
Impact of N-Alkylamino Substituents on Serotonin Receptor (5-HTR) Affinity and Phosphodiesterase 10A (PDE10A) Inhibition of Isoindole-1,3-dione Derivatives

 , , , , , and
, , , , , and
Abstract
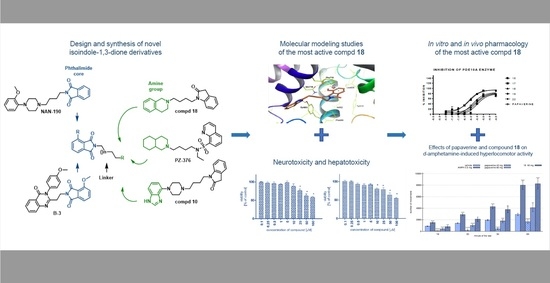
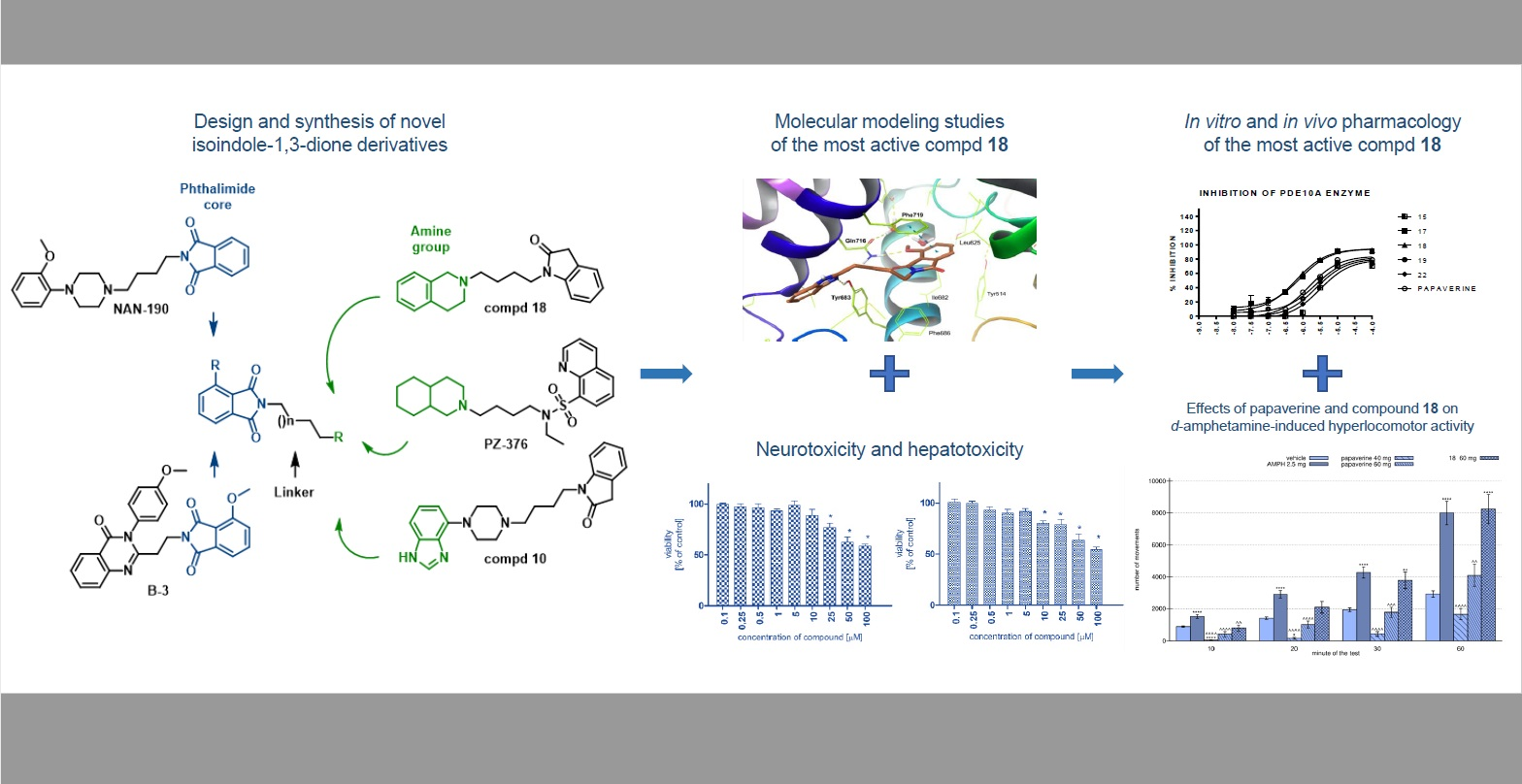
:
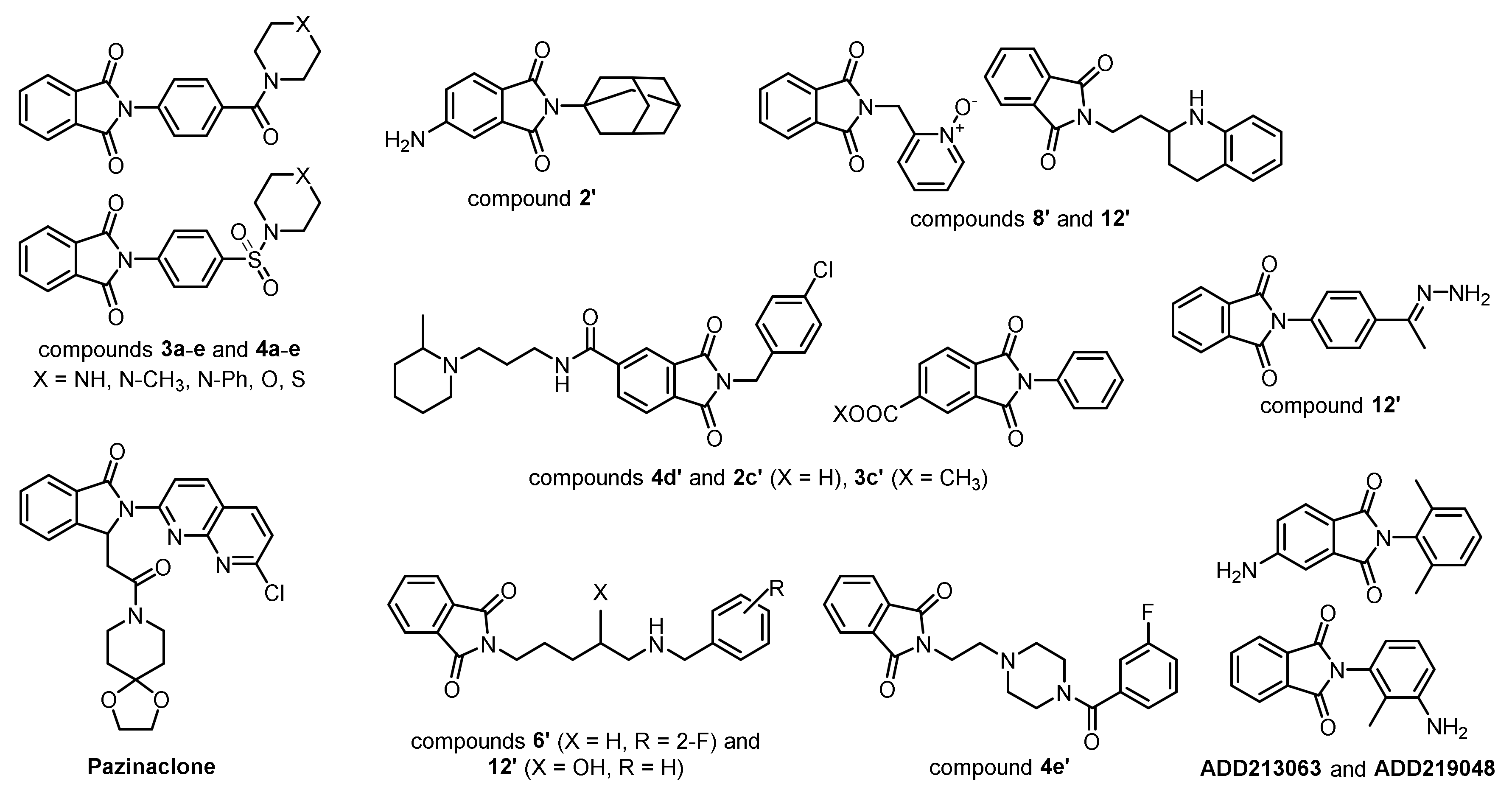
1. Introduction
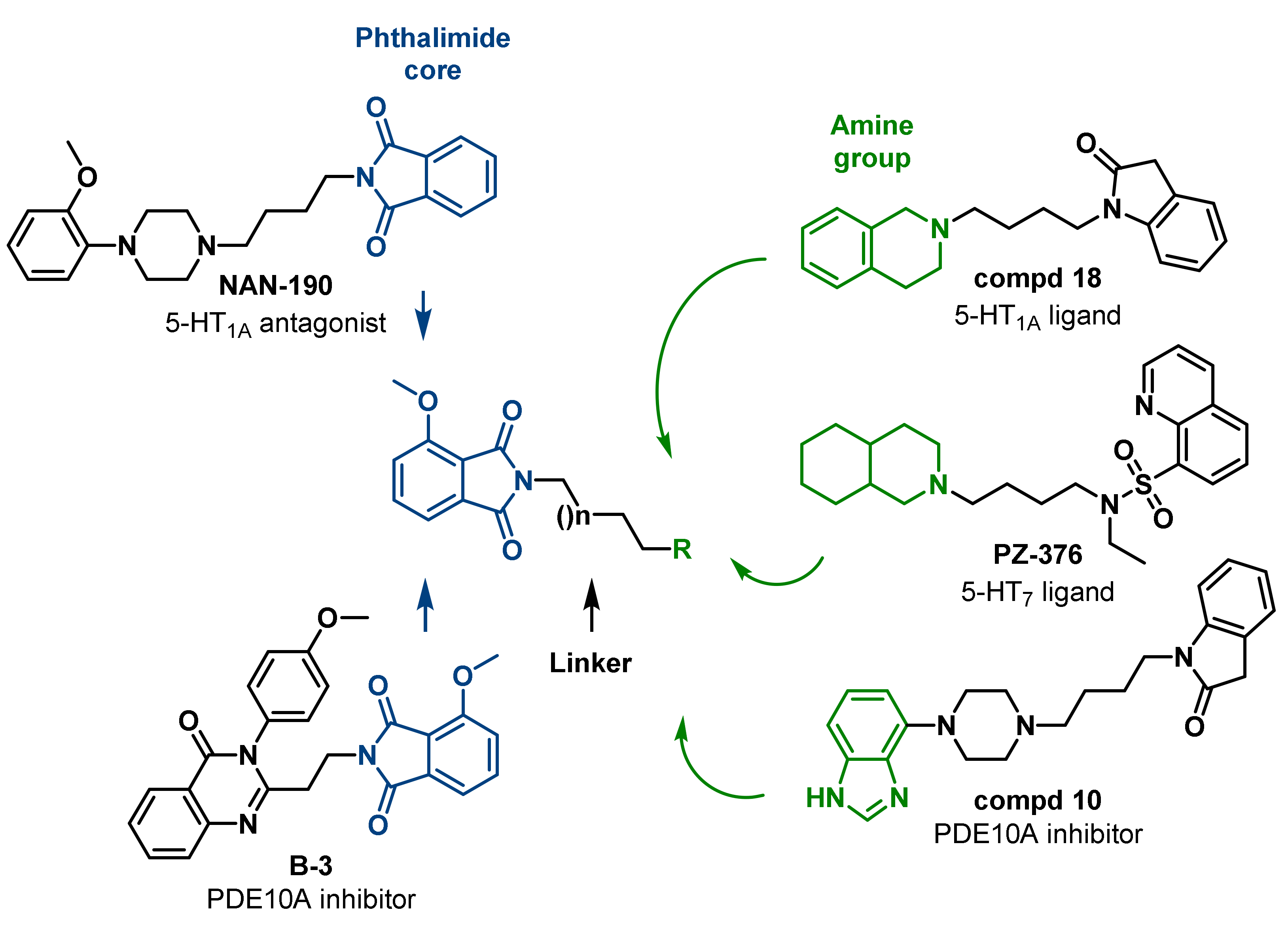
2. Results and Discussion
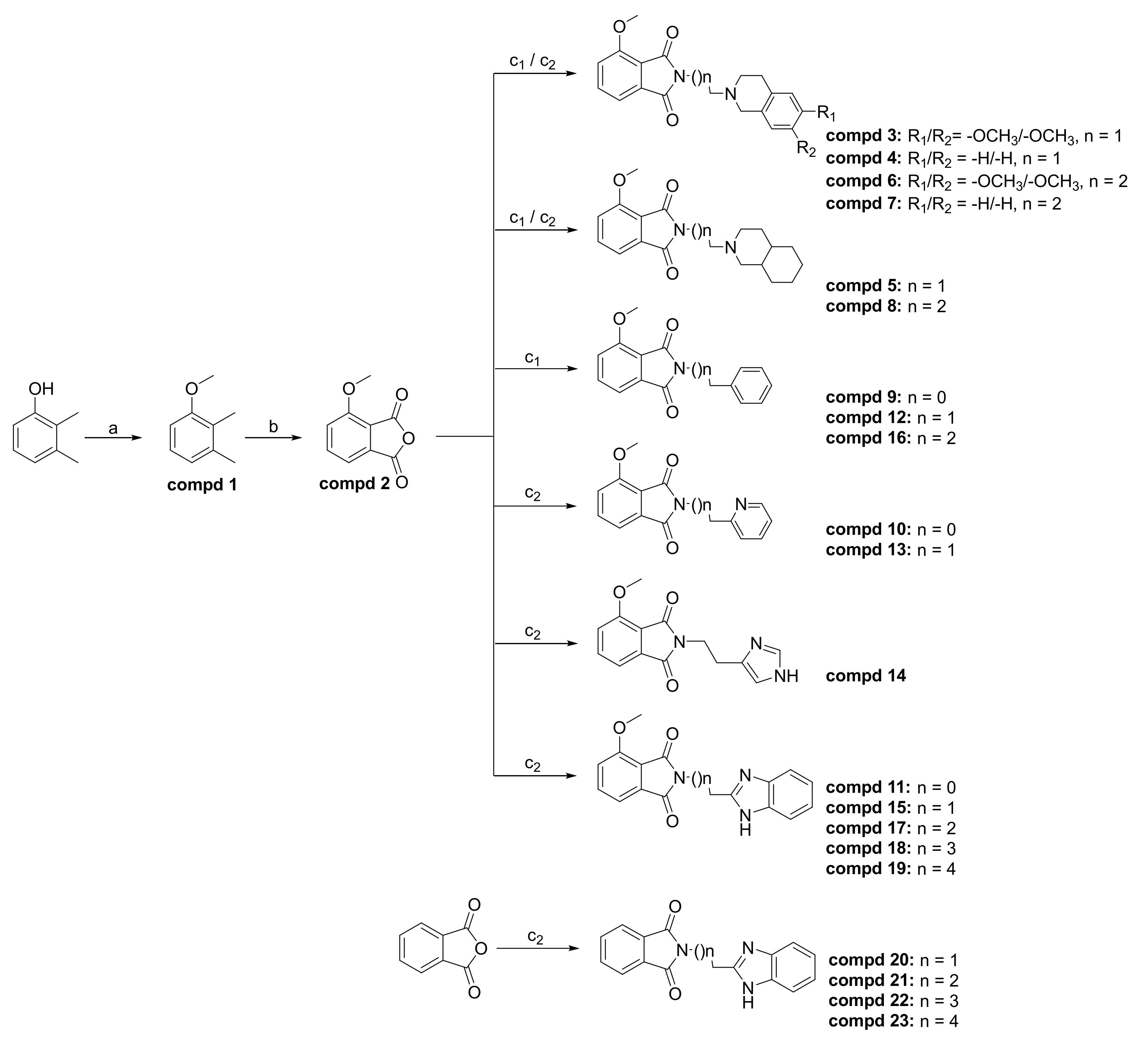
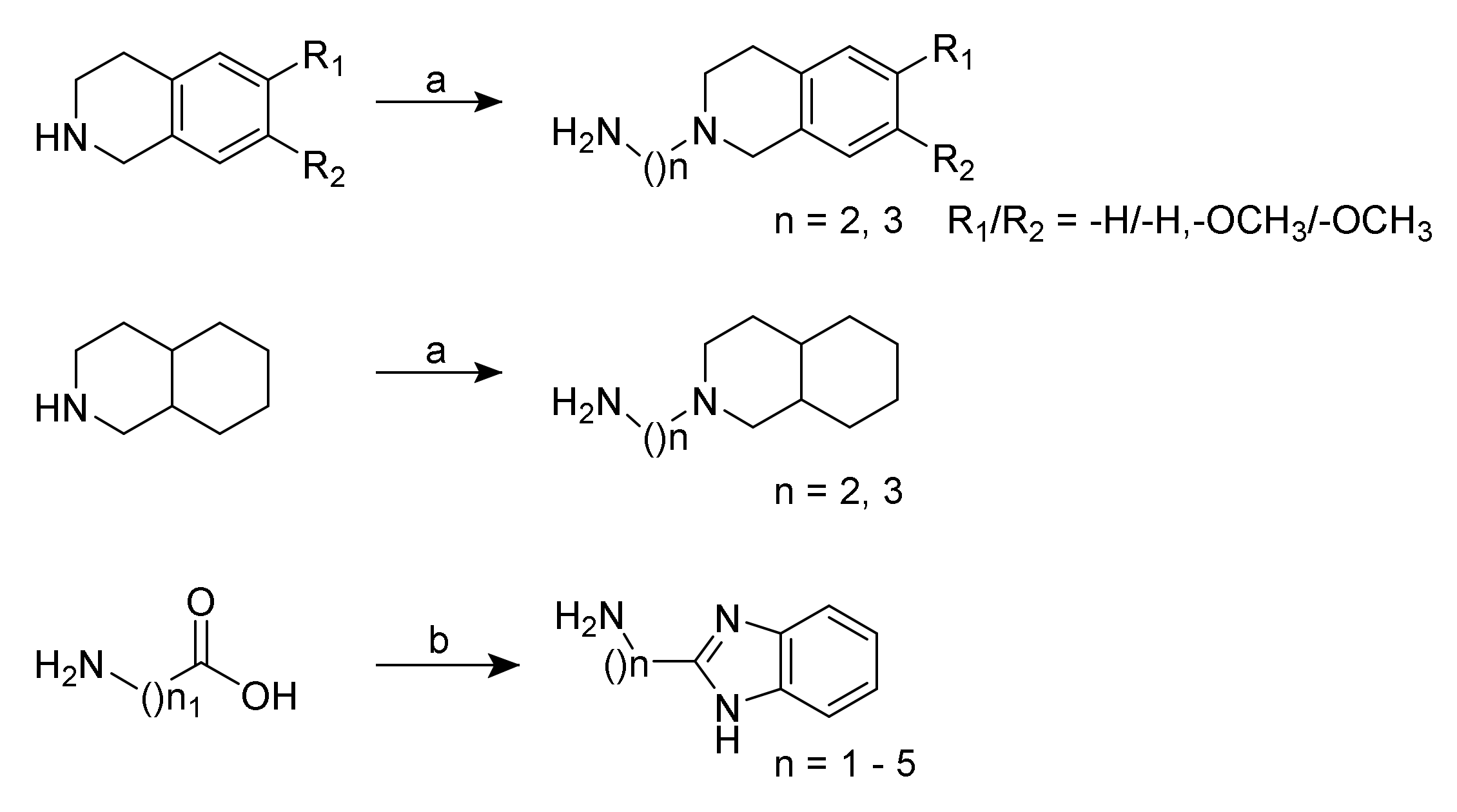
2.1. Chemistry
2.2. In Vitro Activity of Compounds 3–23
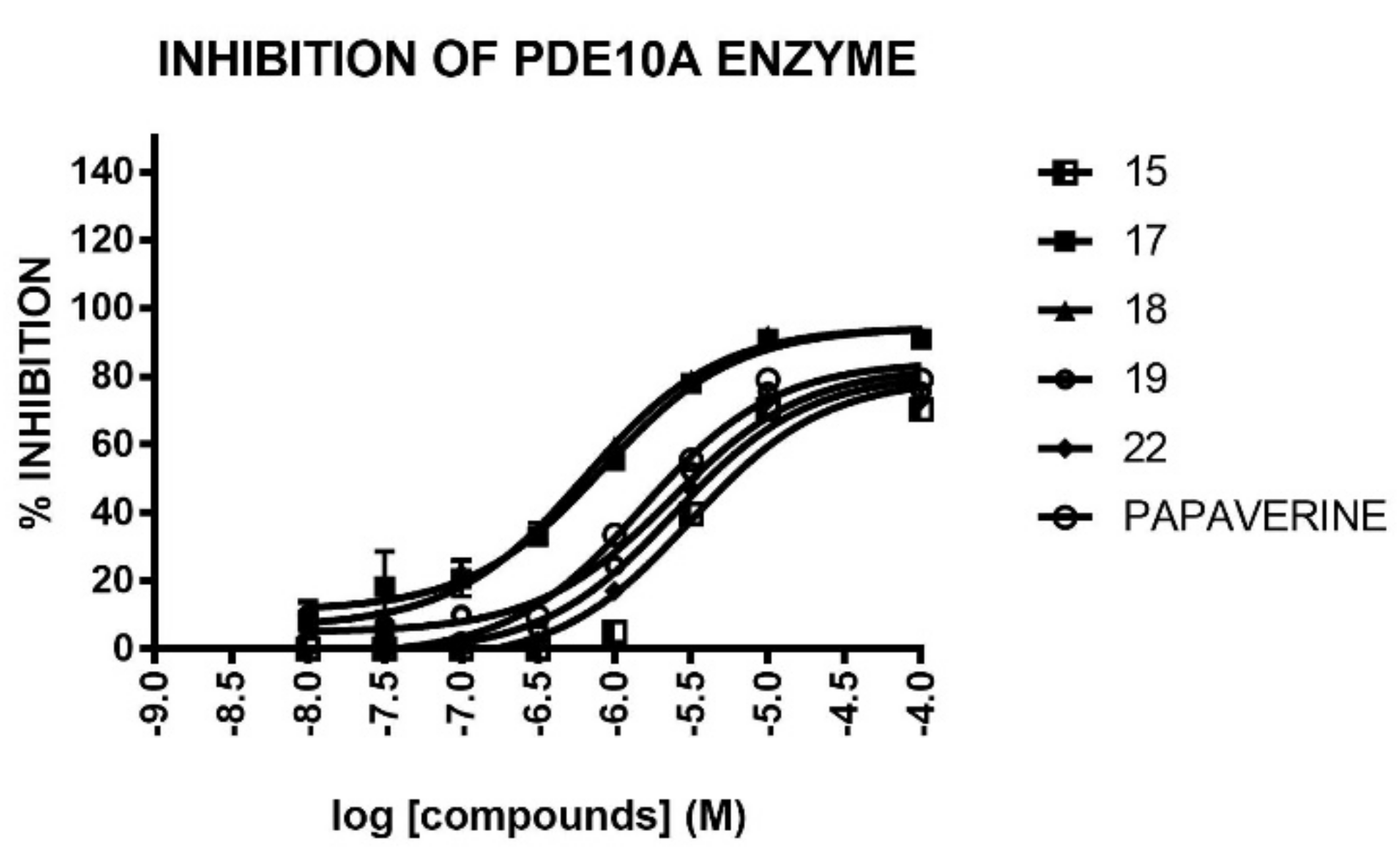
2.2.1. PDE10A Inhibitory Activity
2.2.2. Serotonin Receptor Affinity
2.3. Molecular Modeling Studies
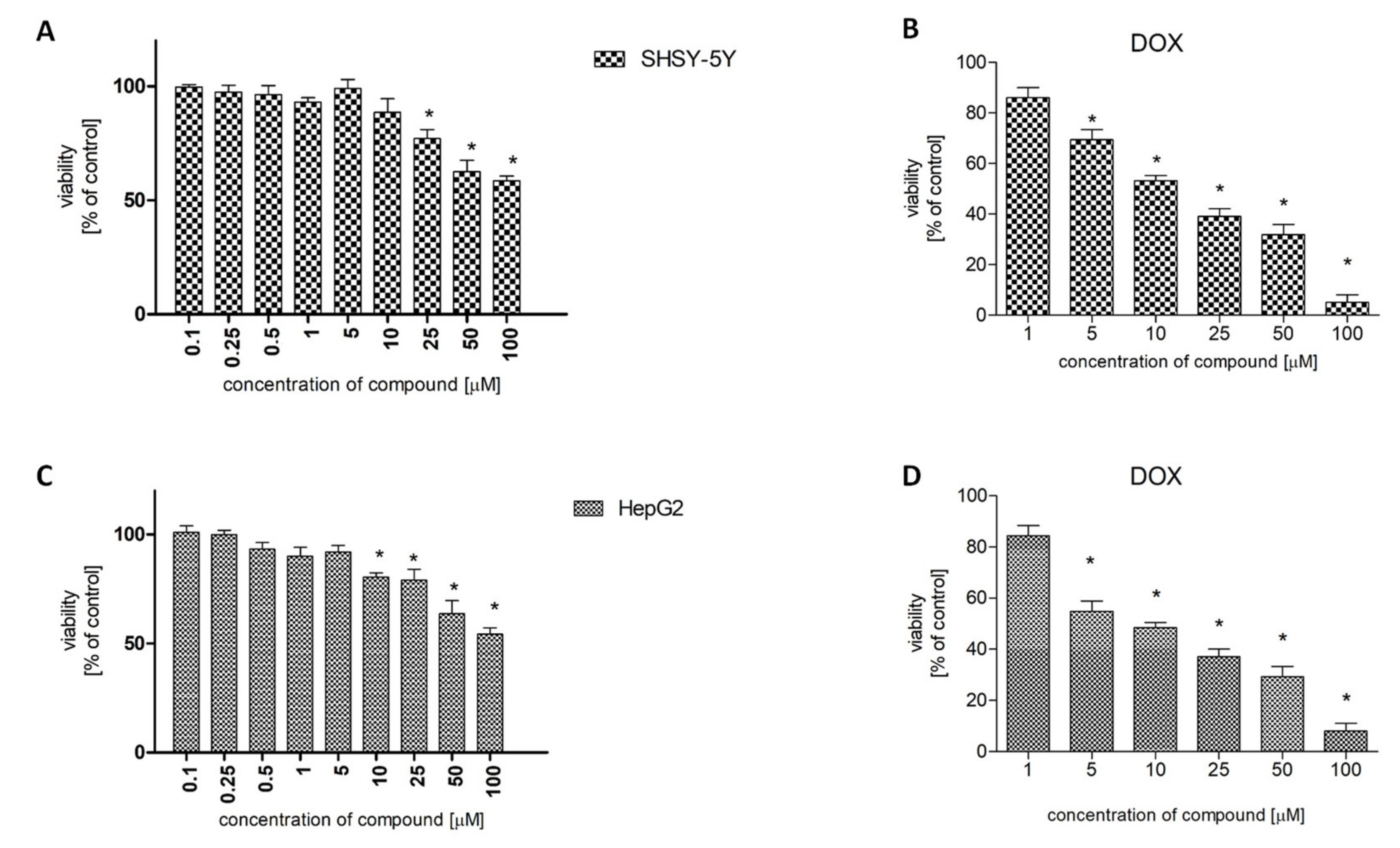
2.4. Cytotoxicity
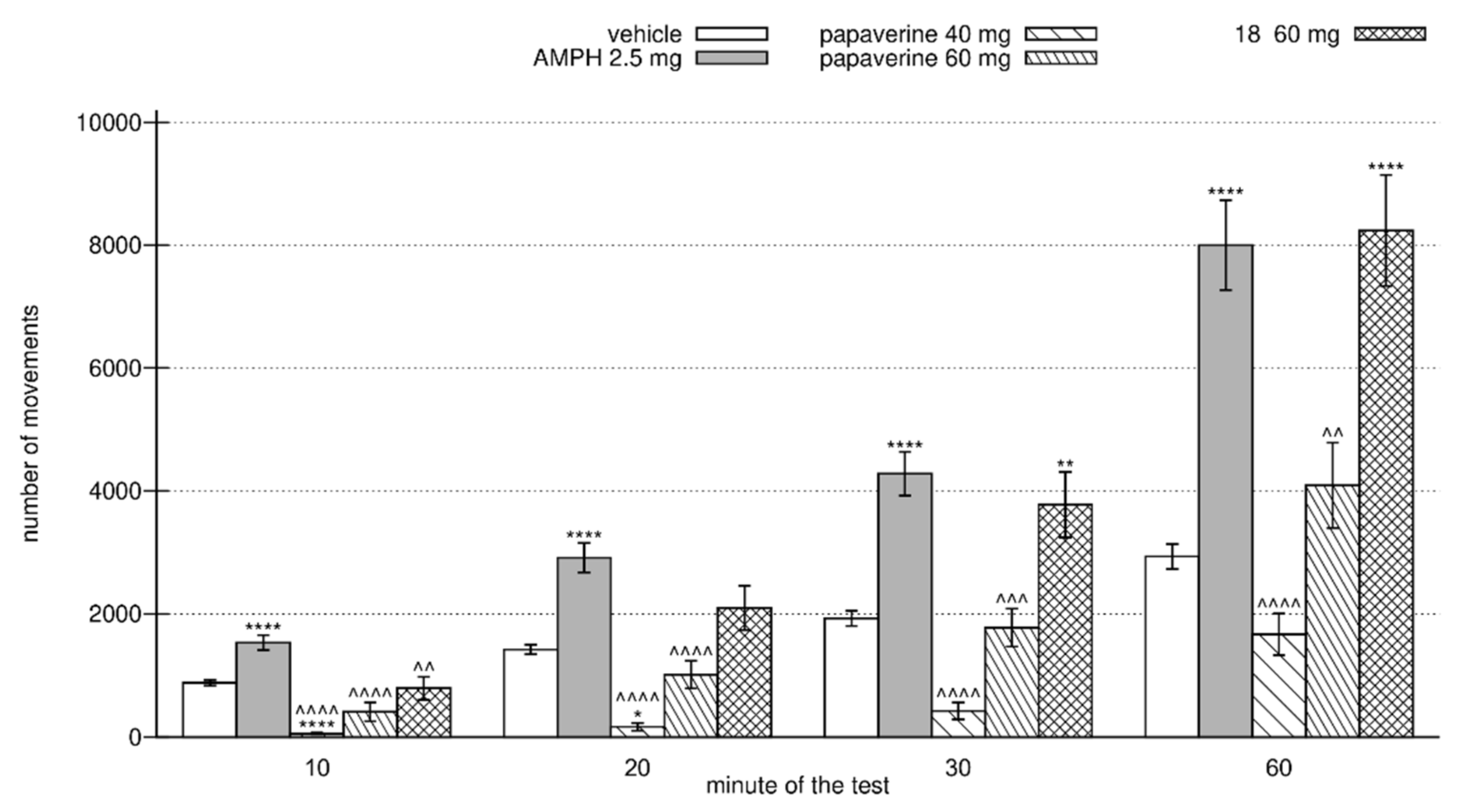
2.5. Behavioral Evaluation
3. Materials and Methods
3.1. Chemistry
3.1.1. General Procedure for Synthesis of 4-Methoxy-2-benzofuran-1,3-dione (2)
3.1.2. General Procedure for Synthesis of the Final Series of Compounds (3–23)
2-[2-(6,7-Dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)ethyl]-4-methoxy-1H-isoindole-1,3(2H)-dione (3)
2-[2-(3,4-Dihydroisoquinolin-2(1H)-yl)ethyl]-4-methoxy-1H-isoindole-1,3(2H)-dione (4)
2-[2-(Octahydroisoquinolin-2(1H)-yl)ethyl]-4-methoxy-1H-isoindole-1,3(2H)-dione (5)
2-[3-(6,7-Dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)propyl]-4-methoxy-1H-isoindole-1,3(2H)-dione (6)
2-[3-(3,4-Dihydroisoquinolin-2(1H)-yl)propyl]-4-methoxy-1H-isoindole-1,3(2H)-dione (7)
2-[3-(Octahydroisoquinolin-2(1H)-yl)propyl]-4-methoxy-1H-isoindole-1,3(2H)-dione (8)
2-Benzyl-4-methoxy-1H-isoindole-1,3(2H)-dione (9)
4-Methoxy-2-(pyridin-2-ylmethyl)-1H-isoindole-1,3(2H)-dione (10)
2-(1H-Benzimidazol-2-ylmethyl)-4-methoxy-1H-isoindole-1,3(2H)-dione (11)
4-Methoxy-2-(2-phenylethyl)-1H-isoindole-1,3(2H)-dione (12)
4-Methoxy-2-[2-(pyridin-2-yl)ethyl]-1H-isoindole-1,3(2H)-dione (13)
2-[2-(1H-Imidazol-4-yl)ethyl]-4-methoxy-1H-isoindole-1,3(2H)-dione (14)
2-[2-(1H-Benzimidazol-2-yl)ethyl]-4-methoxy-1H-isoindole-1,3(2H)-dione (15)
