. Ed. 2018, 57, 3406–3410. [Google Scholar] [CrossRef]
- Fan, H.; Wang, J.; Tao, Z.; Huang, J.; Rao, P.; Kurokawa, T.; Gong, J.P. Adjacent cationic–aromatic sequences yield strong electrostatic adhesion of hydrogels in seawater. Nat. Commun. 2019, 10, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Purcell, B.P.; Lobb, D.; Charati, M.B.; Dorsey, S.M.; Wade, R.J.; Zellars, K.N.; Doviak, H.; Pettaway, S.; Logdon, C.B.; Shuman, J.A.; et al. Injectable and bioresponsive hydrogels for on-demand matrix metalloproteinase inhibition. Nat. Mater. 2014, 13, 653–661. [Google Scholar] [CrossRef] [Green Version]
- Chilin, C.; Metters, A. Hydrogels in controlled release formulations: Network design and mathematical modelling. Adv. Drug Deliv. Rev. 2006, 58, 1379–1408. [Google Scholar]
- Censi, R.; Casadidio, C.; Dubbini, A.; Cortese, M.; Scuri, S.; Grappasonni, I.; Golob, S.; Vojnovic, D.; Sabbieti, M.G.; Agas, D.; et al. Thermosensitive hybrid hydrogels for the controlled release of bioactive vancomycin in the treatment of orthopaedic implant infections. Eur. J. Pharm. Biopharm. 2019, 142, 322–333. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Berry, D.; Moran, A.; He, F.; Tam, T.; Chen, L.; Chen, S. Controlled growth factor release in 3D-printed hydrogels. Adv. Healthc. Mater. 2020, 9, 1900977. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Jiang, S.; Yuan, Q.; Li, G.; Wang, F.; Zhang, Z.; Liu, J. Co-immobilization of multiple enzymes by metal coordinated nucleotide hydrogel nanofibers: Improved stability and an enzyme cascade for glucose detection. Nanoscale 2016, 8, 6071–6078. [Google Scholar] [CrossRef]
- Chatterjee, S.; Hui, P.C.-L. Stimuli-responsive hydrogels: An interdisciplinary overview. In Hydrogels-Smart Materials for Biomedical Applications; IntechOpen: London, UK, 2018. [Google Scholar]
- Koetting, M.C.; Peters, J.T.; Steichen, S.D.; Peppas, N.A. Stimulus-responsive hydrogels: Theory, modern advances, and applications. Mater. Sci. Eng. R Rep. 2015, 93, 1–49. [Google Scholar] [CrossRef] [PubMed]
- Miyata, T. Biomolecule-sensitive hydrogels. Smart Mater. Drug Deliv. 2013, 2, 261. [Google Scholar] [CrossRef]
- Yan, L.; Zhu, Z.; Zou, Y.; Huang, Y.; Liu, D.; Jia, S.; Xu, D.; Wu, M.; Zhou, Y.; Zhou, S.; et al. Target-responsive “sweet” hydrogel with glucometer readout for portable and quantitative detection of non-Glucose targets. J. Am. Chem. Soc. 2013, 135, 3748–3751. [Google Scholar] [CrossRef]
- Knipe, J.M.; Chen, F.; Peppas, N.A. Enzymatic biodegradation of hydrogels for protein delivery targeted to the small intestine. Biomacromolecules 2015, 16, 962–972. [Google Scholar] [CrossRef]
- Liang, J.; Xiao, X.; Chou, T.-M.; Libera, M. Counterion exchange in peptide-complexed core–shell microgels. Langmuir 2019, 35, 9521–9528. [Google Scholar] [CrossRef]
- Huebsch, N.; Kearney, C.J.; Zhao, X.; Kim, J.; Cezar, C.A.; Suo, Z.; Mooney, D.J. Ultrasound-triggered disruption and self-healing of reversibly cross-linked hydrogels for drug delivery and enhanced chemotherapy. Proc. Natl. Acad. Sci. USA 2014, 111, 9762–9767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chhibber, T.; Shinde, R.; Lahooti, B.; Bagchi, S.; Varahachalam, S.P.; Gaddam, A.; Jaiswal, A.K.; Gracia, E.; Chand, H.S.; Kaushik, A. Hydrogels in tissue engineering. In Intelligent Hydrogels in Diagnostics and Therapeutics; CRC Press: Boca Raton, FL, USA, 2020; pp. 105–122. [Google Scholar]
