
Sulfoximines-Assisted Rh(III)-Catalyzed C–H Activation and Intramolecular Annulation for the Synthesis of Fused Isochromeno-1,2-Benzothiazines Scaffolds under Room Temperature

Abstract
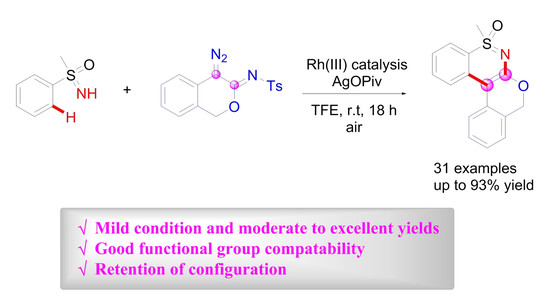
:
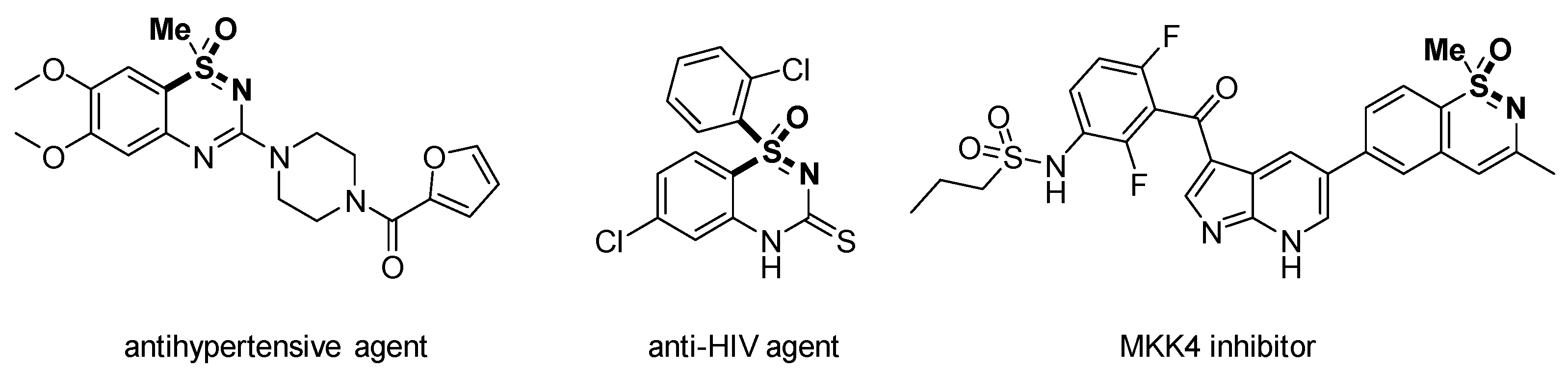
1. Introduction
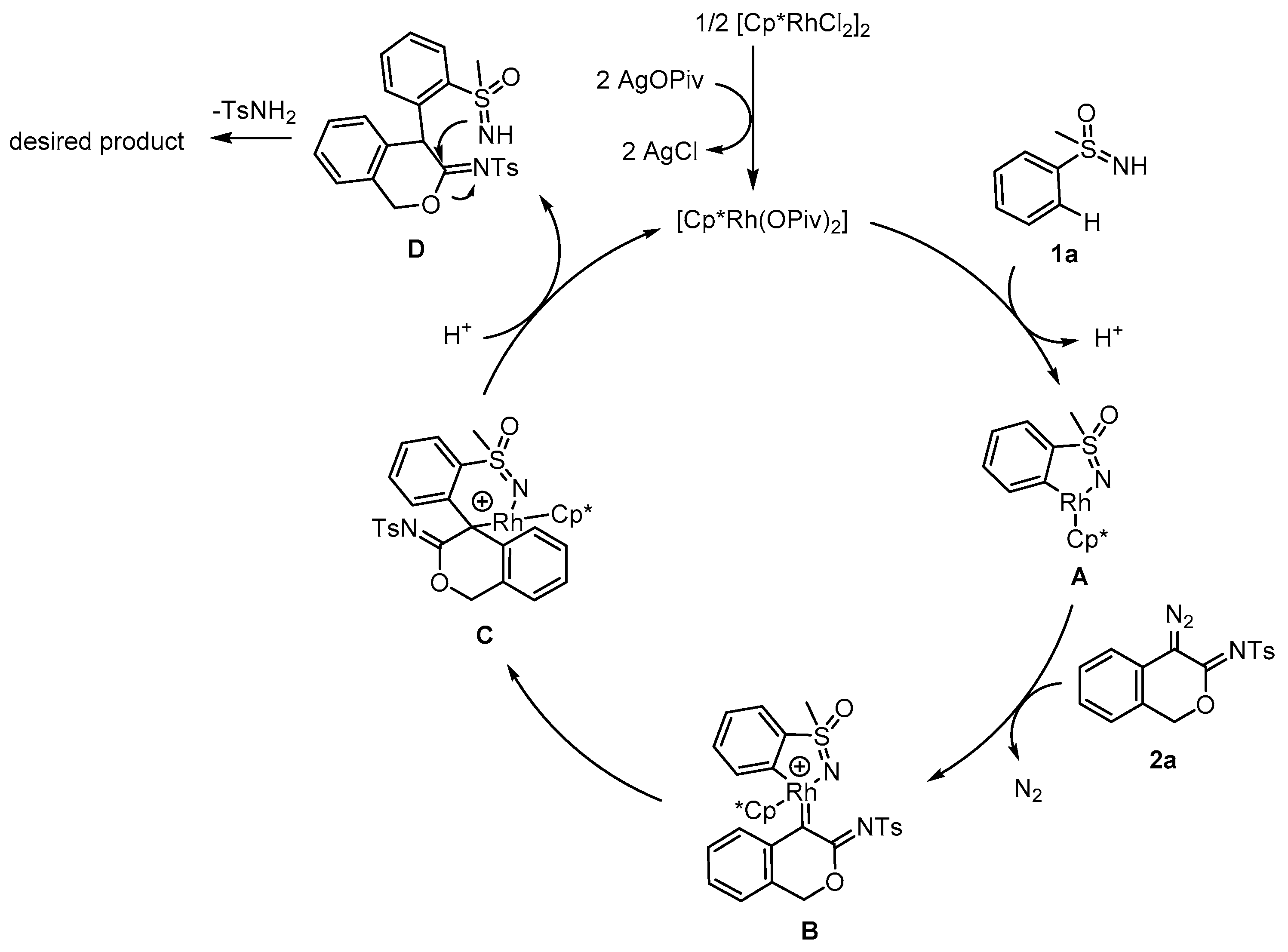
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. Experimental Part Method
3.2.1. General Procedure A for the Synthesis of Substrates 1a–1w
3.2.2. General Procedure for the Synthesis of Substrates 2a–2k
3.2.3. General Procedure for the Synthesis of Compounds 3
3.2.4. Characterization of the Products
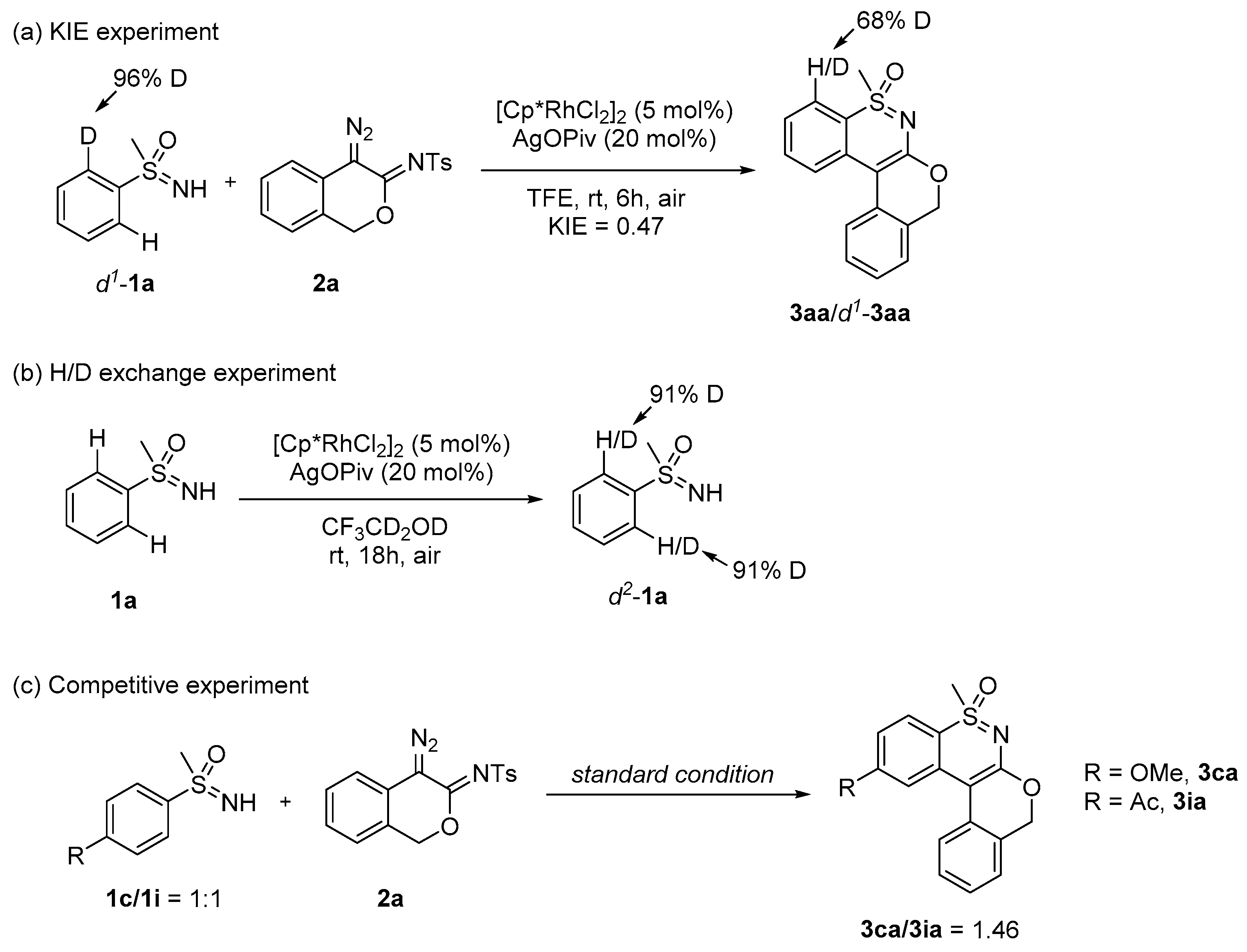
3.2.5. Mechanistic Investigations
4. Conclusions
Supplementary Materials
Supplementary File 1Author Contributions
Funding
Conflicts of Interest
References
- Cho, G.Y.; Bolm, C. Silver-Catalyzed Imination of Sulfoxides and Sulfides. Org. Lett. 2005, 7, 4983–4985. [Google Scholar] [CrossRef]
- Gais, H.-J. Development of New Methods for Asymmetric Synthesis Based on Sulfoximines. Heteroat. Chem. 2007, 18, 472–481. [Google Scholar] [CrossRef]
- Hendriks, C.M.M.; Lamers, P.; Engel, J.; Bolm, C. Sulfoxide-to-Sulfilimine Conversions: Use of Modified Burgess-Type Reagents. Adv. Synth. Catal. 2013, 355, 3363–3368. [Google Scholar] [CrossRef]
- Bizet, V.; Kowalczyk, R.; Bolm, C. Fluorinated sulfoximines: Syntheses, properties and applications. Chem. Soc. Rev. 2014, 43, 2426–2438. [Google Scholar] [CrossRef] [PubMed]
