
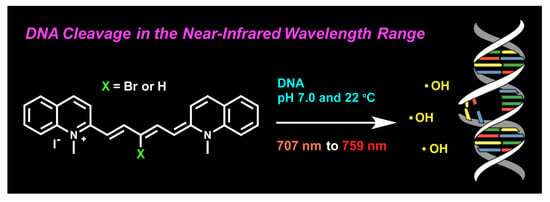
DNA Photocleavage in the Near-Infrared Wavelength Range by 2-Quinolinium Dicarbocyanine Dyes

Abstract
:
1. Introduction
2. Results and Discussion
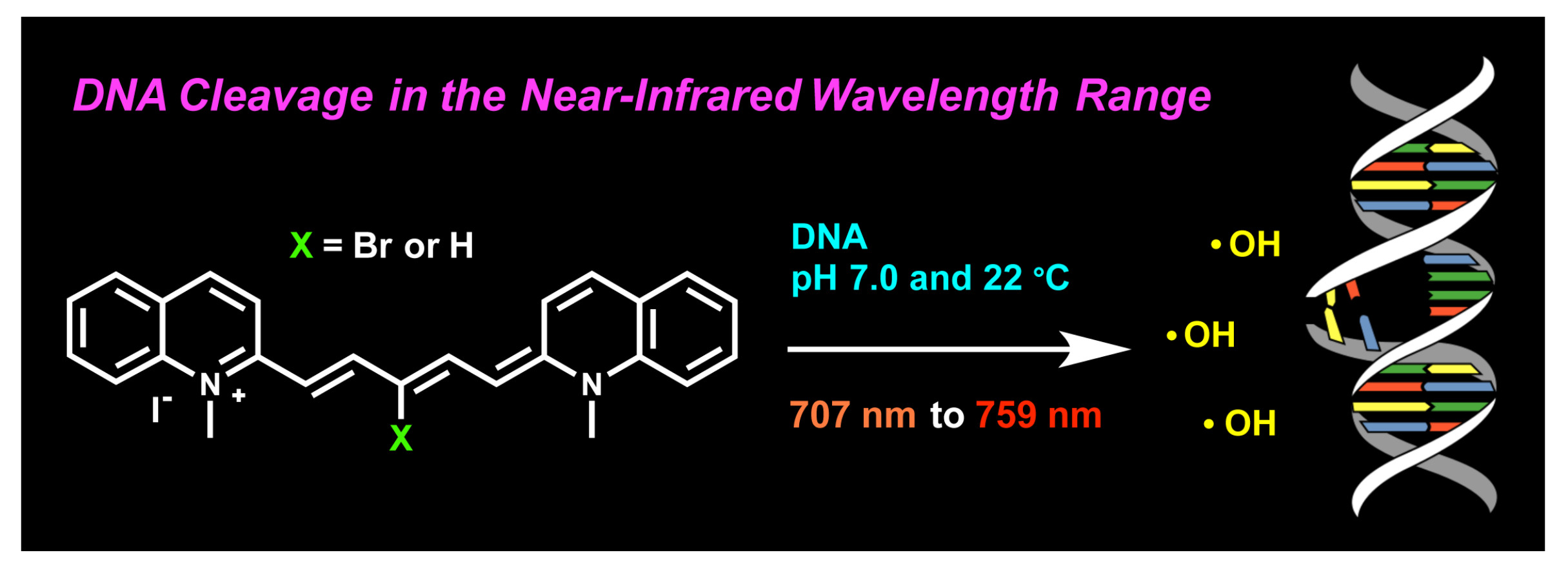
2.1. UV–Visible Spectrophotometry: Dye Stability and DNA Interactions
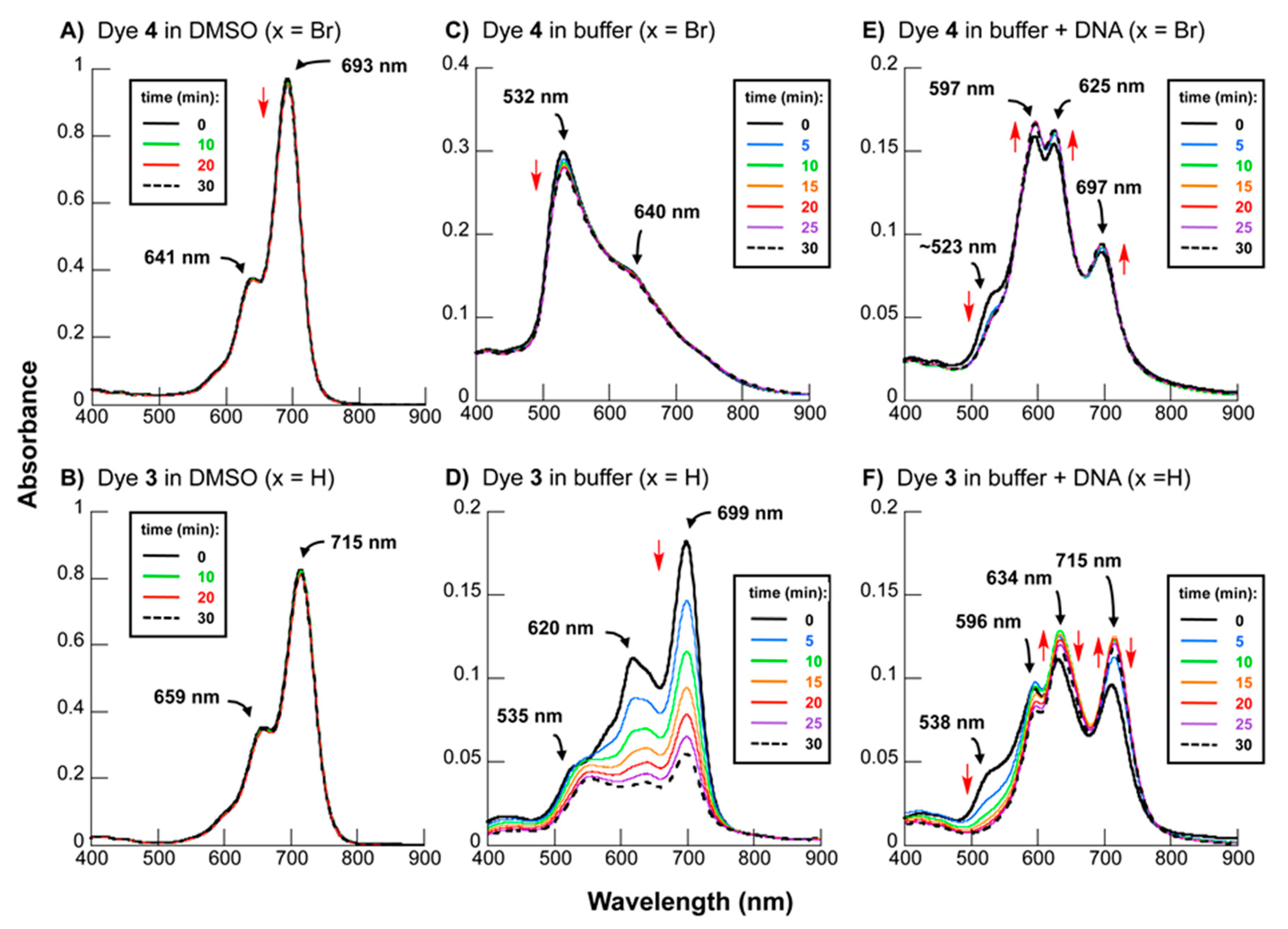
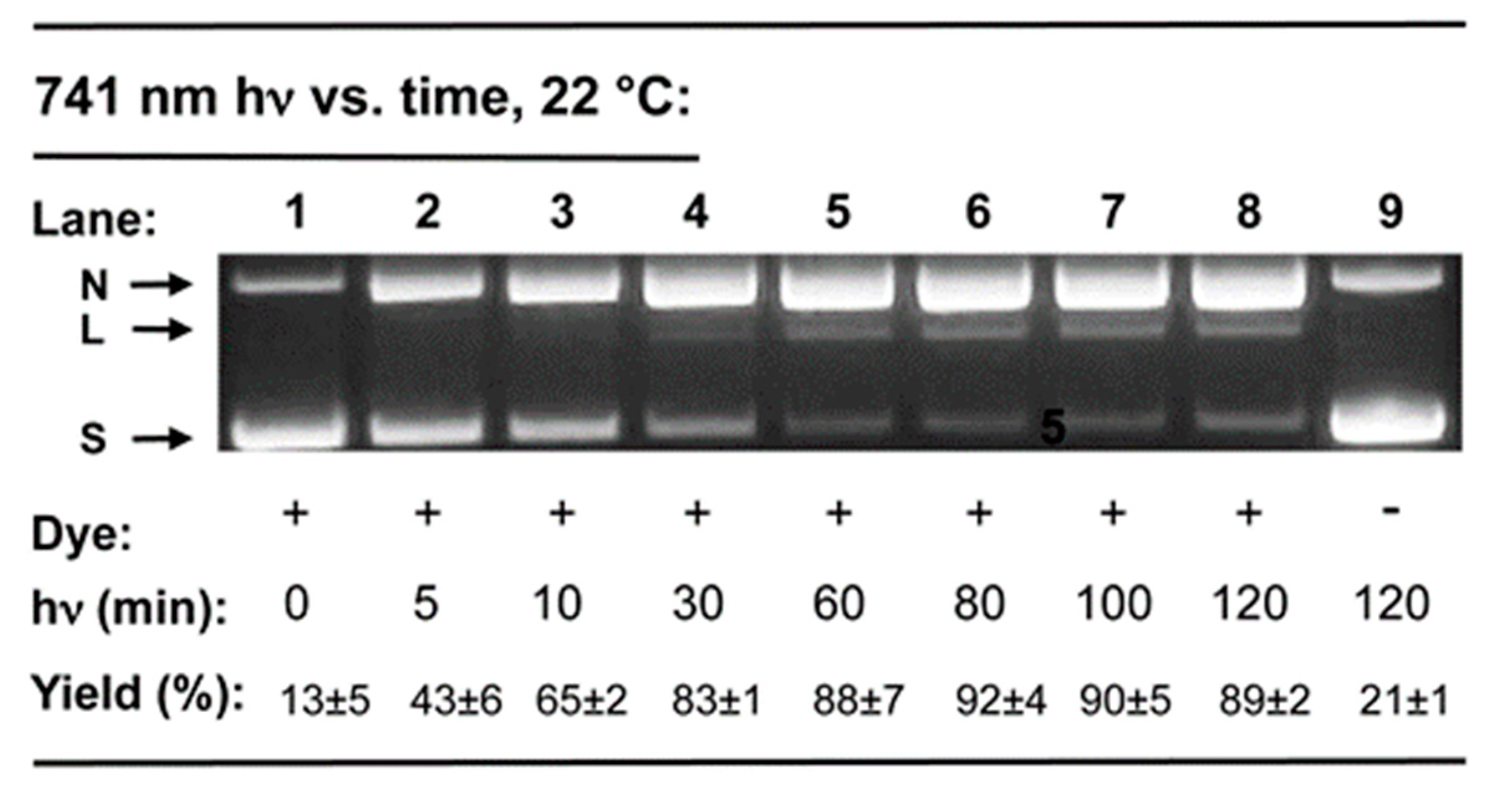
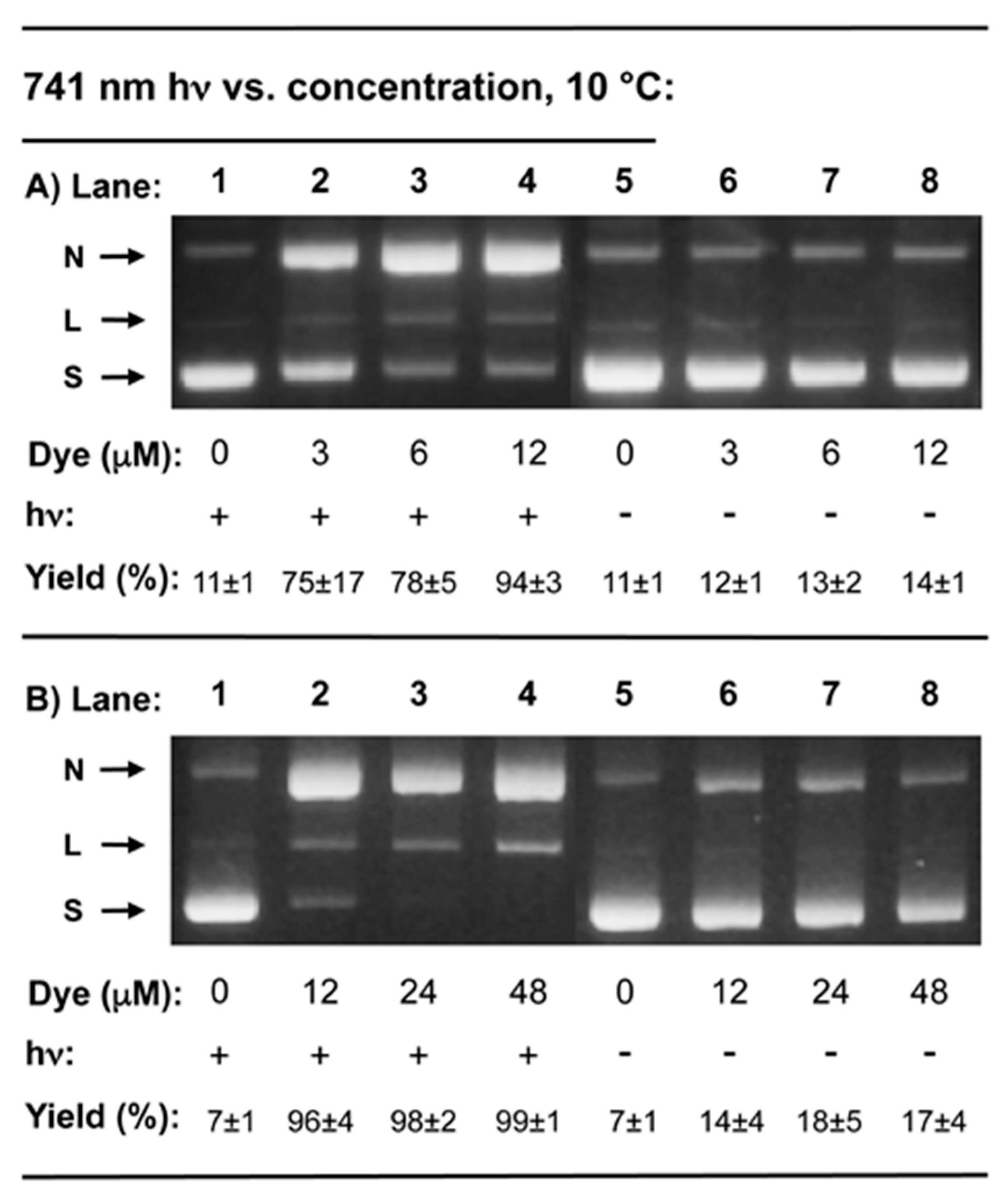
2.2. DNA Photocleavage in the Near-Infrared Wavelength Range
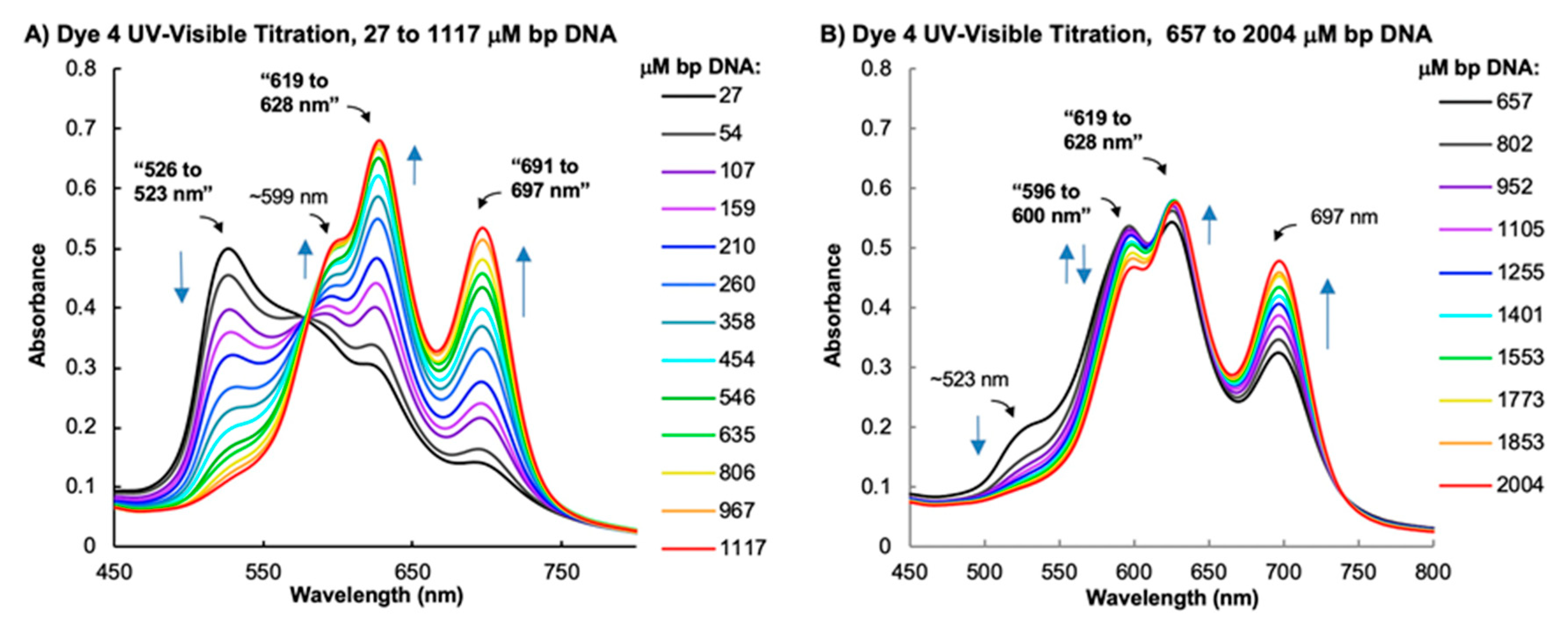
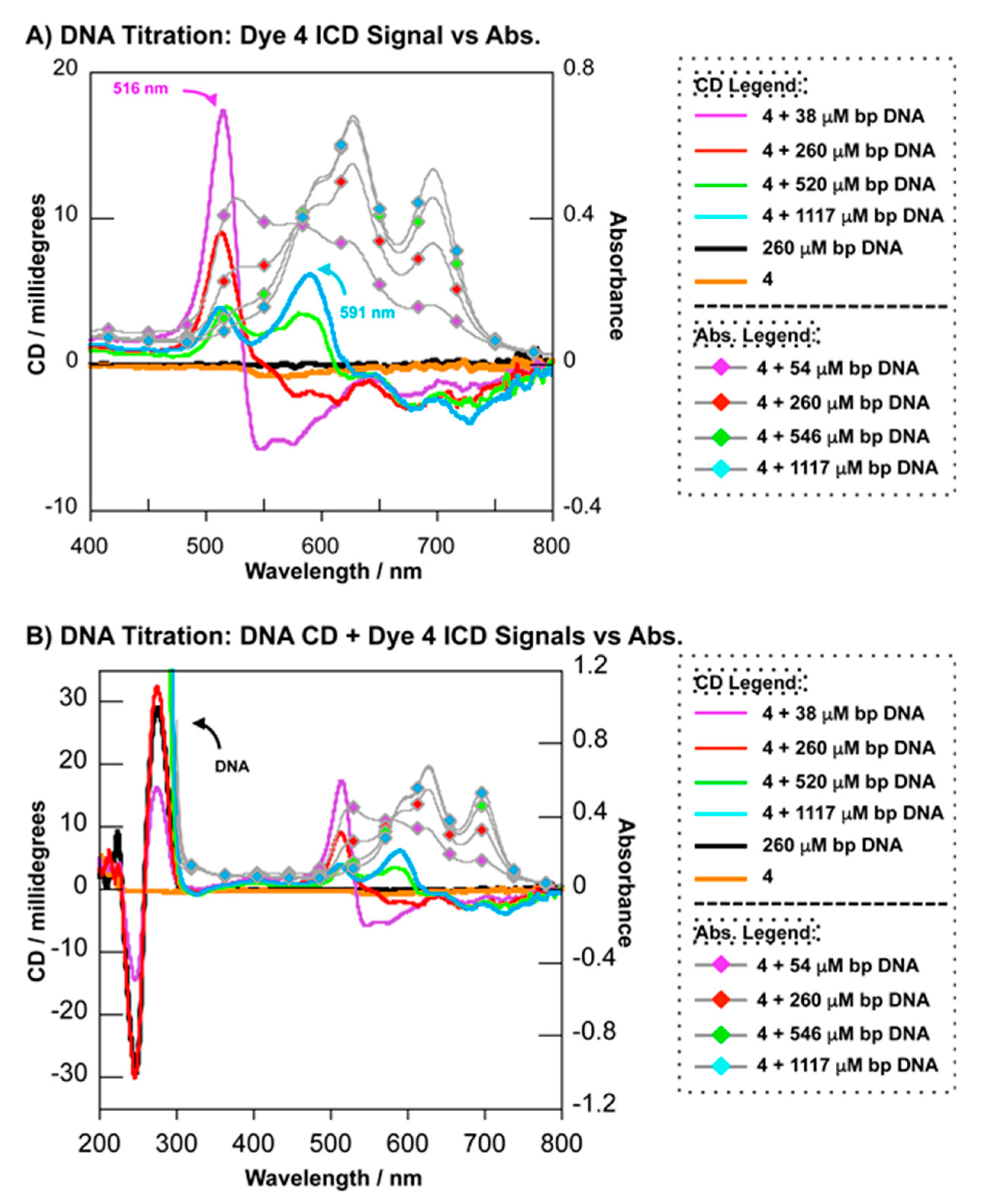
2.3. Spectrophotometric Analyses of DNA Binding Modes
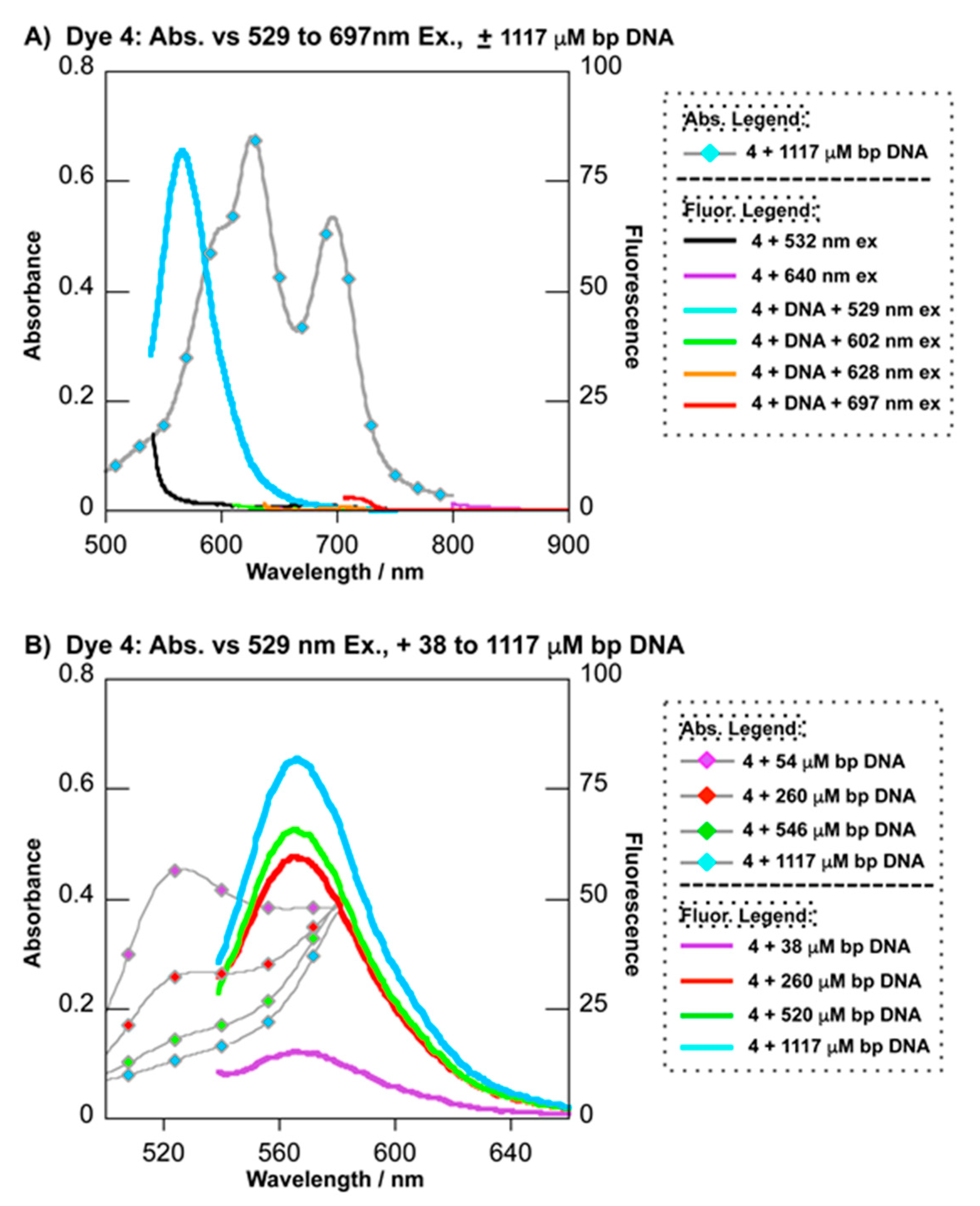
2.3.1. UV–Visible Absorption Spectrophotometry
2.3.2. Circular Dichroism
2.3.3. Fluorescence Spectroscopy
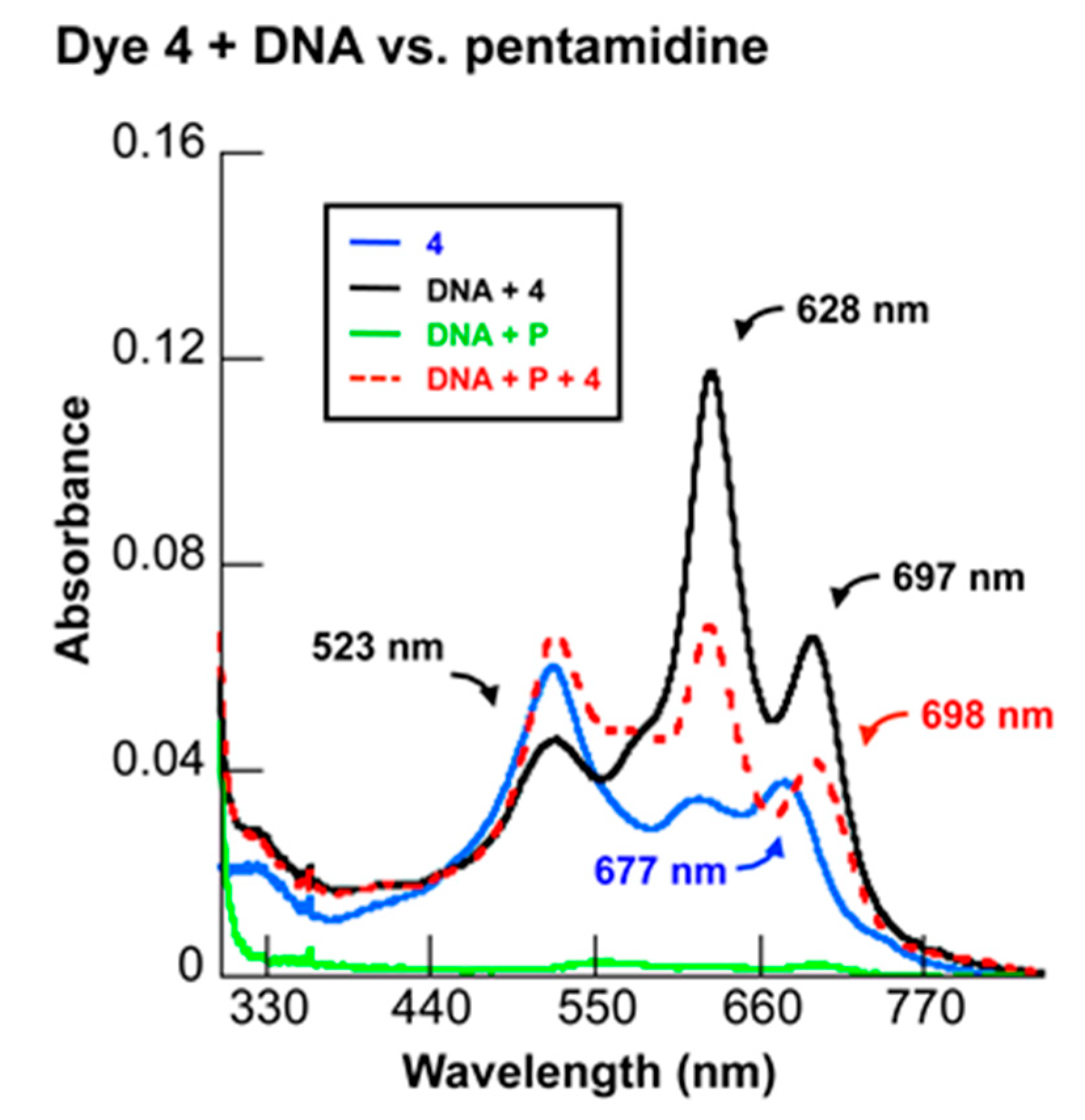
2.3.4. Competitive DNA Binding
2.4. Mechanistic Analysis of Dye-Sensitized DNA Photocleavage
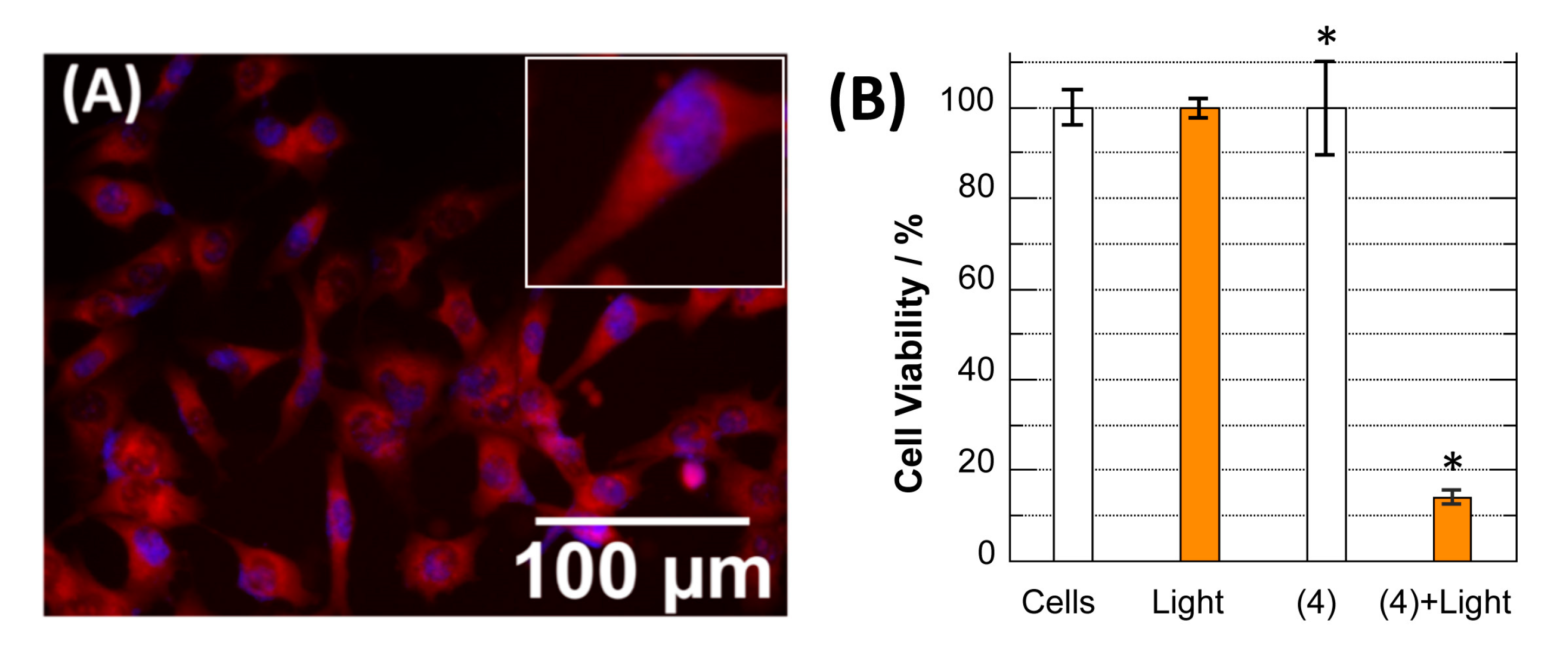
2.5. Uptake, Distribution, and Phototoxicity of Dye 4 in Ovarian Cancer Cells
3. Materials and Methods
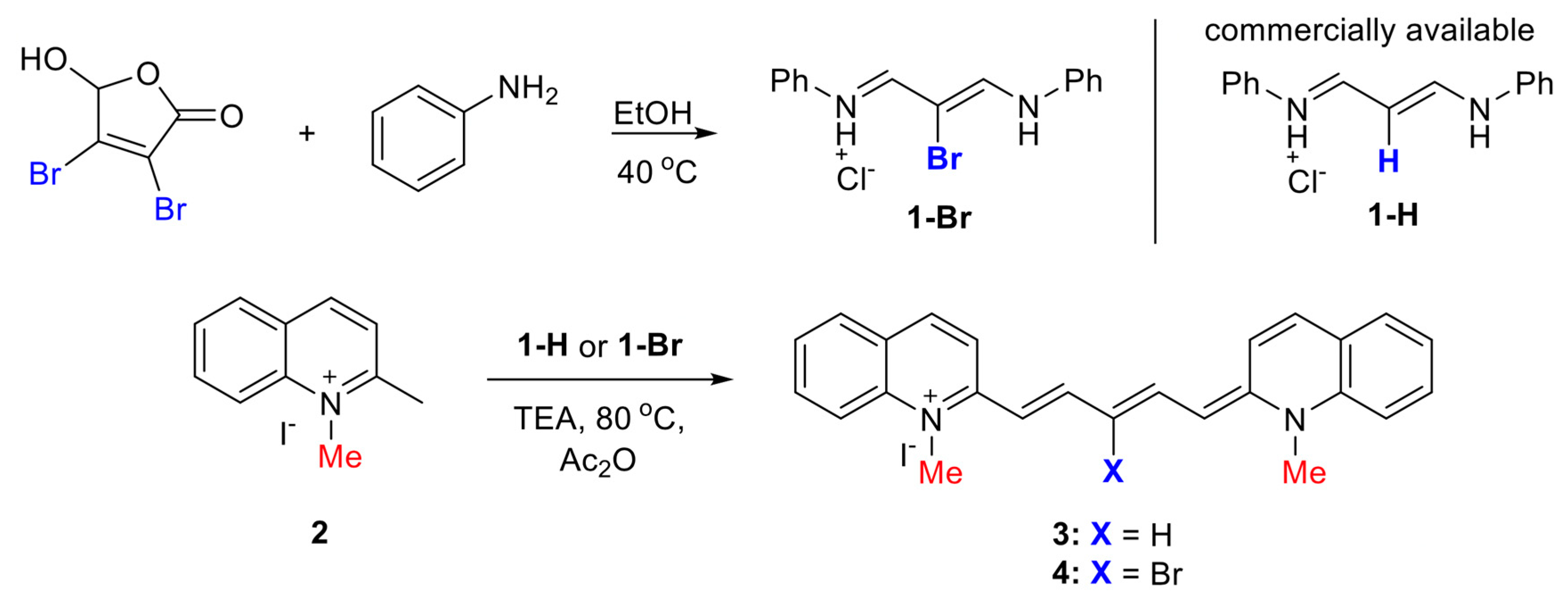
3.1. Synthesis
3.1.1. General
3.1.2. Procedure for the Preparation of Dyes 3 and 4
3.2. Analysis of Dye-DNA Interactions
3.2.1. General
3.2.2. DNA Photocleavage
3.2.3. UV–Visible Spectrophotometry
3.2.4. Circular Dichroism
3.2.5. Fluorescence Spectroscopy
3.2.6. Regent-Induced Changes in DNA Photocleavage
3.3. Cytotoxicity Studies
3.3.1. Cell Line
3.3.2. Cellular Uptake and Fluorescence Imaging
3.3.3. Evaluation of Phototherapeutic Effect
4. Summary and Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| bp | base pair |
| CT DNA | calf thymus DNA |
| DAPI | 4′,6-diamidino-2-phenylindole |
| DMEM | Dulbecco’s Modified Eagle Media |
| DMSO | dimethyl sulfoxide |
| DPBS | Dulbecco’s phosphate-buffered saline |
| EDTA | ethylenediaminetetraacetic acid |
| ICD | induced circular dichroism |
| ICG | indocyanine green |
| OY | oxazole yellow |
| PS | photosensitizing agent |