4-Methoxy-2-(3-phenylpropyl)-1H-isoindole-1,3(2H)-dione (16)
2-[3-(1H-Benzimidazol-2-yl)propyl]-4-methoxy-1H-isoindole-1,3(2H)-dione (17)
2-[4-(1H-Benzimidazol-2-yl)butyl]-4-methoxy-1H-isoindole-1,3(2H)-dione (18)
2-[5-(1H-Benzimidazol-2-yl)pentyl]-4-methoxy-1H-isoindole-1,3(2H)-dione (19)
2-[2-(1H-Benzimidazol-2-yl)ethyl]-1H-isoindole-1,3(2H)-dione (20)
2-[3-(1H-Benzimidazol-2-yl)propyl]-1H-isoindole-1,3(2H)-dione (21)
2-[4-(1H-Benzimidazol-2-yl)butyl]-1H-isoindole-1,3(2H)-dione (22)
2-[5-(1H-Benzimidazol-2-yl)pentyl]-1H-isoindole-1,3(2H)-dione (23)
3.2. Pharmacology
3.2.1. Protocols for Measuring PDE10A Inhibition In Vitro
3.2.2. Radioligand Binding Studies
3.3. Molecular Modeling
3.4. Cytotoxicity
3.4.1. Cells Culture
3.4.2. Cytotoxicity Analysis—MTT
3.5. In Vivo Studies
3.5.1. d-Amphetamine-Induced Hyperlocomotor Activity in CD-1 Mice
3.5.2. Spontaneous Locomotor Activity in CD-1 Mice
3.5.3. Drugs
3.5.4. Statistics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Lima, L.M.; Castro, P.; Machado, A.L.; Fraga, C.A.M.; Lugnier, C.; De Moraes, V.L.G.; Barreiro, E.J. Synthesis and anti-inflammatory activity of phthalimide derivatives, designed as new thalidomide analogues. Bioorg. Med. Chem. 2002, 10, 3067–3073. [Google Scholar] [CrossRef]
- Van Derpoorten, K.; Balzarini, J.; De Clercq, E.; Poupaert, J.H. Anti-HIV activity of N-1-adamantyl-4-aminophthalimide. Biomed. Pharmacother. 1997, 51, 464–468. [Google Scholar] [CrossRef]
- Hall, I.H.; Wong, O.T.; Scovill, J.P. The cytotoxicity of N-pyridinyl and N-quinolinyl substituted derivatives of phthalimide and succinimide. Biomed. Pharmacother. 1995, 49, 251–258. [Google Scholar] [CrossRef]
- Kim, H.S.; Kim, Y.; Doddareddy, M.R.; Seo, S.H.; Rhim, H.; Tae, J.; Pae, A.N.; Choo, H.; Cho, Y.S. Design, synthesis, and biological evaluation of 1,3-dioxoisoindoline-5-carboxamide derivatives as T-type calcium channel blockers. Biorg. Med. Chem. Lett. 2007, 17, 476–481. [Google Scholar] [CrossRef] [PubMed]
