
Alkoxyamines Designed as Potential Drugs against Plasmodium and Schistosoma Parasites


 , and
, and
Abstract
: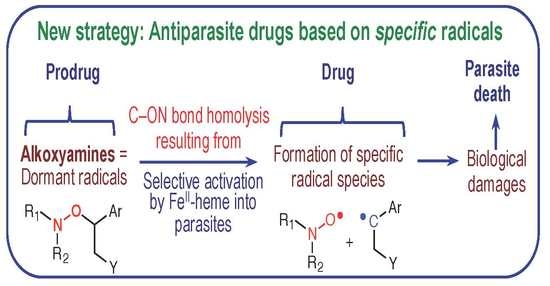
1. Introduction
2. Results and Discussion
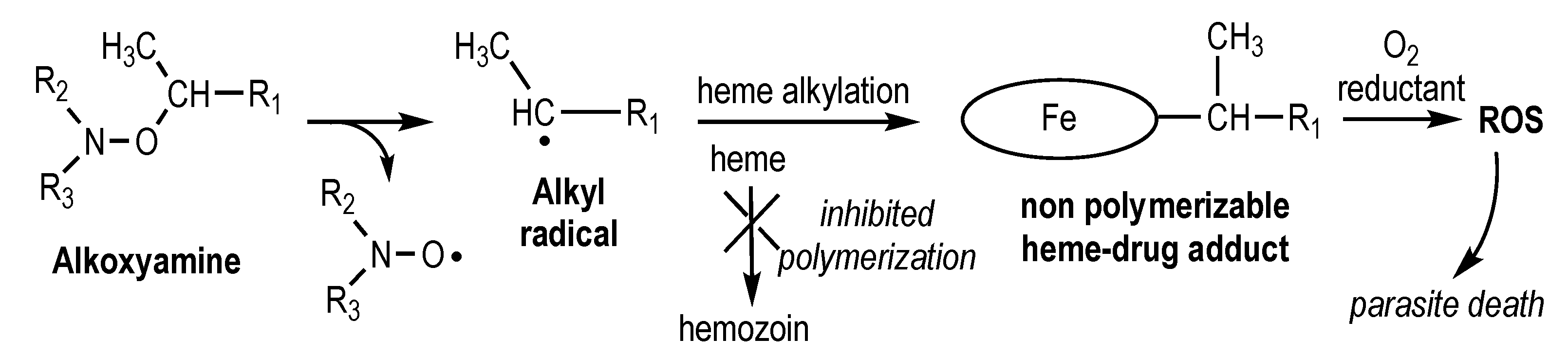
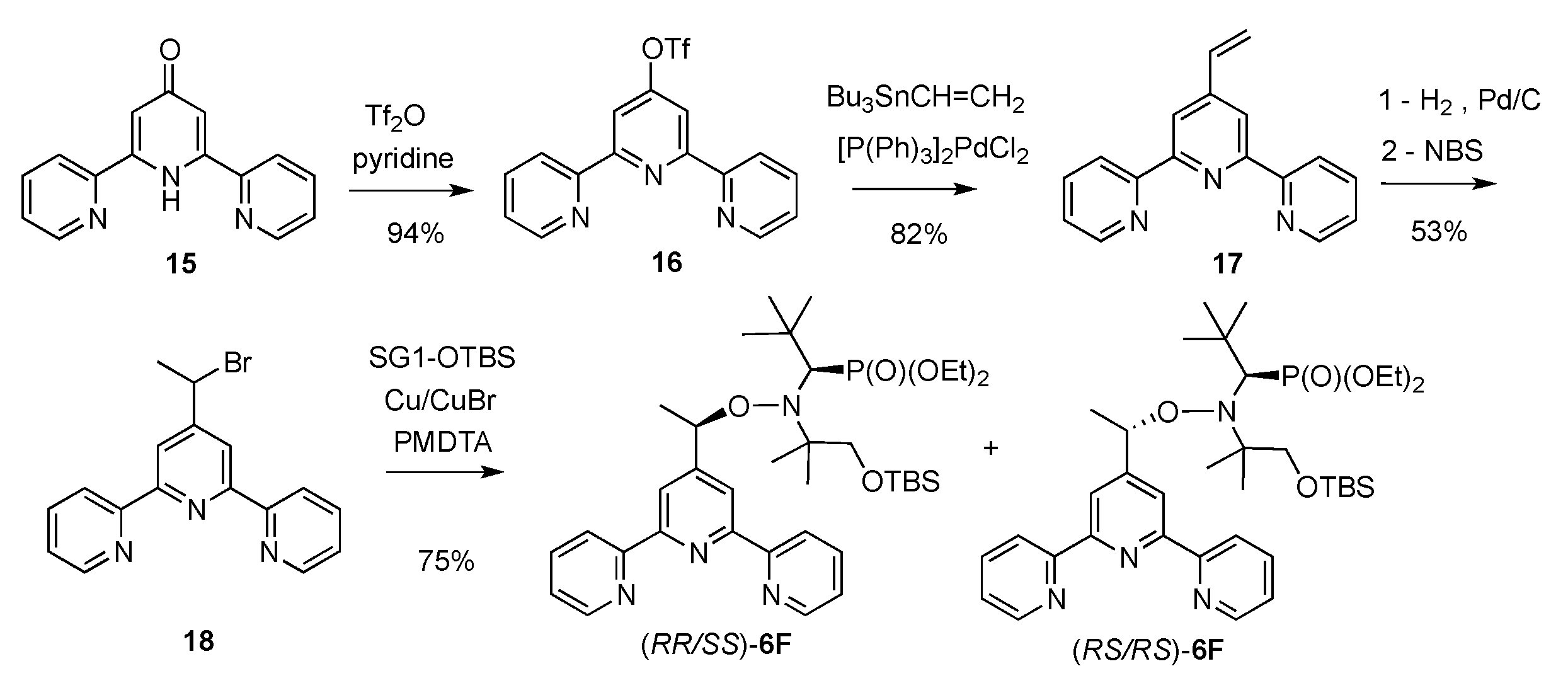
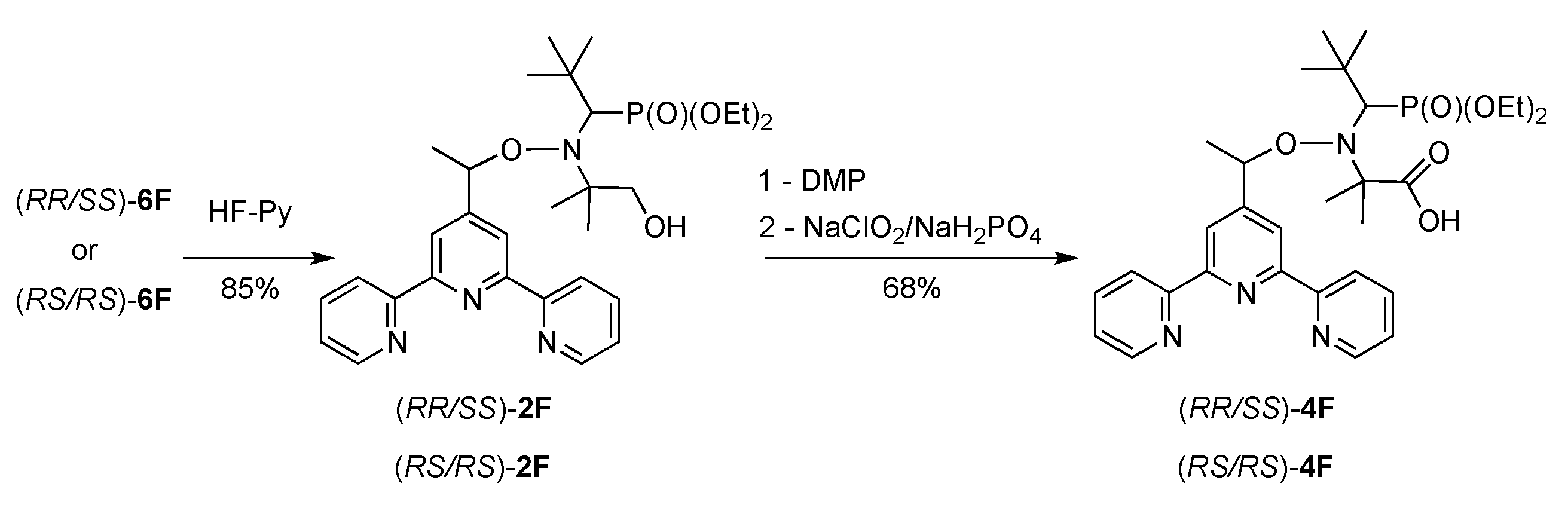
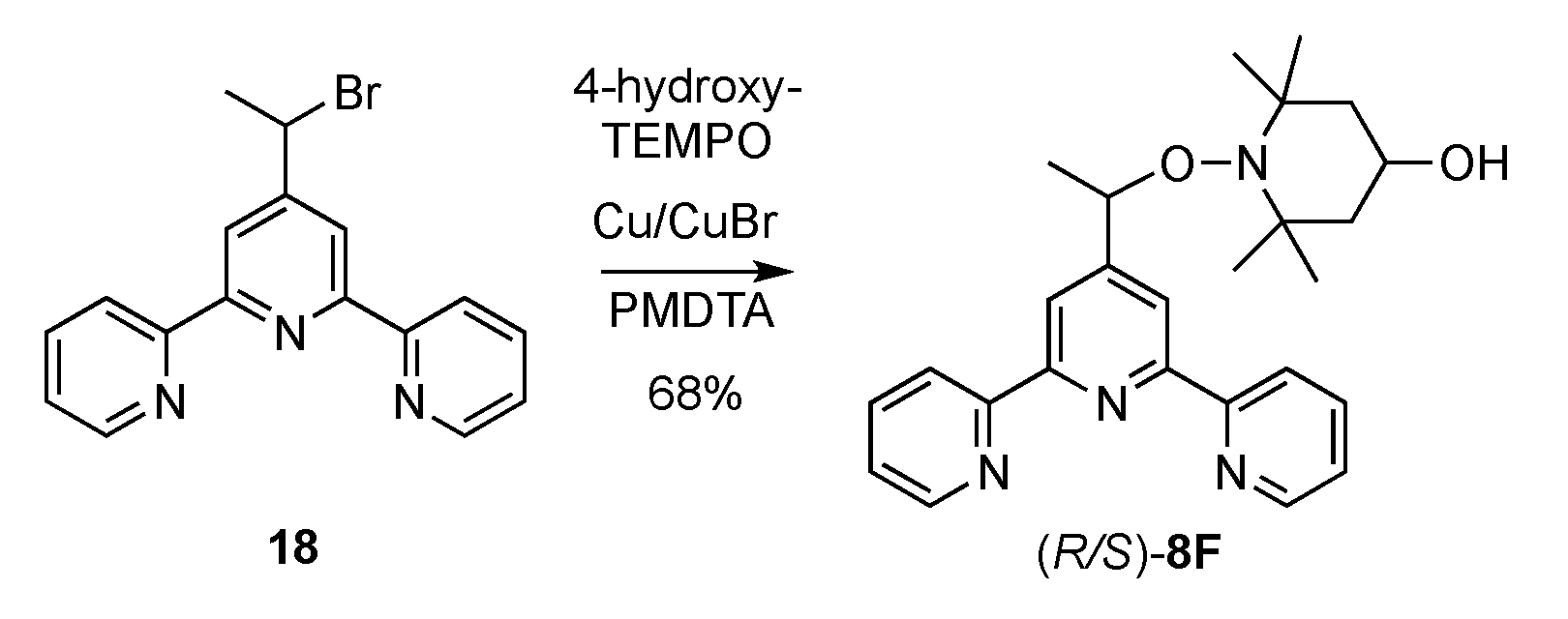
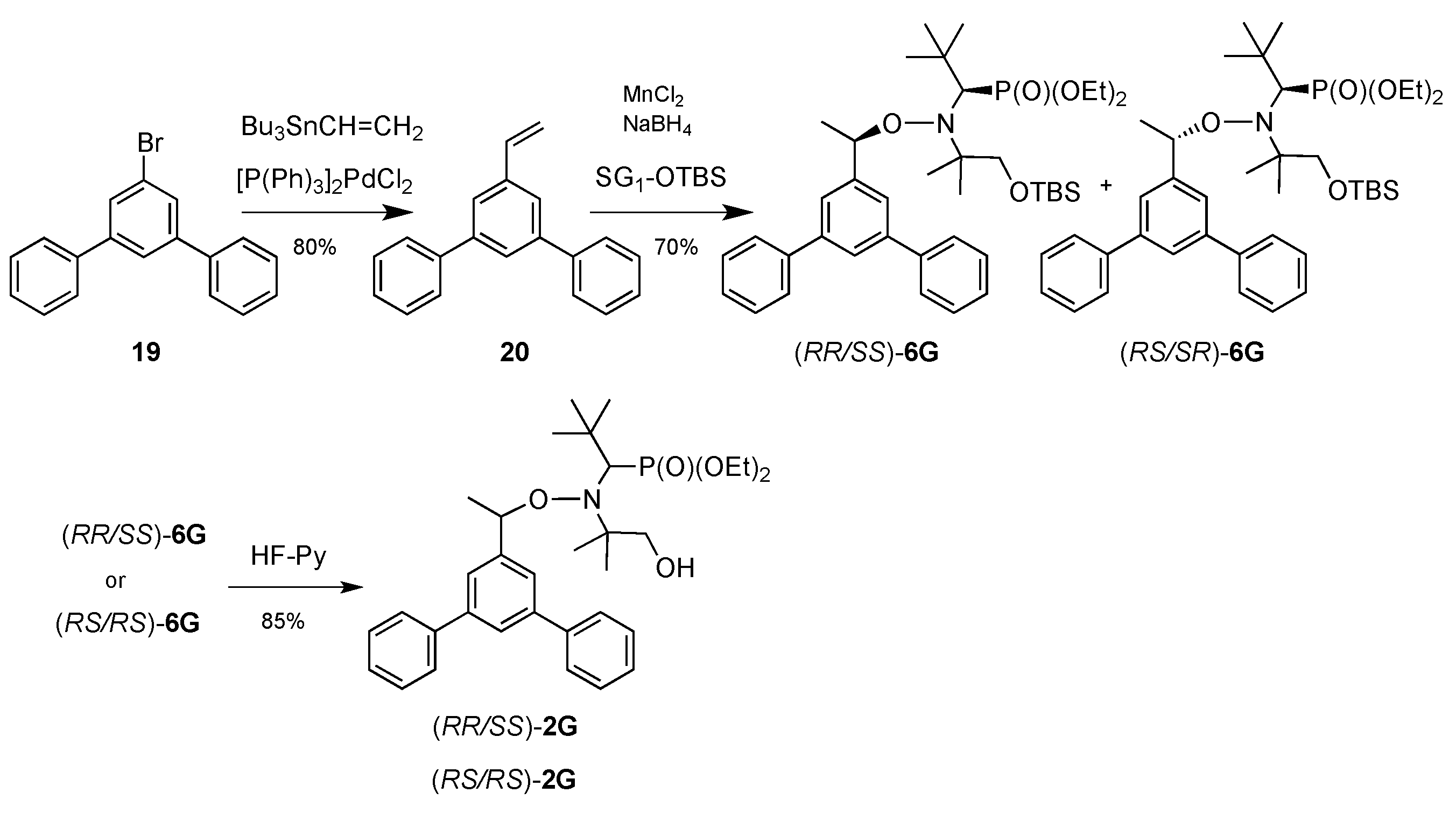
2.1. Synthesis of Alkoxyamines
2.2. Antimalarial Activity
2.3. Antischistosomal Activity
2.4. Cytotoxicity
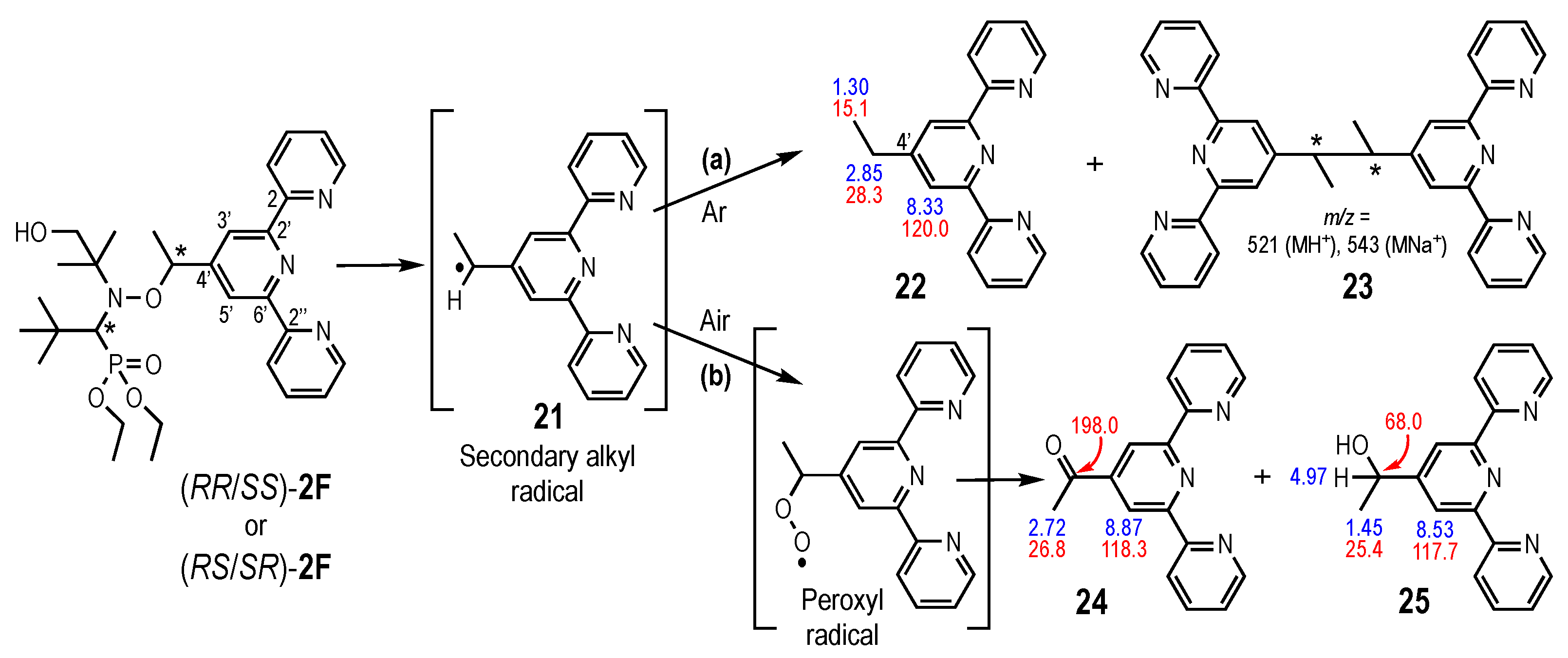
2.5. Thermodynamic and Kinetic Data for the Homolysis of the NO–C Bond
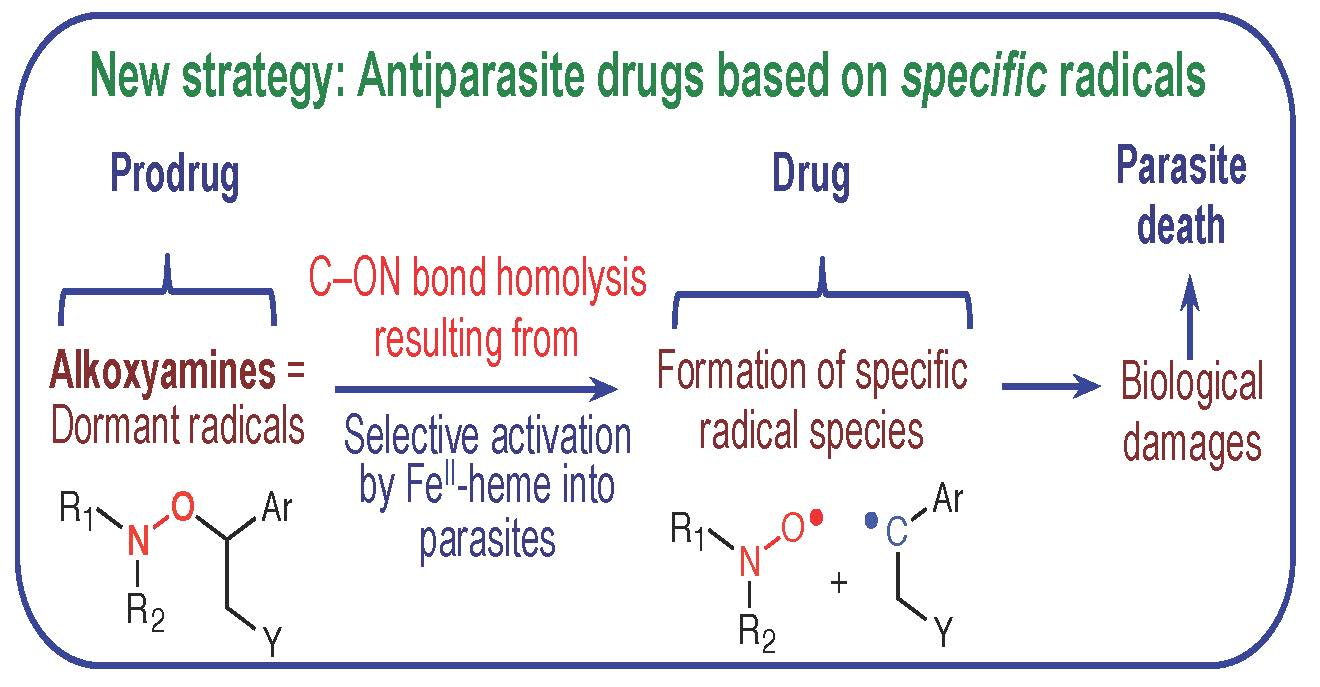
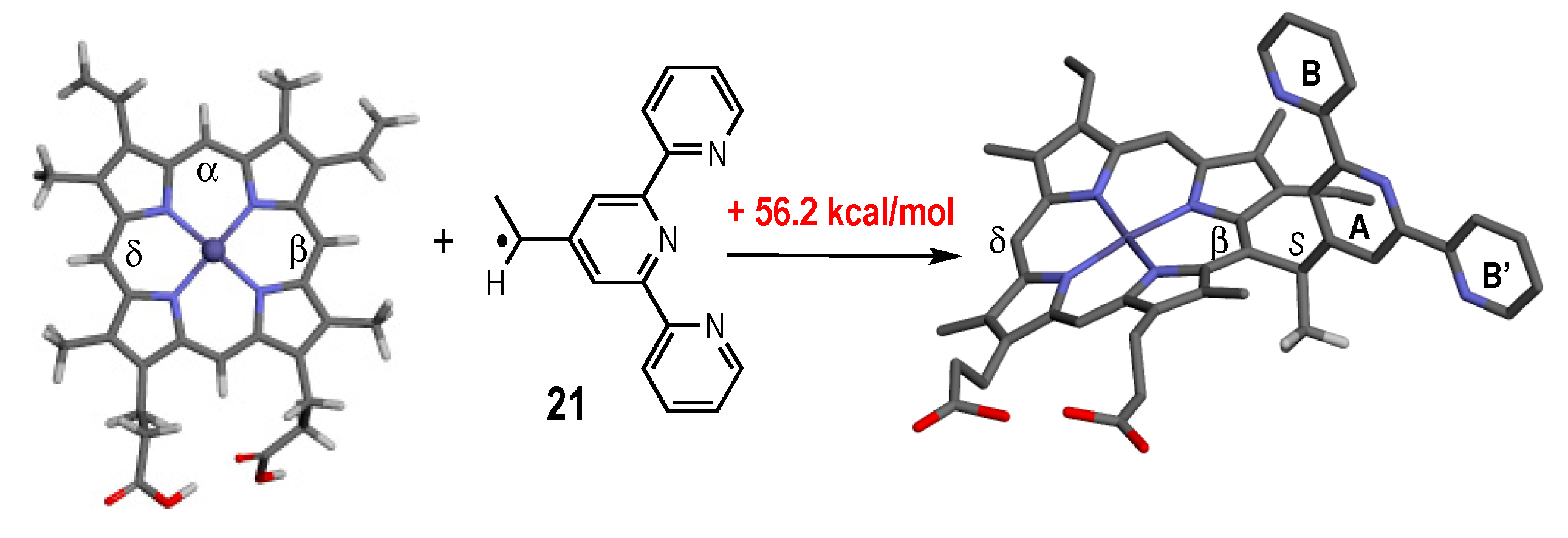
2.6. Reactivity of Alkoxyamines Toward Heme
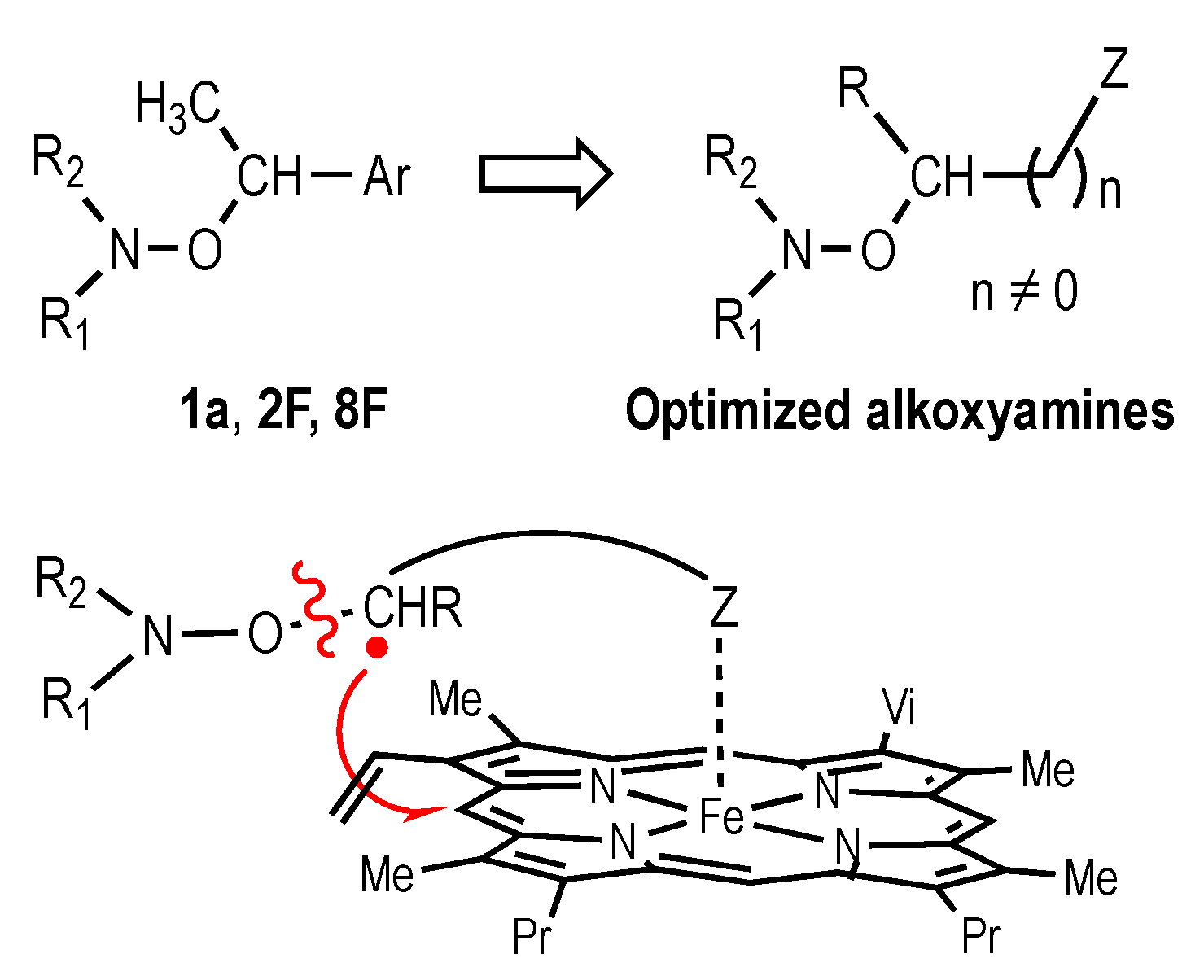
3. Conclusions
4. Materials and Methods
4.1. Synthesis of Alkoxyamines
4.1.1. Methods
4.1.2. Syntheses and Characterization
4.2. Evaluation against P. falciparum
4.2.1. Parasite Culture
4.2.2. In Vitro Antimalarial Activities
4.2.3. Recrudescence Assay
4.3. Evaluation against S. mansoni
4.3.1. Parasite Culture
4.3.2. In Vitro Antischistosomal Activities
4.4. Cytotoxicity Assays
4.5. Computational Studies
4.6. Reactivity of Alkoxyamines toward Heme
4.6.1. Materials and Analytical Conditions
4.6.2. Reaction of (RR/SS)-1a or (RS/SR)-1a with Fe(III)-heme
4.6.3. Reaction of (RS/SR)-2F and (RR/SS)-2F with Fe(III)-heme
4.6.4. Reaction of (R/S)-8F with Fe(III)-heme