| ROS | reactive oxygen species |
| TAE | tris-acetate-EDTA |
| TO | thiazole orange |
References
- Sun, W.; Guo, S.; Hu, C.; Fan, J.; Peng, X. Recent development of chemosensors based on cyanine platforms. Chem. Rev. 2016, 116, 7768–7817. [Google Scholar] [CrossRef]
- Shindy, H.A. Fundamentals in the chemistry of cyanine dyes: A review. Dye. Pigm. 2017, 145, 505–513. [Google Scholar] [CrossRef]
- Armitage, B. Cyanine dye–nucleic acid interactions. In Heterocyclic Polymethine Dyes; Strekowski, L., Ed.; Springer: Berlin/Heidelberg, Germany, 2008; Volume 14, pp. 11–29. [Google Scholar]
- Armitage, B.A. Cyanine dye–DNA interactions: Intercalation, groove binding, and aggregation. In DNA Binders and Related Subjects; Waring, M., Chaires, J., Eds.; Springer: Berlin/Heidelberg, Germany, 2005; Volume 253, pp. 55–76. [Google Scholar]
- Kawabe, Y.; Kato, S. Spectroscopic study of cyanine dyes interacting with the biopolymer, DNA. Dye. Pigm. 2012, 95, 614–618. [Google Scholar] [CrossRef]
- Garoff, R.A.; Litzinger, E.A.; Connor, R.E.; Fishman, I.; Armitage, B.A. Helical aggregation of cyanine dyes on DNA templates: Effect of dye structure on formation of homo- and heteroaggregates. Langmuir 2002, 18, 6330–6337. [Google Scholar] [CrossRef]
- Wang, M.; Silva, G.L.; Armitage, B.A. DNA-templated formation of a helical cyanine dye J-aggregate. J. Am. Chem. Soc. 2000, 122, 9977–9986. [Google Scholar] [CrossRef]
- Boutorine, A.S.; Novopashina, D.S.; Krasheninina, O.A.; Nozeret, K.; Venyaminova, A.G. Fluorescent probes for nucleic acid visualization in fixed and live cells. Molecules 2013, 18, 15357–15397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.; Alexander, S.C.; Jin, S.; Devaraj, N.K. A bioorthogonal near-infrared fluorogenic probe for mRNA detection. J. Am. Chem. Soc. 2016, 138, 11429–11432. [Google Scholar] [CrossRef] [PubMed]
- Jacques, S.L. Optical properties of biological tissues: A review. Phys. Med. Biol. 2013, 58, R37–R61. [Google Scholar] [CrossRef] [PubMed]