- Gunduğdu, Ö.; Noma, S.A.A.; Taskin-Tok, T.; Ateş, B.; Kishali, N. Evaluation of xanthine oxidase inhibitor properties on isoindoline-1,3-dion derivatives and calculation of interaction mechanism. J. Mol. Struct. 2020, 1204, 127523. [Google Scholar] [CrossRef]
- Lamie, P.F.; Philoppes, J.N.; El-Gendy, A.O.; Rarova, L.; Gruz, J. Design, synthesis and evaluation of novel phthalimide derivatives as in vitro anti-microbial, anti-oxidant and anti-inflammatory agents. Molecules 2015, 20, 16620–16642. [Google Scholar] [CrossRef] [Green Version]
- Bailleux, V.; Vallée, L.; Nuyts, J.P.; Vamecq, J. Original anticonvulsant properties of two N-phenylphthalimide derivatives. Biomed. Pharmacother. 1993, 47, 463–464. [Google Scholar] [CrossRef]
- Atack, J.R. The benzodiazepine binding site of GABAA receptors as a target for the development of novel anxiolytics. Expert Opin. Investig. Drugs 2005, 14, 601–618. [Google Scholar] [CrossRef]
- Bajda, M.; Więckowska, A.; Hebda, M.; Guzior, N.; Sotriffer, C.; Malawska, B. Structure-Based Search for New Inhibitors of Cholinesterases. Int. J. Mol. Sci. 2013, 14, 5608–5632. [Google Scholar] [CrossRef] [Green Version]
- Panek, D.; Wiȩckowska, A.; Pasieka, A.; Godyń, J.; Jończyk, J.; Bajda, M.; Knez, D.; Gobec, S.; Malawska, B. Design, synthesis, and biological evaluation of 2-(benzylamino-2-hydroxyalkyl)isoindoline-1,3-diones derivatives as potential disease-modifying multifunctional anti-Alzheimer agents. Molecules 2018, 23, 347. [Google Scholar] [CrossRef] [Green Version]
- Mohammadi-Farani, A.; Abdi, N.; Moradi, A.; Aliabadi, A. 2-(2-(4-Benzoylpiperazin-1-yl)ethyl)isoindoline-1,3-dione derivatives: Synthesis, docking and acetylcholinesterase inhibitory evaluation as anti-alzheimer agents. Iran. J. Basic Med. Sci. 2017, 20, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Foong, J.P.P.; Bornstein, J.C. 5-HT antagonists NAN-190 and SB 269970 block α2-adrenoceptors in the guinea pig. Neuroreport 2009, 20, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Huttunen, M. The evolution of the serotonin-dopamine antagonist concept. J. Clin. Psychopharmacol. 1995, 15, 4S–10S. [Google Scholar] [CrossRef] [PubMed]