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Organization. World Malaria Report 2019; World Health Organization: Geneva, Switzerland, 2019; p. 185. [Google Scholar]
- World Health Organization. Artemisinin and Artemisinin-Based Combination Therapy Resistance: Status Report; 2016. Available online: https://apps.who.int/iris/bitstream/handle/10665/250294/WHO-HTM-GMP-2016.11-eng.pdf?sequence=1&isAllowed=y (accessed on 13 August 2020).
- Duru, V.; Khim, N.; Leang, R.; Kim, S.; Domergue, A.; Kloeung, N.; Ke, S.; Chy, S.; Eam, R.; Khean, C.; et al. Plasmodium falciparum dihydroartemisinin-piperaquine failures in Cambodia are associated with mutant K13 parasites presenting high survival rates in novel piperaquine in vitro assays: Retrospective and prospective investigations. BMC Med. 2015, 13, 305. [Google Scholar] [CrossRef] [Green Version]
- Witkowski, B.; Duru, V.; Khim, N.; Ross, L.S.; Saintpierre, B.; Beghain, J.; Chy, S.; Kim, S.; Ke, S.; Kloeung, N.; et al. A surrogate marker of piperaquine-resistant Plasmodium falciparum malaria: A phenotype-genotype association study. Lancet Infect. Dis. 2017, 17, 174–183. [Google Scholar] [CrossRef] [Green Version]
- Ménard, S.; Ben Haddou, T.; Ramadani, A.P.; Ariey, F.; Iriart, X.; Beghain, J.; Bouchier, C.; Witkowski, B.; Berry, A.; Mercereau-Puijalon, O.; et al. Induction of multidrug tolerance in Plasmodium falciparum by extended artemisinin pressure. Emerg. Infect. Dis. 2015, 21, 1733–1741. [Google Scholar] [CrossRef]