- Nuttelman, C.R.; Rice, M.A.; Rydholm, A.E.; Salinas, C.N.; Shah, D.N.; Anseth, K.S. Macromolecular monomers for the synthesis of hydrogel niches and their application in cell encapsulation and tissue engineering. Prog. Polym. Sci. 2008, 33, 167–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, X.; Kiick, K.L. Hybrid multicomponent hydrogels for tissue engineering. Macromol. Biosci. 2009, 9, 140–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, J.D.; Crick, F.H. Molecular structure of nucleic acids: A structure for deoxyribose nucleic acid. Nature 1953, 171, 737–738. [Google Scholar] [CrossRef] [PubMed]
- Stoleru, E.; Vasile, C. Nucleic acids–based bionanomaterials for drug and gene therapy. In Polymeric Nano Materials in Nanotherapeutics; Elsevier: Amsterdam, The Netherlands, 2019; pp. 235–259. [Google Scholar]
- Nummelin, S.; Kommeri, J.; Kostiainen, M.A.; Linko, V. Evolution of structural DNA nanotechnology. Adv. Mater. 2018, 30, 1703721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chi, J.; Shao, C.; Zhang, Y.; Ni, D.; Kong, T.; Zhao, Y. Magnetically responsive colloidal crystals with angle-independent gradient structural colors in microfluidic droplet arrays. Nanoscale 2019, 11, 12898–12904. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Tang, J.; Geng, J.; Luo, D.; Yang, D. Polymeric DNA hydrogel: Design, synthesis and applications. Prog. Polym. Sci. 2019, 98, 101163. [Google Scholar] [CrossRef]
- Dhandayuthapani, B.; Yoshida, Y.; Maekawa, T.; Kumar, D.S. Polymeric scaffolds in tissue engineering application: A review. Int. J. Polym. Sci. 2011, 2011. [Google Scholar] [CrossRef]
- Shahbazi, M.A.; Bauleth-Ramos, T.; Santos, H.A. DNA hydrogel assemblies: Bridging synthesis principles to biomedical applications. Adv. Ther. 2018, 1, 1800042. [Google Scholar] [CrossRef]
- Song, J.; Gu, Y.C.; Xu, X.; Luo, F.; Tang, X.H.; Xie, P.; Qian, Z.Y. Synthesis and characterization of pH-sensitive hydrogel based on methoxyl poly (ethylene glycol), poly (ε-caprolactone) and itaconic acid for delivery of doxorubicin. Adv. Sci. Lett. 2012, 16, 130–136. [Google Scholar] [CrossRef]
- Guaresti, O.; García–Astrain, C.; Palomares, T.; Alonso–Varona, A.; Eceiza, A.; Gabilondo, N. Synthesis and characterization of a biocompatible chitosan–based hydrogel cross–linked via ‘click’chemistry for controlled drug release. Int. J. Biol. Macromol. 2017, 102, 1–9. [Google Scholar] [CrossRef]
- Daniele, M.A.; Adams, A.A.; Naciri, J.; North, S.H.; Ligler, F.S. Interpenetrating networks based on gelatin methacrylamide and PEG formed using concurrent thiol click chemistries for hydrogel tissue engineering scaffolds. Biomaterials 2014, 35, 1845–1856. [Google Scholar] [CrossRef]
- Zhou, L.; Hu, H.; Zhang, Y.-X.; Meng, Q.-Y.; Yu, B.; Shen, Y.-Q.; Cong, H.-L. A near-infrared triggered intracellular pH regulative PAMAM/O-nitrobenzaldehyde coated UCNPs for cancer therapy. Integr. Ferroelectr. 2019, 199, 85–94. [Google Scholar] [CrossRef]