- Meltzer, H.Y.; Massey, B.W. The role of serotonin receptors in the action of atypical antipsychotic drugs. Curr. Opin. Pharmacol. 2011, 11, 59–67. [Google Scholar] [CrossRef]
- Stahl, S.M. Dopamine system stabilizers, aripiprazole, and the next generation of antipsychotics, part 1: “Goldilocks” actions at dopamine receptors. J. Clin. Psychiatry 2001, 62, 841–842. [Google Scholar] [CrossRef]
- Mailman, R.; Murthy, V. Third Generation Antipsychotic Drugs: Partial Agonism or Receptor Functional Selectivity? Curr. Pharm. Des. 2010, 16, 488–501. [Google Scholar] [CrossRef] [Green Version]
- McCreary, A.; Newman-Tancredi, A. Serotonin 5-HT1A Receptors and Antipsychotics - An Update in Light of New Concepts and Drugs. Curr. Pharm. Des. 2015, 21, 3725–3731. [Google Scholar] [CrossRef]
- Kusumi, I.; Boku, S.; Takahashi, Y. Psychopharmacology of atypical antipsychotic drugs: From the receptor binding profile to neuroprotection and neurogenesis. Psychiatry Clin. Neurosci. 2015, 69, 243–258. [Google Scholar] [CrossRef]
- Siuciak, J.A.; Strick, C.A. Treating neuropsychiatric disorders with PDE10A inhibitors. Drug Discov. Today Ther. Strateg. 2006, 3, 527–532. [Google Scholar] [CrossRef]
- Chappie, T.A.; Helal, C.J.; Hou, X. Current landscape of phosphodiesterase 10A (PDE10A) inhibition. J. Med. Chem. 2012, 55, 7299–7331. [Google Scholar] [CrossRef]
- Zagorska, A.; Partyka, A.; Bucki, A.; Gawalskax, A.; Czopek, A.; Pawlowski, M. Phosphodiesterase 10 Inhibitors—Novel Perspectives for Psychiatric and Neurodegenerative Drug Discovery. Curr. Med. Chem. 2018, 25, 3455–3481. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Harada, A.; Suzuki, H.; Capuani, C.; Ugolini, A.; Corsi, M.; Kimura, H. Combined treatment with a selective PDE10A inhibitor TAK-063 and either haloperidol or olanzapine at subeffective doses produces potent antipsychotic-like effects without affecting plasma prolactin levels and cataleptic responses in rodents. Pharmacol. Res. Perspect. 2018, 6. [Google Scholar] [CrossRef] [PubMed]