- Keller, D.S.; Ishizawa, T.; Cohen, R.; Chand, M. Indocyanine green fluorescence imaging in colorectal surgery: Overview, applications, and future directions. Lancet Gastroenterol. Hepatol. 2017, 2, 757–766. [Google Scholar] [CrossRef] [Green Version]
- Giraudeau, C.; Moussaron, A.; Stallivieri, A.; Mordon, S.; Frochot, C. Indocyanine green: Photosensitizer or chromophore? Still a debate. Curr. Med. Chem. 2014, 21, 1871–1897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Q.; Chen, T.; Chen, S. Fluorescent contrast agents for tumor surgery. Exp. Med. 2018, 16, 1577–1585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.H.; Schreiber, C.L.; Smith, B.D. Sterically shielded heptamethine cyanine dyes for bioconjugation and high performance near-infrared fluorescence imaging. Angew. Chem. Int. Ed. Engl. 2020. [Google Scholar] [CrossRef]
- Atchison, J.; Kamila, S.; Nesbitt, H.; Logan, K.A.; Nicholas, D.M.; Fowley, C.; Davis, J.; Callan, B.; McHale, A.P.; Callan, J.F. Iodinated cyanine dyes: A new class of sensitisers for use in NIR activated photodynamic therapy (PDT). Chem. Commun. 2017, 53, 2009–2012. [Google Scholar] [CrossRef] [PubMed]
- Jing, T.; Fu, L.; Liu, L.; Yan, L. A reduction-responsive polypeptide nanogel encapsulating NIR photosensitizer for imaging guided photodynamic therapy. Polym. Chem. 2016, 7, 951–957. [Google Scholar] [CrossRef]
- Cao, J.; Chi, J.; Xia, J.; Zhang, Y.; Han, S.; Sun, Y. Iodinated cyanine dyes for fast near-infrared-guided deep tissue synergistic phototherapy. ACS Appl. Mater. Interfaces 2019, 11, 25720–25729. [Google Scholar] [CrossRef]
- Siriwibool, S.; Kaekratoke, N.; Chansaenpak, K.; Siwawannapong, K.; Panajapo, P.; Sagarik, K.; Noisa, P.; Lai, R.Y.; Kamkaew, A. Near-infrared fluorescent pH responsive probe for targeted photodynamic cancer therapy. Sci. Rep. 2020, 10, 1283. [Google Scholar] [CrossRef] [Green Version]