- Heinen, L.; Heuser, T.; Steinschulte, A.; Walther, A. Antagonistic enzymes in a biocatalytic pH feedback system program autonomous DNA hydrogel life cycles. Nano Lett. 2017, 17, 4989–4995. [Google Scholar] [CrossRef] [PubMed]
- Lau, H.K.; Kiick, K.L. Opportunities for multicomponent hybrid hydrogels in biomedical applications. Biomacromolecules 2015, 16, 28–42. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.; Yao, C.; Gu, Z.; Jung, S.; Luo, D.; Yang, D. Super-soft and super-elastic DNA robot with magnetically driven navigational locomotion for cell delivery in confined space. Angew. Chem. Int. Ed. 2020, 59, 2490–2495. [Google Scholar] [CrossRef] [PubMed]
- Mengatto, L.; Ferreyra, M.G.; Rubiolo, A.; Rintoul, I.; Luna, J. Hydrophilic and hydrophobic interactions in cross-linked chitosan membranes. Mater. Chem. Phys. 2013, 139, 181–186. [Google Scholar] [CrossRef]
- Fan, H.; Wang, J.; Jin, Z. Tough, swelling-resistant, self-healing, and adhesive dual-cross-linked hydrogels based on polymer–tannic acid multiple hydrogen bonds. Macromolecules 2018, 51, 1696–1705. [Google Scholar] [CrossRef]
- Xu, Y.; Wu, Q.; Sun, Y.; Bai, H.; Shi, G. Three-dimensional self-assembly of graphene oxide and DNA into multifunctional hydrogels. ACS Nano 2010, 4, 7358–7362. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Duan, X.; Feng, X.; Liu, L.; Wang, S.; Li, Y.; Zhu, D. Fluorescent DNA–poly (phenylenevinylene) hybrid hydrogels for monitoring drug release. Chem. Commun. 2009, 6, 641–643. [Google Scholar] [CrossRef]
- Wang, S.; Fan, W.; Liu, Z.; Yu, A.; Jiang, X. Advances on tungsten oxide based photochromic materials: Strategies to improve their photochromic properties. J. Mater. Chem. C 2018, 6, 191–212. [Google Scholar] [CrossRef]
- Shao, Y.; Jia, H.; Cao, T.; Liu, D. Supramolecular hydrogels based on DNA self-assembly. Acc. Chem. Res. 2017, 50, 659–668. [Google Scholar] [CrossRef]
- Wang, P.; Huang, S.; Hu, Z.; Yang, W.; Lan, Y.; Zhu, J.; Hancharou, A.; Guo, R.; Tang, B. In situ formed anti-inflammatory hydrogel loading plasmid DNA encoding VEGF for burn wound healing. Acta Biomater. 2019, 100, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, S.; Benoit, D.S. Degradable poly (ethylene glycol)(PEG)-based hydrogels for spatiotemporal control of siRNA/nanoparticle delivery. J. Control. Release 2018, 287, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, V.; Naskar, D.; Kinnear, B.F.; Grenik, E.; Dye, D.E.; Grounds, M.D.; Kundu, S.C.; Coombe, D.R. Silk fibroin scaffolds with muscle-like elasticity support in vitro differentiation of human skeletal muscle cells. J. Tissue Eng. Regen. Med. 2017, 11, 3178–3192. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Heilshorn, S.C. Adaptable hydrogel networks with reversible linkages for tissue engineering. Adv. Mater. 2015, 27, 3717–3736. [Google Scholar] [CrossRef]
- Wang, L.L.; Burdick, J.A. Engineered Hydrogels for local and sustained delivery of RNA-interference therapies. Adv. Healthc. Mater. 2017, 6, 1601041. [Google Scholar] [CrossRef]