- Proschak, E.; Stark, H.; Merk, D. Polypharmacology by Design: A Medicinal Chemist’s Perspective on Multitargeting Compounds. J. Med. Chem. 2019, 62, 420–444. [Google Scholar] [CrossRef] [PubMed]
- Kondej, M.; Stępnicki, P.; Kaczor, A.A. Multi-target approach for drug discovery against schizophrenia. Int. J. Mol. Sci. 2018, 19, 3105. [Google Scholar] [CrossRef] [Green Version]
- Christopher, D.C.; BUNDA, J.L.; Broc, A.F.; SHIPE, W. Pyrimidinones as AS PDE10 Inhibitors. U.S. Patent WO2010138585, 12 February 2010. [Google Scholar]
- Medina, R.A.; Sallander, J.; Benhamú, B.; Porras, E.; Campillo, M.; Pardo, L.; López-Rodríguez, M.L. Synthesis of new serotonin 5-HT7 receptor ligands. Determinants of 5-ht7/5-ht1a receptor selectivity. J. Med. Chem. 2009, 52, 2384–2392. [Google Scholar] [CrossRef]
- Zagórska, A.; Bucki, A.; Kołaczkowski, M.; Siwek, A.; Głuch-Lutwin, M.; Starowicz, G.; Kazek, G.; Partyka, A.; Wesołowska, A.; Słoczyńska, K.; et al. Synthesis and biological evaluation of 2-fluoro and 3-trifluoromethyl-phenyl-piperazinylalkyl derivatives of 1H-imidazo [2,1-f]purine-2,4(3H,8H)-dione as potential antidepressant agents. J. Enzyme Inhib. Med. Chem. 2016, 31, 10–24. [Google Scholar] [CrossRef] [Green Version]
- Zajdel, P.; Marciniec, K.; Maślankiewicz, A.; Satała, G.; Duszyńska, B.; Bojarski, A.J.; Partyka, A.; Jastrzbska-Wisek, M.; Wróbel, D.; Wesołowska, A.; et al. Quinoline- and isoquinoline-sulfonamide derivatives of LCAP as potent CNS multi-receptor—5-HT 1A/5-HT 2A/5-HT 7 and D 2/D 3/D 4-Agents: The synthesis and pharmacological evaluation. Bioorg. Med. Chem. 2012, 20, 1545–1556. [Google Scholar] [CrossRef]
- Kehler, J.; Ritzen, A.; Langgård, M.; Petersen, S.L.; Farah, M.M.; Bundgaard, C.; Christoffersen, C.T.; Nielsen, J.; Kilburn, J.P. Triazoloquinazolines as a novel class of phosphodiesterase 10A (PDE10A) inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 3738–3742. [Google Scholar] [CrossRef]