- Boissier, J.; Portela, J.; Pradines, V.; Coslédan, F.; Robert, A.; Meunier, B. Activity of trioxaquine PA1259 in mice infected by Schistosoma mansoni. C. R. Chimie 2012, 15, 75–78. [Google Scholar] [CrossRef]
- Portela, J.; Boissier, J.; Gourbal, B.; Pradines, V.; Collière, V.; Coslédan, F.; Meunier, B.; Robert, A. Antischistosomal activity of trioxaquines: In vivo efficacy and mechanism of action on Schistosoma mansoni. PLoS Negl. Trop. Dis. 2012, 6, e1474. [Google Scholar] [CrossRef] [Green Version]
- Robert, A.; Meunier, B. Characterization of the first covalent adduct between artemisinin and a heme model. J. Am. Chem. Soc. 1997, 119, 5968–5969. [Google Scholar] [CrossRef]
- Robert, A.; Cazelles, J.; Meunier, B. Characterization of the alkylation product of heme by the antimalarial drug artemisinin. Angew. Chem. Int. Ed. 2001, 40, 1954–1957. [Google Scholar] [CrossRef]
- Meunier, B.; Robert, A. Heme as triger and target for trioxane containing antimalarial drugs. Acc. Chem. Res. 2010, 43, 1444–1451. [Google Scholar] [CrossRef]
- Cazelles, J.; Robert, A.; Meunier, B. Alkylating capacity and reaction products of antimalarial trioxanes after activation by a heme model. J. Org. Chem. 2002, 67, 609–619. [Google Scholar] [CrossRef]
- Dechy-Cabaret, O.; Benoit-Vical, F.; Loup, C.; Robert, A.; Gornitzka, H.; Bonhoure, A.; Vial, H.; Magnaval, J.F.; Séguéla, J.P.; Meunier, B. Synthesis and antimalarial activity of trioxaquine derivatives. Chem. Eur. J. 2004, 10, 1625–1636. [Google Scholar] [CrossRef]