- Miao, J.; Richards, N.G.J.; Ge, H. Rhodium-catalyzed direct synthesis of unprotected NH-sulfoximines from sulfoxides. Chem. Commun. 2014, 50, 9687–9689. [Google Scholar] [CrossRef] [PubMed]
- Bizet, V.; Hendriks, C.M.M.; Bolm, C. Sulfur imidations: Access to sulfimides and sulfoximines. Chem. Soc. Rev. 2015, 44, 3378–3390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tota, A.; Zenzola, M.; Chawner, S.J.; John-Campbell, S.S.; Carlucci, C.; Romanazzi, G.; DeGennaro, L.; Bull, J.A.; Luisi, R. Synthesis of NH-sulfoximines from sulfides by chemoselective one-pot N- and O-transfers. Chem. Commun. 2017, 53, 348–351. [Google Scholar] [CrossRef] [Green Version]
- Davies, T.; Hall, A.; Willis, M.C. One-Pot, Three-Component Sulfonimidamide Synthesis Exploiting the Sulfinylamine Reagent N -Sulfinyltritylamine, TrNSO. Angew. Chem. Int. Ed. 2017, 56, 14937–14941. [Google Scholar] [CrossRef]
- Yu, H.; Li, Z.; Bolm, C. Iron(II)-Catalyzed Direct Synthesis of NH Sulfoximines from Sulfoxides. Angew. Chem. Int. Ed. 2017, 57, 324–327. [Google Scholar] [CrossRef]
- Lücking, U. Sulfoximines: A Neglected Opportunity in Medicinal Chemistry. Angew. Chem. Int. Ed. 2013, 52, 9399–9408. [Google Scholar] [CrossRef]
- MarcusFrings; CarstenBolm; AndreasBlum; ChristianGnamm, Sulfoximines from a Medicinal Chemist’s Perspective: Physicochemical and in vitro Parameters Relevant for Drug Discovery. Eur. J. Med. Chem. 2017, 126, 225–245. [CrossRef] [PubMed]