- Tang, Z.; Chen, Q.; Chen, F.; Zhu, L.; Lu, S.; Ren, B.; Zhang, Y.; Yang, J.; Zheng, J. General principle for fabricating natural globular protein-based double-network hydrogels with integrated highly mechanical properties and surface adhesion on solid surfaces. Chem. Mater. 2018, 31, 179–189. [Google Scholar] [CrossRef]
- Mouw, J.K.; Ou, G.; Weaver, V.M. Extracellular matrix assembly: A multiscale deconstruction. Nat. Rev. Mol. Cell Biol. 2014, 15, 771–785. [Google Scholar] [CrossRef]
- Yang, Z.; Kou, S.; Wei, X.; Zhang, F.; Li, F.; Wang, X.-W.; Lin, Y.; Wan, C.; Zhang, W.-B.; Sun, F. Genetically programming stress-relaxation behavior in entirely protein-based molecular networks. ACS Macro Lett. 2018, 7, 1468–1474. [Google Scholar] [CrossRef]
- Sun, W.; Duan, T.; Cao, Y.; Li, H. An injectable self-healing protein hydrogel with multiple dissipation modes and tunable dynamic response. Biomacromolecules 2019, 20, 4199–4207. [Google Scholar] [CrossRef] [PubMed]
- Lv, S.; Dudek, D.M.; Cao, Y.; Balamurali, M.; Gosline, J.; Li, H. Designed biomaterials to mimic the mechanical properties of muscles. Nature 2010, 465, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Kharkar, P.M.; Rehmann, M.S.; Skeens, K.M.; Maverakis, E.; Kloxin, A.M. Thiol–ene click hydrogels for therapeutic delivery. ACS Biomater. Sci. Eng. 2016, 2, 165–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gramlich, W.M.; Kim, I.L.; Burdick, J.A. Synthesis and orthogonal photopatterning of hyaluronic acid hydrogels with thiol-norbornene chemistry. Biomaterials 2013, 34, 9803–9811. [Google Scholar] [CrossRef] [Green Version]
- Nair, D.P.; Podgorski, M.; Chatani, S.; Gong, T.; Xi, W.; Fenoli, C.R.; Bowman, C.N. The thiol-Michael addition click reaction: A powerful and widely used tool in materials chemistry. Chem. Mater. 2014, 26, 724–744. [Google Scholar] [CrossRef]
- Kim, Y.; Ho, S.O.; Gassman, N.R.; Korlann, Y.; Landorf, E.V.; Collart, F.R.; Weiss, S. Efficient site-specific labeling of proteins via cysteines. Bioconjugate Chem. 2008, 19, 786–791. [Google Scholar] [CrossRef] [Green Version]
- DeForest, C.A.; Anseth, K.S. Back Cover: Photoreversible patterning of biomolecules within click-based hydrogels. Angew. Chem. Int. Ed. 2012, 51, 1978. [Google Scholar] [CrossRef]
- Hoyle, C.E.; Bowman, C.N. Thiol-ene click chemistry. Angew. Chem. Int. Ed. 2010, 49, 1540–1573. [Google Scholar] [CrossRef]
- Sun, F.; Zhang, W.-B. Unleashing chemical power from protein sequence space toward genetically encoded “click” chemistry. Chin. Chem. Lett. 2017, 28, 2078–2084. [Google Scholar] [CrossRef]
- Zakeri, B.; Fierer, J.O.; Celik, E.; Chittock, E.C.; Schwarz-Linek, U.; Moy, V.T.; Howarth, M. Peptide tag forming a rapid covalent bond to a protein, through engineering a bacterial adhesin. Proc. Natl. Acad. Sci. USA 2012, 109, E690–E697. [Google Scholar] [CrossRef] [Green Version]
- Keppler, A.; Gendreizig, S.; Gronemeyer, T.; Pick, H.; Vogel, H.; Johnsson, K. A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat. Biotechnol. 2003, 21, 86–89. [Google Scholar] [CrossRef] [PubMed]