- Bousejra-El Garah, F.; Claparols, C.; Benoit-Vical, F.; Meunier, B.; Robert, A. The antimalarial trioxaquine DU1301 alkylates heme in malaria-infected mice. Antimicrob. Agents Chemother. 2008, 52, 2966–2969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, A.; Benoit-Vical, F.; Claparols, C.; Meunier, B. The antimalarial drug artemisinin alkylates heme in infected mice. Proc. Natl. Acad. Sci. USA 2005, 102, 13676–13680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, A.; Claparols, C.; Witkowski, B.; Benoit-Vical, F. Correlation between plasmodium yoelii nigeriensis susceptibility to artemisinin and alkylation of heme by the drug. Antimicrob. Agents Chemother. 2013, 57, 3998–4000. [Google Scholar] [CrossRef] [Green Version]
- Pradines, V.; Portela, J.; Boissier, J.; Coslédan, F.; Meunier, B.; Robert, A. Trioxaquine PA1259 alkylates heme in the blood-feeding parasite Schistosoma mansoni. Antimicrob. Agents Chemother. 2011, 55, 2403–2405. [Google Scholar] [CrossRef] [Green Version]
- Audran, G.; Brémond, P.; Marque, S.R.A.; Obame, G. Chemically triggered C–ON bond homolysis of alkoxyamines. 5. Cybotactic Effect. J. Org. Chem. 2012, 77, 9634–9640. [Google Scholar] [CrossRef]
- Audran, G.; Brémond, P.; Marque, S.R.A.; Obame, G. Chemically triggered C–ON bond homolysis of alkoxyamines. Part 4. Solvent Effect. Polym. Chem. 2012, 3, 2901–2908. [Google Scholar] [CrossRef]