- Sirvent, J.A.; Lücking, U. Novel Pieces for the Emerging Picture of Sulfoximines in Drug Discovery: Synthesis and Evaluation of Sulfoximine Analogues of Marketed Drugs and Advanced Clinical Candidates. ChemMedChem 2017, 12, 487–501. [Google Scholar] [CrossRef]
- Lücking, U. Neglected sulfur(vi) pharmacophores in drug discovery: Exploration of novel chemical space by the interplay of drug design and method development. Org. Chem. Front. 2019, 6, 1319–1324. [Google Scholar] [CrossRef] [Green Version]
- Dillard, R.D.; Yen, T.T.; Stark, P.; Pavey, D.E. Synthesis and Blood Pressure Lowering Activity of 3-(Substituted-amino)-l,2,4-benzothiadiazine 1-Oxide Derivatives. J. Med. Chem. 1980, 23, 717–722. [Google Scholar] [CrossRef]
- Buckheit, R.W.; Fliakas-Boltz, V.; Decker, W.; Roberson, J.L.; Pyle, C.A.; White, E.; Bowdon, B.J.; McMahon, J.B.; Boyd, M.R.; Bader, J.P.; et al. Biological and biochemical anti-HIV activity of the benzothiadiazine class of nonnucleoside reverse transcriptase inhibitors. Antivir. Res. 1994, 25, 43–56. [Google Scholar] [CrossRef]
- Okamura, H.; Bolm, C. Rhodium-Catalyzed Imination of Sulfoxides and Sulfides: Efficient Preparation of N-Unsubstituted Sulfoximines and Sulfilimines. Org. Lett. 2004, 6, 1305–1307. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Zhou, B.; Zhou, S.; Wei, W.; Liu, J.; Zhan, Y.; Cheng, D.; Chen, M.; Li, Y.; Wang, B.; et al. Sulfimine-Promoted Fast O Transfer: One-step Synthesis of Sulfoximine from Sulfide. ChemistrySelect 2017, 2, 1620–1624. [Google Scholar] [CrossRef]