- Chino, A.; Masuda, N.; Amano, Y.; Honbou, K.; Mihara, T.; Yamazaki, M.; Tomishima, M. Novel benzimidazole derivatives as phosphodiesterase 10A (PDE10A) inhibitors with improved metabolic stability. Bioorg. Med. Chem. 2014, 22, 3515–3526. [Google Scholar] [CrossRef]
- Bieszczad, B.; Barbasiewicz, M. The Key Role of the Nonchelating Conformation of the Benzylidene Ligand on the Formation and Initiation of Hoveyda-Grubbs Metathesis Catalysts. Chem. A Eur. J. 2015, 21, 10322–10325. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, N.J.; Lyons, J.F.; Thompson, N.T.; Yyle, S.M.; Stephen, M.M.; Christopher, W. Pharmaceutical Combinations. U.S. Patent WO2008044041, 17 April 2008. [Google Scholar]
- Vooturi, S.K.; Firestine, S.M. Solution-phase parallel synthesis of novel membrane-targeted antibiotics. J. Comb. Chem. 2010, 12, 151–160. [Google Scholar] [CrossRef]
- Elshihawy, H.; Helal, M.A.; Said, M.; Hammad, M.A. Design, synthesis, and enzyme kinetics of novel benzimidazole and quinoxaline derivatives as methionine synthase inhibitors. Bioorg. Med. Chem. 2014, 22, 550–558. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Venkataramana, M.; Chandra Nayaka, S.; Anand, T.; Rajesh, R.; Aiyaz, M.; Divakara, S.T.; Murali, H.S.; Prakash, H.S.; Lakshmana Rao, P.V. Zearalenone induced toxicity in SHSY-5Y cells: The role of oxidative stress evidenced by N-acetyl cysteine. Food Chem. Toxicol. 2014, 65, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.; Breier, M.; Ko, D.; Thangaraj, N.; Marzan, D.E.; Swerdlow, N.R. Evaluating the antipsychotic profile of the preferential PDE10A inhibitor, papaverine. Psychopharmacology 2009, 203, 723–735. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, C.J.; Chapin, D.S.; Cianfrogna, J.; Corman, M.L.; Hajos, M.; Harms, J.F.; Hoffman, W.E.; Lebel, L.A.; McCarthy, S.A.; Nelson, F.R.; et al. Preclinical characterization of selective phosphodiesterase 10A inhibitors: A new therapeutic approach to the treatment of schizophrenia. J. Pharmacol. Exp. Ther. 2008, 325, 681–690. [Google Scholar] [CrossRef] [Green Version]
- Kurys, B.E.; Fink, D.M.; Fink, D.M.; Freed, B.S.; Merriman, G.H. N-(pyridinylamino)isoindolines and Related Compounds. U.S. Patent No 6,004,977, 29 October 1997. [Google Scholar]