- Ormond, A.B.; Freeman, H.S. Dye sensitizers for photodynamic therapy. Materials 2013, 6, 817–840. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.Y.; Grant, K.B. DNA photo-cleaving agents in the far-red to near-infrared range-a review. RSC Adv. 2016, 6, 24617–24634. [Google Scholar] [CrossRef]
- Luby, B.M.; Walsh, C.D.; Zheng, G. Advanced photosensitizer activation strategies for smarter photodynamic therapy beacons. Angew. Chem. Int. Ed. Engl. 2019, 58, 2558–2569. [Google Scholar] [CrossRef]
- Bonnett, R. Photosensitizers of the porphyrin and phthalocyanine series for photodynamic therapy. Chem. Soc. Rev. 1995, 24, 19–33. [Google Scholar] [CrossRef]
- Ochsner, M. Photophysical and photobiological processes in the photodynamic therapy of tumours. J. Photochem. Photobiol. B 1997, 39, 1–18. [Google Scholar] [CrossRef]
- Hatz, S.; Poulsen, L.; Ogilby, P.R. Time-resolved singlet oxygen phosphorescence measurements from photosensitized experiments in single cells: Effects of oxygen diffusion and oxygen concentration. Photochem. Photobiol. 2008, 84, 1284–1290. [Google Scholar] [CrossRef] [PubMed]
- Makrigiorgos, G.M. Detection of chromatin-associated hydroxyl radicals generated by DNA-bound metal compounds and antitumor antibiotics. Met. Ions Biol. Syst. 1999, 36, 521–545. [Google Scholar] [PubMed]
- Pogozelski, W.K.; Tullius, T.D. Oxidative strand scission of nucleic acids: Routes initiated by hydrogen abstraction from the sugar moiety. Chem. Rev. 1998, 98, 1089–1108. [Google Scholar] [CrossRef]
- Agostinis, P.; Berg, K.; Cengel, K.A.; Foster, T.H.; Girotti, A.W.; Gollnick, S.O.; Hahn, S.M.; Hamblin, M.R.; Juzeniene, A.; Kessel, D.; et al. Photodynamic therapy of cancer: An update. CA Cancer J. Clin. 2011, 61, 250–281. [Google Scholar] [CrossRef]
- Mapp, C.T.; Owens, E.A.; Henary, M.; Grant, K.B. Oxidative cleavage of DNA by pentamethine carbocyanine dyes irradiated with long-wavelength visible light. Bioorg. Med. Chem. Lett. 2014, 24, 214–219. [Google Scholar] [CrossRef]