- Veggiani, G.; Nakamura, T.; Brenner, M.D.; Gayet, R.V.; Yan, J.; Robinson, C.V.; Howarth, M. Programmable polyproteams built using twin peptide superglues. Proc. Natl. Acad. Sci. USA 2016, 113, 1202–1207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gautier, A.; Juillerat, A.; Heinis, C.; Corrêa Jr, I.R.; Kindermann, M.; Beaufils, F.; Johnsson, K. An engineered protein tag for multiprotein labeling in living cells. Chem. Biol. 2008, 15, 128–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyu, S.; Fang, J.; Duan, T.; Fu, L.; Liu, J.; Li, H. Optically controlled reversible protein hydrogels based on photoswitchable fluorescent protein Dronpa. Chem. Commun. 2017, 53, 13375–13378. [Google Scholar] [CrossRef] [PubMed]
- Ehrick, J.D.; Deo, S.K.; Browning, T.W.; Bachas, L.G.; Madou, M.J.; Daunert, S. Genetically engineered protein in hydrogels tailors stimuli-responsive characteristics. Nat. Mater. 2005, 4, 298–302. [Google Scholar] [CrossRef]
- Murphy, W.L.; Dillmore, W.S.; Modica, J.; Mrksich, M. Dynamic hydrogels: Translating a protein conformational change into macroscopic motion. Angew. Chem. Int. Ed. 2007, 46, 3066–3069. [Google Scholar] [CrossRef]
- Miyata, T.; Asami, N.; Uragami, T. A reversibly antigen-responsive hydrogel. Nature 1999, 399, 766–769. [Google Scholar] [CrossRef]
- Yuan, W.; Yang, J.; Kopečková, P.; Kopecek, J. Smart hydrogels containing adenylate kinase: Translating substrate recognition into macroscopic motion. J. Am. Chem. Soc. 2008, 130, 15760–15761. [Google Scholar] [CrossRef] [Green Version]
- Lorden, E.R.; Levinson, H.M.; Leong, K.W. Integration of drug, protein, and gene delivery systems with regenerative medicine. Drug Deliv. Transl. Res. 2015, 5, 168–186. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, P.K.; Sarkar, B.; Siddiqui, Z.; McGowan, M.; Iglesias-Montoro, P.; Rachapudi, S.; Kim, S.; Gao, W.; Lee, E.J.; Kumar, V.A. Self-assembly of an antiangiogenic nanofibrous peptide hydrogel. ACS Appl. Biomater. 2018, 1, 865–870. [Google Scholar] [CrossRef]
- Huynh, V.; Wylie, R.G. Displacement affinity release of antibodies from injectable hydrogels. ACS Appl. Mater. Interfaces 2019, 11, 30648–30660. [Google Scholar] [CrossRef] [PubMed]
- Selvan, N.K.; Shanmugarajan, T.; Uppuluri, V.N.V.A. Hydrogel based scaffolding polymeric biomaterials: Approaches towards skin tissue regeneration. J. Drug Deliv. Sci. Technol. 2020, 55, 101456. [Google Scholar] [CrossRef]
- Xu, F.; Zou, D.; Dai, T.; Xu, H.; An, R.; Liu, Y.; Liu, B. Effects of incorporation of granule-lyophilised platelet-rich fibrin into polyvinyl alcohol hydrogel on wound healing. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef]
- Latifi, N.; Asgari, M.; Vali, H.; Mongeau, L. A tissue-mimetic nano-fibrillar hybrid injectable hydrogel for potential soft tissue engineering applications. Sci. Rep. 2018, 8, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calori, G.M.; Donati, D.; Di Bella, C.; Tagliabue, L. Bone morphogenetic proteins and tissue engineering: Future directions. Injury 2009, 40, S67–S76. [Google Scholar] [CrossRef]
- Zhang, X.; Kang, X.; Jin, L.; Bai, J.; Liu, W.; Wang, Z. Stimulation of wound healing using bioinspired hydrogels with basic fibroblast growth factor (bFGF). Int. J. Nanomed. 2018, 13, 3897. [Google Scholar] [CrossRef] [Green Version]