- Dhanya, R.P.; Sidique, S.; Sheffler, D.J.; Nickols, H.H.; Herath, A.; Yang, L.; Dahl, R.; Ardecky, R.; Semenova, S.; Markou, A.; et al. Design and synthesis of an orally active metabotropic glutamate receptor subtype-2 (mGluR2) positive allosteric modulator (PAM) that decreases cocaine self-administration in rats. J. Med. Chem. 2011, 54, 342–353. [Google Scholar] [CrossRef] [Green Version]
- Czopek, A.; Zagorska, A.; Kolaczkowski, M.; Bucki, A.; Gryzlo, B.; Rychtyk, J.; Pawlowsk, M.; Siwek, A.; Satala, G.; Bojarski, A.; et al. New Spirohydanoin Derivatives-Synthesis, Pharmacological Evaluation, and Molecular Modeling Study. Acta Pol. Pharm. 2016, 73, 1545–1554. [Google Scholar]
- Intagliata, S.; Modica, M.N.; Pittalà, V.; Salerno, L.; Siracusa, M.A.; Cagnotto, A.; Salmona, M.; Kurczab, R.; Romeo, G. New N- and O-arylpiperazinylalkyl pyrimidines and 2-methylquinazolines derivatives as 5-HT7and 5-HT1Areceptor ligands: Synthesis, structure-activity relationships, and molecular modeling studies. Bioorg. Med. Chem. 2017, 25, 1250–1259. [Google Scholar] [CrossRef] [PubMed]
- Malamas, M.S.; Stange, H.; Schindler, R.; Lankau, H.J.; Grunwald, C.; Langen, B.; Egerland, U.; Hage, T.; Ni, Y.; Erdei, J.; et al. Novel triazines as potent and selective phosphodiesterase 10A inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 5876–5884. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 3–23 are available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compd | R | n | PDE10A [%] 1 | IC50 PDE10A [µM] ± SEM 3 | 5-HT1A | 5-HT7 | ||
|---|---|---|---|---|---|---|---|---|
| 10µM | 3µM | 1 µM 2 ± SEM 3 | ||||||
 | 3 | A | 2 | 0 | 0 | - | 27 ± 3 | 0 ± 0 |
| 4 | B | 2 | 0 | 0 | - | 12 ± 4 | 0 ± 0 | |
| 5 | C | 2 | 0 | 0 | - | 25 ± 3 | 0 ± 0 | |
| 6 | A | 3 | 0 | 0 | - | 32 ± 4 | 0 ± 0 | |
| 7 | B | 3 | 19 | 18 | - | 33 ± 2 | 51 ± 2 | |
| 8 | C | 3 | 7 | 7 | - | 28 ± 3 | 0 ± 0 | |
| 9 | D | 1 | 0 | 0 | - | 6 ± 2 | 0 ± 0 | |
| 10 | E | 1 | 0 | 0 | - | 10 ± 1 | 0 ± 2 | |