- Basnet, K.; Fatemipouya, T.; St Lorenz, A.; Nguyen, M.; Taratula, O.; Henary, M.; Grant, K.B. Single photon DNA photocleavage at 830 nm by quinoline dicarbocyanine dyes. Chem. Commun. 2019, 55, 12667–12670. [Google Scholar] [CrossRef]
- Åkerman, B.; Tuite, E. Single- and double-strand photocleavage of DNA by YO, YOYO and TOTO. Nucleic Acids Res. 1996, 24, 1080–1090. [Google Scholar] [CrossRef] [Green Version]
- Kanony, C.; Akerman, B.; Tuite, E. Photobleaching of asymmetric cyanines used for fluorescence imaging of single DNA molecules. J. Am. Chem. Soc. 2001, 123, 7985–7995. [Google Scholar] [CrossRef]
- Mahon, K.P.; Ortiz-Meoz, R.F.; Prestwich, E.G.; Kelley, S.O. Photosensitized DNA cleavage promoted by amino acids. Chem. Commun. 2003, 1956–1957. [Google Scholar] [CrossRef]
- Biton, A.; Ezra, A.; Kasparkova, J.; Brabec, V.; Yavin, E. DNA photocleavage by DNA and DNA-LNA amino acid-dye conjugates. Bioconjugate Chem. 2010, 21, 616–621. [Google Scholar] [CrossRef] [PubMed]
- Nanjunda, R.; Owens, E.A.; Mickelson, L.; Alyabyev, S.; Kilpatrick, N.; Wang, S.; Henary, M.; Wilson, W.D. Halogenated pentamethine cyanine dyes exhibiting high fidelity for G-quadruplex DNA. Bioorg. Med. Chem. 2012, 20, 7002–7011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parton, R.L.; Lenhard, J.R. Dimerization reactions of cyanine radical dications. J. Org. Chem. 1990, 55, 49–57. [Google Scholar] [CrossRef]
- Miyazoe, Y.; Maeda, M. Stimulated emission from 19 polymethine dyes-laser action over the continuous range 710–1060 mμ. Appl. Phys. Lett. 1968, 12, 206–208. [Google Scholar] [CrossRef]
- Patnaik, L.N.; Misra, P.K.; Rout, M.K. Application of a free electron molecular orbital model: Absorption spectra of cyanine dyes. J. Indian Chem. Soc. 1976, 53, 391–392. [Google Scholar]
- Owens, E.; Hyun, H.; Tawney, J.G.; Choi, H.; Henary, M. Correlating molecular character of NIR imaging agents with tissue-specific uptake. J. Med. Chem. 2015, 58, 4348–4356. [Google Scholar] [CrossRef]