- Chong, B.F.; Blank, L.M.; Mclaughlin, R.; Nielsen, L.K. Microbial hyaluronic acid production. Appl. Microbiol. Biotechnol. 2005, 66, 341–351. [Google Scholar] [CrossRef]
- Serban, M.A.; Prestwich, G.D. Modular extracellular matrices: Solutions for the puzzle. Methods 2008, 45, 93–98. [Google Scholar] [CrossRef] [Green Version]
- Jonker, A.M.; Löwik, D.W.; Van Hest, J.C. Peptide-and protein-based hydrogels. Chem. Mater. 2012, 24, 759–773. [Google Scholar] [CrossRef]
- Worthington, P.; Pochan, D.J.; Langhans, S.A. Peptide hydrogels–versatile matrices for 3D cell culture in cancer medicine. Front. Oncol. 2015, 5, 92. [Google Scholar] [CrossRef] [Green Version]
- Loo, Y.; Hauser, C.A. Bioprinting synthetic self-assembling peptide hydrogels for biomedical applications. Biomed. Mater. 2015, 11, 014103. [Google Scholar] [CrossRef] [PubMed]
- Cobo, I.; Li, M.; Sumerlin, B.S.; Perrier, S. Smart hybrid materials by conjugation of responsive polymers to biomacromolecules. Nat. Mater. 2015, 14, 143–159. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, N.; Zhang, L.; Chae, B.-S.; Palla, C.S.; Furst, E.M.; Kiick, K.L. Growth factor mediated assembly of cell receptor-responsive hydrogels. J. Am. Chem. Soc. 2007, 129, 3040–3041. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.J.; Lee, J.-S.; Chansakul, T.; Yu, C.; Elisseeff, J.H.; Seungju, M.Y. Collagen mimetic peptide-conjugated photopolymerizable PEG hydrogel. Biomaterials 2006, 27, 5268–5276. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Stewart, R.J.; KopeČek, J. Hybrid hydrogels assembled from synthetic polymers and coiled-coil protein domains. Nature 1999, 397, 417–420. [Google Scholar] [CrossRef] [PubMed]
- Stoica, F.; Alexander, C.; Tirelli, N.; Miller, A.F.; Saiani, A. Selective synthesis of double temperature-sensitive polymer–peptide conjugates. Chem. Commun. 2008, 37, 4433–4435. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; McRae, N.L.; McCulloch, D.R.; Boyd-Moss, M.; Barrow, C.J.; Nisbet, D.R.; Stupka, N.; Williams, R.J. Large and small assembly: Combining functional macromolecules with small peptides to control the morphology of skeletal muscle progenitor cells. Biomacromolecules 2018, 19, 825–837. [Google Scholar] [CrossRef] [PubMed]
- Diaferia, C.; Ghosh, M.; Sibillano, T.; Gallo, E.; Stornaiuolo, M.; Giannini, C.; Morelli, G.; Adler-Abramovich, L.; Accardo, A. Fmoc-FF and hexapeptide-based multicomponent hydrogels as scaffold materials. Soft Matter 2019, 15, 487–496. [Google Scholar] [CrossRef]
- Ghosh, M.; Halperin-Sternfeld, M.; Grigoriants, I.; Lee, J.; Nam, K.T.; Adler-Abramovich, L. Arginine-presenting peptide hydrogels decorated with hydroxyapatite as biomimetic scaffolds for bone regeneration. Biomacromolecules 2017, 18, 3541–3550. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer | Abbreviation | Structure | Properties | Applications | Refs. |
|---|---|---|---|---|---|
| Poly(2-hydroxyethyl methacrylate) | PHEMA |  | Hydrophilic, biocompatible | cell growth; tissue engineering; regenerative medicine | [134,135,136] |
| Poly(2-hydroxypropyl methacrylate) | PHPMA |  | Hydrophilic, biocompatible | cell growth; tissue regeneration | [125] |