- Audran, G.; Brémond, P.; Joly, J.P.; Marque, S.R.A.; Yamasaki, T. C–ON Bond homolysis of alkoxyamines. Part 12: Effect of the para-substituent in the 1-Phenylethyl fragment. Org. Biomol. Chem. 2016, 14, 3574–3583. [Google Scholar] [CrossRef]
- Audran, G.; Brémond, P.; Marque, S.R.A.; Yamasaki, T. C–ON bond homolysis of alkoxyamines. Part 11: Activation of the nitroxyl fragment. J. Org. Chem. 2016, 81, 1981–1988. [Google Scholar] [CrossRef] [Green Version]
- Audran, G.; Bikanga, R.; Brémond, P.; Joly, J.P.; Marque, S.R.A.; Nkolo, P. C–ON bond homolysis of alkoxyamines: Activation of the nitroxyl fragment. Normal, leveled, and enhanced steric effects in alkoxyamines carrying α,β-phosphorylated nitroxyl fragment. J. Org. Chem. 2017, 82, 5702–5709. [Google Scholar] [CrossRef]
- Nkolo, P.; Audran, G.; Bikanga, R.; Brémond, P.; Marque, S.R.A.; Roubaud, V. C–ON Bond homolysis of alkoxyamine: When too high polarity is detrimental. Org. Biomol. Chem. 2017, 15, 6167–6176. [Google Scholar] [CrossRef]
- Audran, G.; Bikanga, R.; Brémond, P.; Edeleva, M.; Marque, S.R.A.; Nkolo, P.; Roubaud, V. How intramolecular hydrogen bonding (IHB) controls the C–ON bond homolysis in alkoxyamines. Org. Biomol. Chem. 2017, 15, 8425–8439. [Google Scholar] [CrossRef] [Green Version]
- Yamasaki, T.; Buric, D.; Chacon, C.; Audran, G.; Braguer, D.; Marque, S.; Carré, M.; Brémond, P. Chemical modifications of imidazole-containing alkoxyamines increase C–ON bond homolysis rate: Effects on their cytotoxic properties in glioblastoma cells. Bioorg. Med. Chem. 2019, 27, 1942–1951. [Google Scholar] [CrossRef]