- Kodama, M.; Tagashira, Y.; Nagata, C. The interaction of pinacyanol with nucleic acids. Biochim. Biophys. Acta 1996, 129, 638–640. [Google Scholar] [CrossRef]
- Barros, T.C.; Toma, S.H.; Toma, H.E.; Bastos, E.L.; Baptista, M.S. Polymethine cyanine dyes in beta-cyclodextrin solution: Multiple equilibria and chemical oxidation. J. Phys. Org. Chem. 2010, 23, 893–903. [Google Scholar] [CrossRef]
- Gorman, A.; Killoran, J.; O’Shea, C.; Kenna, T.; Gallagher, W.M.; O’Shea, D.F. In vitro demonstration of the heavy-atom effect for photodynamic therapy. J. Am. Chem. Soc. 2004, 126, 10619–10631. [Google Scholar] [CrossRef]
- Seifert, J.L.; Connor, R.E.; Kushon, S.A.; Wang, M.; Armitage, B.A. Spontaneous assembly of helical cyanine dye aggregates on DNA nanotemplates. J. Am. Chem. Soc. 1999, 121, 2987–2995. [Google Scholar] [CrossRef]
- Petty, J.T.; Bordelon, J.A.; Robertson, M.E. Thermodynamic characterization of the association of cyanine dyes with DNA. J. Phys. Chem. B 2000, 104, 7221–7227. [Google Scholar] [CrossRef]
- Beckford, G.; Owens, E.; Henary, M.; Patonay, G. The solvatochromic effects of side chain substitution on the binding interaction of novel tricarbocyanine dyes with human serum albumin. Talanta 2012, 92, 45–52. [Google Scholar] [CrossRef]
- Karlsson, H.J.; Bergqvist, M.H.; Lincoln, P.; Westman, G. Syntheses and DNA-binding studies of a series of unsymmetrical cyanine dyes: Structural influence on the degree of minor groove binding to natural DNA. Bioorg. Med. Chem. 2004, 12, 2369–2384. [Google Scholar] [CrossRef] [PubMed]
- Norden, B.; Tjerneld, F. Optical studies on complexes between DNA and pseudoisocyanine. Biophys. Chem. 1976, 6, 31–45. [Google Scholar] [CrossRef]
- Garbett, N.C.; Ragazzon, P.A.; Chaires, J.B. Circular dichroism to determine binding mode and affinity of ligand-DNA interactions. Nat. Protoc. 2007, 2, 3166–3172. [Google Scholar] [CrossRef] [PubMed]