- Alberico, D.; Scott, M.E.; Lautens, M. Aryl−Aryl Bond Formation by Transition-Metal-Catalyzed Direct Arylation. Chem. Rev. 2007, 107, 174–238. [Google Scholar] [CrossRef]
- Chen, X.; Engle, K.M.; Wang, N.-H.; Yu, J.-Q. ChemInform Abstract: Palladium(II)-Catalyzed C-H Activation/C-C Cross-Coupling Reactions: Versatility and Practicality. Chemin 2009, 40, 5094–5115. [Google Scholar] [CrossRef]
- Cho, S.H.; Kim, J.Y.; Kwak, J.; Chang, S. ChemInform Abstract: Recent Advances in the Transition Metal-Catalyzed Twofold Oxidative C-H Bond Activation Strategy for C-C and C-N Bond Formation. Chemin 2011, 43, 5068–5083. [Google Scholar] [CrossRef]
- Colby, D.A.; Tsai, A.S.; Bergman, R.G.; Ellman, J.A. Rhodium Catalyzed Chelation-Assisted C–H Bond Functionalization Reactions. Accounts Chem. Res. 2011, 45, 814–825. [Google Scholar] [CrossRef]
- Song, G.; Wang, F.; Li, X. ChemInform Abstract: C-C, C-O and C-N Bond Formation via Rhodium(III)-Catalyzed Oxidative C-H Activation. Chemin 2012, 43, 3651–3678. [Google Scholar] [CrossRef]
- Rouquet, G.; Chatani, N. Catalytic Functionalization of C(sp 2 )?H and C(sp 3 )?H Bonds by Using Bidentate Directing Groups. Angew. Chem. Int. Ed. 2013, 52, 11726–11743. [Google Scholar] [CrossRef]
- Wencel-Delord, J.; Glorius, F. ChemInform Abstract: C-H Bond Activation Enables the Rapid Construction and Late-Stage Diversification of Functional Molecules. Chemin 2013, 44, 369–375. [Google Scholar] [CrossRef]