- Nkolo, P. Synthesis and Physico-Chemical Study of New Activatable Alkoxyamines to Fight Malaria. Ph.D. Thesis, University of Aix-Marseille, Marseille, France, 2017. [Google Scholar]
- To, H.T. Development of New Alkoxyamines Releasing Free Radicals against the Two Major Parasitic Diseases Malaria and Schistosomiasis. Ph.D. Thesis, University of Aix-Marseille, Marseille, France, 2019. [Google Scholar]
- To, H.T.; Audran, G.; Marque, S.R.A.; N’kolo, P.; Edeleva, M.; Cherkasov, S.; Bagryanskaya, E.; Bikanga, R.; Joly, J.-P. Synthesis of alkoxyamines based on bipyridine and acridine fragments. Org Biomol. Chem. 2021. in preparation. [Google Scholar]
- Potts, K.T.; Konwar, D. Synthesis of 4′-Vinyl-2,2′:6′,2′′-terpyridine. J. Org. Chem. 1991, 56, 4815–4816. [Google Scholar] [CrossRef]
- Bertin, D.; Gigmes, D.; Marque, S.R.A. Trialkylhydroxylamines (alkoxyamines) in radical chemistry: Preparation, stability and applications. Recent Res. Devel. Org. Chem. 2006, 10, 63–121. [Google Scholar]
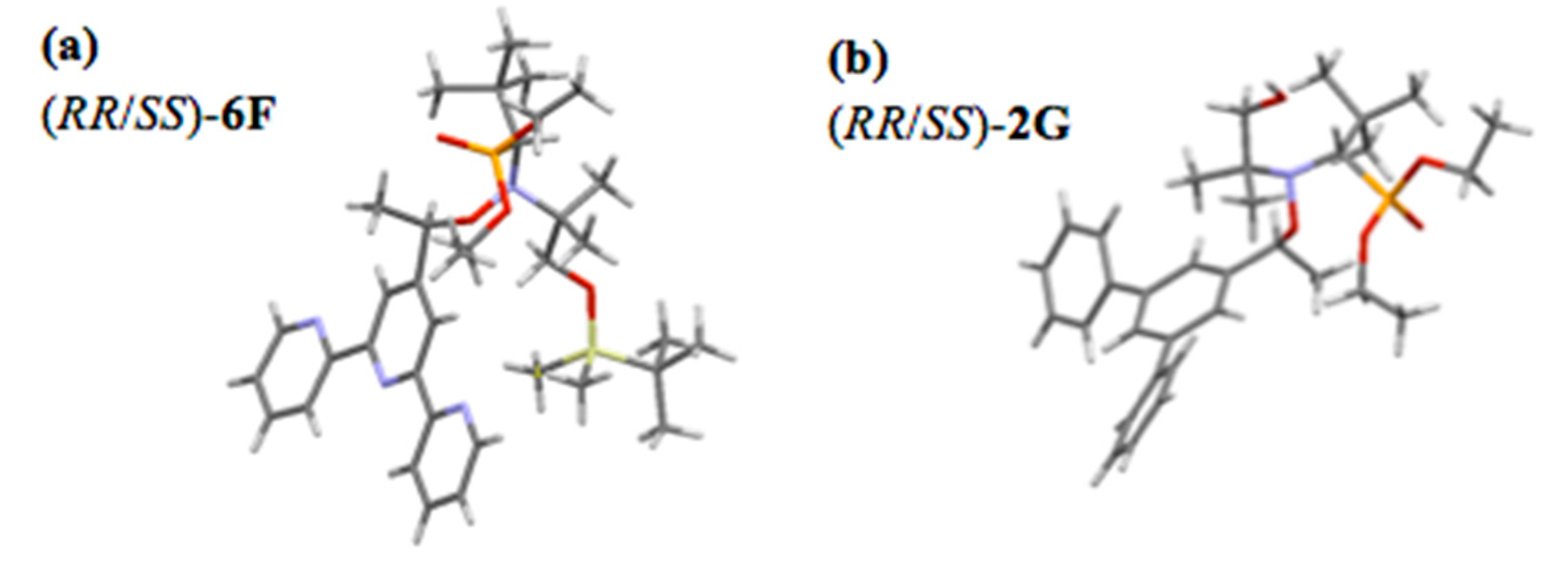
- Crystallographic Data for the Structure of (RR/SS)-2G and (RR/SS)-6F Were Deposited with the Cambridge Crystallographic Data Centre (CCDC) as Supplementary Publication for the Present Article, With No. CCDC 1938691 and CCDC 1938657, Respectively. Copies of the Data Can Be Obtained Free of Charge from the CCDC (12 Union Road, Cambridge CB2 1EZ, UK). Available online: http://www.ccdc.cam.ac.uk (accessed on 10 August 2020).