| 11 | F | 1 | 15 | 7 | - | 39 ± 3 | 0 ± 1 | |
| 12 | D | 2 | 10 | 5 | - | 13 ± 2 | 0 ± 0 | |
| 13 | E | 2 | 0 | 0 | - | 59 ± 4 | 0 ± 2 | |
| 14 | G | 2 | 0 | 0 | - | 21 ± 1 | 0 ± 0 | |
| 15 | F | 2 | 70 | 40 | 6.710 ± 0.195 | 44 ± 5 | 0 ± 3 | |
| 16 | D | 3 | 13 | 11 | - | 32 ± 3 | 14 ± 0 | |
| 17 | F | 3 | 86 | 73 | 1.001 ± 0.009 | 16 ± 5 | 10 ± 3 | |
| 18 4 | F | 4 | 92 | 79 | 0.886 ± 0.017 | 28 ± 3 | 3 ± 1 | |
| 19 | F | 5 | 75 | 52 | 4.305 ± 0.365 | 28 ± 2 | 6 ± 0 | |
 | 20 | F | 2 | 1 | 0 | - | 26 ± 2 | 36 ± 0 |
| 21 | F | 3 | 45 | 16 | - | 27 ± 2 | 14 ± 2 | |
| 22 | F | 4 | 73 | 47 | 5.645 ± 0.495 | 20 ± 3 | 15 ± 2 | |
| 23 | F | 5 | 58 | 26 | - | 48 ± 3 | 34 ± 2 | |
| Papaverine | 75 | 54 | 5.755 ± 0.055 | - | - | |||
| Methiothiepin | - | - | - | 100 ± 0 | 99 ± 1 | |||
| DMSO | 0 | 0 | - | - | - | |||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Czopek, A.; Partyka, A.; Bucki, A.; Pawłowski, M.; Kołaczkowski, M.; Siwek, A.; Głuch-Lutwin, M.; Koczurkiewicz, P.; Pękala, E.; Jaromin, A.; et al. Impact of N-Alkylamino Substituents on Serotonin Receptor (5-HTR) Affinity and Phosphodiesterase 10A (PDE10A) Inhibition of Isoindole-1,3-dione Derivatives. Molecules 2020, 25, 3868. https://doi.org/10.3390/molecules25173868
Czopek A, Partyka A, Bucki A, Pawłowski M, Kołaczkowski M, Siwek A, Głuch-Lutwin M, Koczurkiewicz P, Pękala E, Jaromin A, et al. Impact of N-Alkylamino Substituents on Serotonin Receptor (5-HTR) Affinity and Phosphodiesterase 10A (PDE10A) Inhibition of Isoindole-1,3-dione Derivatives. Molecules. 2020; 25(17):3868. https://doi.org/10.3390/molecules25173868
Chicago/Turabian StyleCzopek, Anna, Anna Partyka, Adam Bucki, Maciej Pawłowski, Marcin Kołaczkowski, Agata Siwek, Monika Głuch-Lutwin, Paulina Koczurkiewicz, Elżbieta Pękala, Anna Jaromin, and et al. 2020. "Impact of N-Alkylamino Substituents on Serotonin Receptor (5-HTR) Affinity and Phosphodiesterase 10A (PDE10A) Inhibition of Isoindole-1,3-dione Derivatives" Molecules 25, no. 17: 3868. https://doi.org/10.3390/molecules25173868