| Poly(ethylene glycol)/poly(ethylene oxide) | PEG/PEO |  | Hydrophilic, biocompatible | cell growth; tissue engineering; regenerative medicine | [137,138] |
| Poly(2-methyl-2-oxazoline) | PMOXA |  | Hydrophilic, biocompatible | cell growth; tissue regeneration | [139,140] |
| Poly(vinyl alcohol) | PVA |  | Hydrophilic, biocompatible | tissue engineering; regenerative medicine | [141,142,143] |
| Poly(acrylic acid) | PAAc |  | Hydrophilic, pH-responsive | cell growth | [144] |
| Poly [2-(dimethylamino)ethyl methacrylate] | PDMAEMA |  | Hydrophilic, pH-responsive | biomolecule delivery, regenerative medicine | [145,146] |
| Poly(ethylenimine) | PEI |  | Hydrophilic, pH-responsive | biomolecule delivery | [147,148] |
| Poly(acrylamide) | PAAm |  | Hydrophilic, biocompatible | antigen sensing, regenerative medicine | [149] |
| Poly(N-vinyl pyrrolidone) | PNVP |  | Hydrophilic, biocompatible | biomolecule delivery; tissue engineering | [150] |
| Poly(N-isopropyl acrylamide) | PNIPAM |  | Hydrophilic, biocompatible, temperature responsive | biomolecule delivery; cell growth; tissue engineering | [145,151] |
| Poly(ε-caprolactone) | PCL |  | Hydrophobic, biodegradable | cell growth; tissue engineering | [152,153] |
| Poly(lactic acid) | PLA |  | Hydrophobic, biodegradable, biocompatible | biomolecule delivery; tissue engineering | [154] |
| Poly(glycolic acid) | PGA |  | Slightly hydrophilic, biodegradable | Biomolecule delivery; tissue engineering | [155,156,157] |
| Poly(N-vinylcaprolactam) | PVCL |  | Hydrophobic, biocompatible, temperature responsive | Biomolecule delivery; theranostic; tissue engineering | [158,159,160] |
| Biomolecule | Conjugation Methods | Applications | Refs. |
|---|---|---|---|
| Oligonucleotides | Covalent attachment, non-covalent crosslinking, electrostatic interactions | Drug delivery, cell culture | [171,172,173,174] |
| Proteins (GFs, enzymes) | Covalent attachment, non-covalent crosslinking (host-guest chemistry), electrostatic interactions | tissue regeneration | [175,176,177,178] |
| Carbohydrates | Covalent attachment, non-covalent attachment | tissue regeneration | [179,180] |
| Peptides | Covalent attachment | tissue adhesives, regeneration | [181,182,183,184] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chimisso, V.; Aleman Garcia, M.A.; Yorulmaz Avsar, S.; Dinu, I.A.; Palivan, C.G. Design of Bio-Conjugated Hydrogels for Regenerative Medicine Applications: From Polymer Scaffold to Biomolecule Choice. Molecules 2020, 25, 4090. https://doi.org/10.3390/molecules25184090
Chimisso V, Aleman Garcia MA, Yorulmaz Avsar S, Dinu IA, Palivan CG. Design of Bio-Conjugated Hydrogels for Regenerative Medicine Applications: From Polymer Scaffold to Biomolecule Choice. Molecules. 2020; 25(18):4090. https://doi.org/10.3390/molecules25184090
Chicago/Turabian StyleChimisso, Vittoria, Miguel Angel Aleman Garcia, Saziye Yorulmaz Avsar, Ionel Adrian Dinu, and Cornelia G. Palivan. 2020. "Design of Bio-Conjugated Hydrogels for Regenerative Medicine Applications: From Polymer Scaffold to Biomolecule Choice" Molecules 25, no. 18: 4090. https://doi.org/10.3390/molecules25184090