- Trost, B.M.; Caldwell, G.; Murayama, D.; Heissler, D. Sulfur-substituted dienes and the silylene protecting group in synthesis. Deoxypillaromycinone. J. Org. Chem. 1983, 48, 3252–3265. [Google Scholar] [CrossRef]
- Dess, D.B.; Martin, J.C. A useful 12-I-5 triacetoxyperiodinane (the Dess-Martin periodinane) for the selective oxidation of primary or secondary alcohols and a variety of related 12-I-5 species. J. Am. Chem. Soc. 1991, 113, 7277–7287. [Google Scholar] [CrossRef]
- Bal, B.S.; Childers, W.E.J.; Pinnick, H.W. Oxidation of α,β-unsaturated aldehydes. Tetrahedron 1981, 37, 2091–2096. [Google Scholar] [CrossRef]
- Sun, Q.; Mao, R.; Wang, D.; Hu, C.; Zheng, Y.; Sun, D. The cytotoxicity study of praziquantel enantiomers. Drug Des. Dev. Ther. 2016, 10, 2061–2068. [Google Scholar] [CrossRef] [Green Version]
- Witkowski, B.; Lelièvre, J.; López Barragán, M.J.; Laurent, V.; Su, X.; Berry, A.; Benoit-Vical, F. Increased tolerance to artemisinin in Plasmodium falciparum is mediated by a quiescence mechanism. Antimicrob. Agents Chemother. 2010, 54, 1872–1877. [Google Scholar] [CrossRef] [Green Version]
- Krȩżel, A.; Bal, W. A formula for correlating pKa values determined in D2O and H2O. J. Inorg. Biochem. 2004, 98, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, J.; Groth, N.; Herrling, T.; Zimmer, G. Electron paramagnetic resonance studies on nitroxide radical 2,2,5,5-tetramethyl-4-piperidin-1-oxyl (TEMPO) redox reactions in human skin. Free Radic. Biol. Med. 1997, 22, 967–976. [Google Scholar] [CrossRef]
- Ingold, K.U. Peroxy radicals. Acc. Chem. Res. 1969, 2, 1–9. [Google Scholar] [CrossRef]
- Soule, B.P.; Hyodo, F.; Matsumoto, K.I.; Simone, N.L.; Cook, J.A.; Krishna, M.C.; Mitchell, J.B. The chemistry and biology of nitroxide compounds. Free Radic. Biol. Med. 2007, 42, 1632–1650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewandowski, M.; Gwozdzinski, K. Nitroxides as antioxidants and anticancer drugs. Int. J. Mol. Sci. 2017, 18, 2490. [Google Scholar] [CrossRef] [Green Version]
- Wilcox, C.S.; Pearlman, A. Chemistry and antihypertensive effects of tempol and other nitroxides. Pharm. Rev. 2008, 60, 418–469. [Google Scholar] [CrossRef] [Green Version]
- Ariey, F.; Witkowski, B.; Amaratunga, C.; Beghain, J.; Langlois, A.C.; Khim, N.; Kim, S.; Duru, V.; Bouchier, C.; Ma, L.; et al. A molecular marker of artemisinin-resistant Plasmodium falciparum malaria. Nature 2014, 505, 50–55. [Google Scholar] [CrossRef]
- Trager, W.; Jensen, J.B. Human malaria parasites in continuous culture. Science 1976, 19, 673–675. [Google Scholar] [CrossRef]
- Smilkstein, M.; Sriwilaijaroen, N.; Kelly, J.X.; Wilairat, P.; Riscoe, M. Simple and inexpensive fluorescence-based technique for high-throughput antimalarial drug screening. Antimicrob. Agents Chemother. 2004, 48, 1803–1806. [Google Scholar] [CrossRef] [Green Version]
- Lambros, C.; Vanderberg, J.P. Synchronization of Plasmodium falciparum erythrocytic stages in culture. J. Parasitol. 1979, 65, 418–420. [Google Scholar] [CrossRef]
- Dumont, M.; Moné, H.; Mouahid, G.; Idris, M.A.; Shaban, M.; Boissier, J. Influence of pattern of exposure, parasite genetic diversity and sex on the degree of protection against reinfection with Schistosoma mansoni. Parasitol. Res. 2007, 101, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Ammerman, N.C.; Beier-Sexton, M.; Azad, A.F. Growth and maintenance of Vero cell lines. Curr. Protoc. Microbiol. 2008, 11, A.4E.1–A.4E.7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Gaussian 09; Revision D.01; Software for Quantum Chemical Calculations; Gaussian, Inc.: Wallingford, CT, USA, 2013.