- Maj, M.; Jeon, J.; Gora, R.W.; Cho, M. Induced optical activity of DNA-templated cyanine dye aggregates: Exciton coupling theory and TD-DFT studies. J. Phys. Chem. A 2013, 117, 5909–5918. [Google Scholar] [CrossRef]
- Zhegalova, N.G.; He, S.; Zhou, H.; Kim, D.M.; Berezin, M.Y. Minimization of self-quenching fluorescence on dyes conjugated to biomolecules with multiple labeling sites via asymmetrically charged NIR fluorophores. Contrast Media Mol. Imaging 2014, 9, 355–362. [Google Scholar] [CrossRef] [Green Version]
- Swiecicki, J.M.; Thiebaut, F.; Di Pisa, M.; Gourdin-Bertin, S.; Tailhades, J.; Mansuy, C.; Burlina, F.; Chwetzoff, S.; Trugnan, G.; Chassaing, G.; et al. How to unveil self-quenched fluorophores and subsequently map the subcellular distribution of exogenous peptides. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, M.; Harada, Y. Direct observation of the reversible unwinding of a single DNA molecule caused by the intercalation of ethidium bromide. Nucleic Acids Res. 2007, 35, e125. [Google Scholar] [CrossRef] [PubMed]
- Edwards, K.J.; Jenkins, T.C.; Neidle, S. Crystal-structure of a pentamidine oligonucleotide complex-implications for DNA-binding properties. Biochemistry 1992, 31, 7104–7109. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, T.C.; Lane, A.N. AT selectivity and DNA minor groove binding: Modelling, NMR and structural studies of the interactions of propamidine and pentamidine with d(CGCGAATTCGCG)(2). BBA-Gene Struct. Expr. 1997, 1350, 189–204. [Google Scholar] [CrossRef]
- Merkel, P.B.; Kearns, D.R. Radiationless decay of singlet molecular oxygen in solution. Experimental and theoretical study of electronic-to-vibrational energy transfer. J. Am. Chem. Soc. 1972, 94, 7244–7253. [Google Scholar] [CrossRef]
- Li, X.; Schumann, C.; Albarqi, H.A.; Lee, C.J.; Alani, A.W.G.; Bracha, S.; Milovancev, M.; Taratula, O.; Taratula, O. A tumor-activatable theranostic nanomedicine platform for NIR fluorescence-guided surgery and combinatorial phototherapy. Theranostics 2018, 8, 767–784. [Google Scholar] [CrossRef]