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. mansoni Mean Survival Time (h) | P. falciparum Strains Mean IC50 (µM) | Cytotoxicity Mean IC50 (µM) | Selectivity Index 4 | ||
|---|---|---|---|---|---|
| Adult S. mansoni | F32-TEM | F32-ART5 | Vero cell lines | ||
| (RR/SS)-1a | >8 | 15 2 | 16 2 | 17 ± 7.0 | 1 |
| (RS/SR)-1a | 1.2 ± 0.1 | 7.6 2 | 11 2 | 28 ± 29 | 3 |
| (RS/SR)-2F | 2.1 ± 0.1 | 0.6 ± 0.1 | 0.5 ± 0.1 | 0.8 ± 0.3 | 1 |
| (RR/SS)-2F | 2.5 ± 0.2 | 0.5 ± 0.1 | 0.4 ± 0.1 | 0.7 ± 0.3 | 1 |
| (RS/SR)-4F (RR/SS)-4F | 5.2 ± 0.2 >8 | 2.9 2 4.6 1 | 4.7 2 4.6 1 | 15 ± 5.0 8.1 ± 3.3 | 4 2 |
| (R/S)-8F | 5.0 ± 0.0 | 1.3 2 | 1.1 2 | 0.7 ± 0.4 | 0.5 |
| (RS/SR)-7F | 2.7 ± 0.1 | 1.3 2 | 1.4 2 | 0.9 ± 0.4 | 0.6 |
| (RR/SS)-7F | 2.5 ± 0.1 | 0.94 2 | 0.85 2 | 1.2 ± 1.3 | 1 |
| 1J | 1.1 ± 0.1 | 2.9 1 | 2.3 1 | 6.2 ± 1.2 | 2 |
| 1K | 1.1 ± 0.0 | 1.1 1 | 1.6 1 | 1.1 ± 0.7 | 1 |
| 2J | 1.0 ± 0.0 | 5.0 1 | 4.6 1 | 17 ± 6.3 | 3 |
| (RS/SR)-1L | ND 3 | 11 2 | 18 2 | ND | ND |
| (RR/SS)-1L | ND | 20 2 | 26 2 | 28 | 1 |
| (RS/SR)-1M | ND | 5.8 2 | 4.9 2 | 13 ± 1.5 | 2 |
| (RR/SS)-1M | ND | 4.8 2 | 6 2 | 6.5 ± 0.3 | 1 |
| 22 | >8 | 0.24 1 | 0.23 1 | 5.3 ± 4.5 | 22 |
| 9a | 3.0 ± 0.0 | 7.5 1 | 13 1 | 40 ± 13 | 3 |
| (RS/SR)-1A | 1.0 ± 0.05 | ND | ND | 21 | |
| (RR/SS)-1A | 1.0 ± 0.05 | ND | ND | >100 | |
| (RR/SS)-1p | 1.0 ± 0.05 | ND | ND | >100 | |
| (RR/SS)-1k | 1.0 ± 0.05 | ND | ND | ||
| Praziquantel | 1.0 ± 0.05 | ND | ND | >>100 5 | >>100 |
| Artemisinin | ND | 0.02 ± 0.001 6 | 0.02 ± 0.002 6 | 160 ± 12 | 8000 |
| Chloroquine | ND | 0.06 ± 0.02 | 0.06 ± 0.03 | 190 ± 56 | 3000 |
| Drug (Dose) | Median (Range) Recrudescence Time (Days) | Mean ± SEM Difference of Recrudescence Time (Days) | p-value | |
|---|---|---|---|---|
| F32-ART5 | F32-TEM | |||
| Artemisinin (18 µM) (RS/SR)-2F (5 µM) | 9 (7–11) 16 (15–17) | >30 (16→30) 1 >30 1 | >16.3 ± 3.7 >14 ± 0.6 | 0.048 0.0017 |
| Solvent | T (°C) a | kd′ (10−4s−1) b | Ea (kJ/mol) c,d | kd (10−3 s−1) d | t1/2 (day) e | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| RS/SR f | RR/SS f | RS/SR f | RR/SS f | RS/SR f | RR/SS f | RS/SR f | RR/SS f | |||
| 6F | t-BuPh | 81 | 2.7 | 1.4 | 121.7 | 123.8 | 16.3 | 8.5 | 10 | 24 |
| 2F | t-BuPh | 81 | 5.3 | 7.0 | 119.7 | 118.9 | 29.9 | 38.4 | 4.8 | 3.5 |
| 2FH+ | t-BuPh + 2eq TFA g | 80 | 8.7 | 6.7 | 117.9 | 118.2 | 51.8 | 40.9 | 2.4 | 3.3 |
| 2F | MeOH/water 1:1 pH = 7.4 h | 81 | 8.7 | 7.1 | 118.2 | 118.8 | 46.7 | 38.9 | 2.7 | 3.5 |
| 2FH+ | MeOH/water 1:1 pH = 1.0 h | 51 | 2.1 | 12.0 | 111.9 | 107.4 | 310.1 | 1305 | 5.6 h | 1.0 h |
| 2FFe2+ | MeOH/water 1:1 pH = 7.4 h FeCl2 | 60 | 17.8 | -i | 109.2 | -j | 732.0 | -j | 2 h | -j |
| 2FFe2+ | MeOH/water 1:1 pH = 7.4 h FeCl2 | 51 | -i | 6.5 | -j | 109.0 | -j | 787.0 | -j | 1.8 h |
| 2FFe3+ | MeOH/water 1:1 pH = 7.4 h FeCl3 | 51 | 3.9 | -i | 116.4 | -j | 83.4 | -j | 1.3 | -j |
| 2FFe3+ | MeOH/water 1:1 pH = 7.4 h FeCl3 | 61 | -i | 2.0 | -j | 115.1 | -j | 120.6 | -j | 8.1 |
| 9F | t-BuPh | 111 | 3.7 k | 131.0 k | 0.95 k | 385 k | ||||
| 9FH+ | t-BuPh + 2 eq. TFA g | 101 | 3.0 k | 128.2 k | 2.2 k | 385 k | ||||
| 9F | MeOH/water 1:1 pH = 7.4 h | 90 | 0.25 k | 131.9 k | 0.7 k | 385 k | ||||
| 8F | t-BuPh | 111 | 1.9 | 133.3 k | 0.46 | 941k | ||||
| 6G | t-BuPh | 92 | 3.5 | 2.7 | 124.7 | 125.5 | 6.50 | 5.10 | 45.3 | 33.4 |
| 2G | t-BuPh | 81 | 3.7 | 3.8 | 120.8 | 120.8 | 21.6 | 21.1 | 7.5 | 7.5 |
| 2G | MeOH/water 2:1 pH = 7.0 h | 81 | 2.3 | 3.3 | 122.1 | 121.1 | 14.4 | 19.5 | 12.7 | 12.3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reyser, T.; To, T.H.; Egwu, C.; Paloque, L.; Nguyen, M.; Hamouy, A.; Stigliani, J.-L.; Bijani, C.; Augereau, J.-M.; Joly, J.-P.; et al. Alkoxyamines Designed as Potential Drugs against Plasmodium and Schistosoma Parasites. Molecules 2020, 25, 3838. https://doi.org/10.3390/molecules25173838
Reyser T, To TH, Egwu C, Paloque L, Nguyen M, Hamouy A, Stigliani J-L, Bijani C, Augereau J-M, Joly J-P, et al. Alkoxyamines Designed as Potential Drugs against Plasmodium and Schistosoma Parasites. Molecules. 2020; 25(17):3838. https://doi.org/10.3390/molecules25173838
Chicago/Turabian StyleReyser, Thibaud, Tung H. To, Chinedu Egwu, Lucie Paloque, Michel Nguyen, Alexandre Hamouy, Jean-Luc Stigliani, Christian Bijani, Jean-Michel Augereau, Jean-Patrick Joly, and et al. 2020. "Alkoxyamines Designed as Potential Drugs against Plasmodium and Schistosoma Parasites" Molecules 25, no. 17: 3838. https://doi.org/10.3390/molecules25173838