- Schumann, C.; Taratula, O.; Khalimonchuk, O.; Palmer, A.L.; Cronk, L.M.; Jones, C.V.; Escalante, C.A.; Taratula, O. ROS-induced nanotherapeutic approach for ovarian cancer treatment based on the combinatorial effect of photodynamic therapy and DJ-1 gene suppression. Nanomedicine 2015, 11, 1961–1970. [Google Scholar] [CrossRef] [PubMed]
- Dani, R.K.; Schumann, C.; Taratula, O.; Taratula, O. Temperature-tunable iron oxide nanoparticles for remote-controlled drug release. AAPS PharmSciTech 2014, 15, 963–972. [Google Scholar] [CrossRef] [Green Version]
- Sambrook, J.; Fritsch, E.F.; Maniatis, T. Molecular Cloning, A Laboratory Manual; Cold Spring Harbor Press: Cold Spring Harbor, NY, USA, 1989. [Google Scholar]
Sample Availability: Not available. |












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
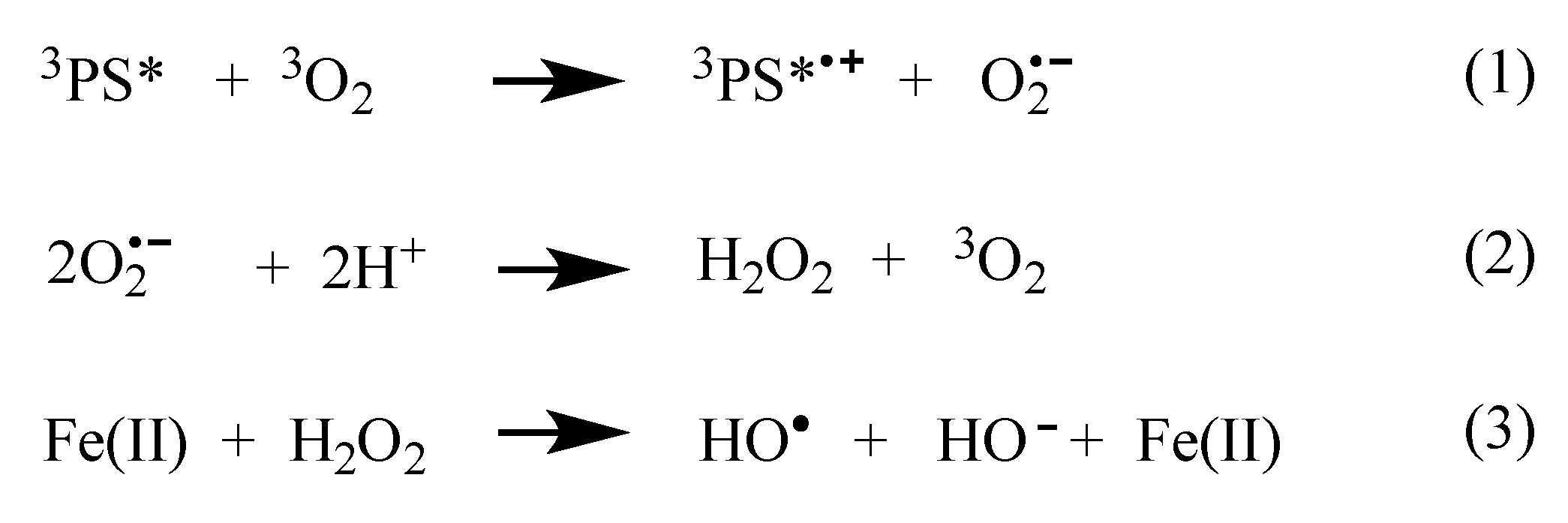{kind=link}
| Reagents Added | Target | Cleavage Inhibition (%) |
|---|---|---|
| Na benzoate | •OH | 77 ± 9 |
| D2O | 1O2 | 11 ± 0.4 |
| Catalase | H2O2 | 30 ± 10 |
| EDTA | Fe2+, Fe3+/Cu1+, Cu2+ | 73 ± 8 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahoulou, E.O.; Drinkard, K.K.; Basnet, K.; St. Lorenz, A.; Taratula, O.; Henary, M.; Grant, K.B. DNA Photocleavage in the Near-Infrared Wavelength Range by 2-Quinolinium Dicarbocyanine Dyes. Molecules 2020, 25, 2926. https://doi.org/10.3390/molecules25122926
Ahoulou EO, Drinkard KK, Basnet K, St. Lorenz A, Taratula O, Henary M, Grant KB. DNA Photocleavage in the Near-Infrared Wavelength Range by 2-Quinolinium Dicarbocyanine Dyes. Molecules. 2020; 25(12):2926. https://doi.org/10.3390/molecules25122926
Chicago/Turabian StyleAhoulou, Effibe O., Kaitlyn K. Drinkard, Kanchan Basnet, Anna St. Lorenz, Oleh Taratula, Maged Henary, and Kathryn B. Grant. 2020. "DNA Photocleavage in the Near-Infrared Wavelength Range by 2-Quinolinium Dicarbocyanine Dyes" Molecules 25, no. 12: 2926. https://doi.org/10.3390/molecules25122926