- Song, G.; Li, X. Substrate Activation Strategies in Rhodium(III)-Catalyzed Selective Functionalization of Arenes. Acc. Chem. Res. 2015, 48, 1007–1020. [Google Scholar] [CrossRef]
- Hummel, J.R.; Boerth, J.A.; Ellman, J.A. Transition-Metal-Catalyzed C–H Bond Addition to Carbonyls, Imines, and Related Polarized π Bonds. Chem. Rev. 2016, 117, 9163–9227. [Google Scholar] [CrossRef]
- Xia, Y.; Qiu, D.; Wang, J. Transition-Metal-Catalyzed Cross-Couplings through Carbene Migratory Insertion. Chem. Rev. 2017, 117, 13810–13889. [Google Scholar] [CrossRef]
- Fernández-Rodríguez, M.A.; Shen, Q.; Hartwig, J.F. A General and Long-Lived Catalyst for the Palladium-Catalyzed Coupling of Aryl Halides with Thiols. J. Am. Chem. Soc. 2006, 128, 2180–2181. [Google Scholar]
- Platon, M.; Wijaya, N.; Rampazzi, V.; Cui, L.; Rousselin, Y.; Saeys, M.; Hierso, J.-C. Thioetherification of Chloroheteroarenes: A Binuclear Catalyst Promotes Wide Scope and High Functional-Group Tolerance. Chem. A Eur. J. 2014, 20, 12584–12594. [Google Scholar] [CrossRef]
- Guilbaud, J.; Labonde, M.; Selmi, A.; Kammoun, M.; Cattey, H.; Pirio, N.; Roger, J.; Hierso, J.-C. Palladium-catalyzed heteroaryl thioethers synthesis overcoming palladium dithiolate resting states inertness: Practical road to sulfones and NH-sulfoximines. Catal. Commun. 2018, 111, 52–58. [Google Scholar] [CrossRef]
- Yu, D.-G.; De Azambuja, F.; Glorius, F. α-MsO/TsO/Cl Ketones as Oxidized Alkyne Equivalents: Redox-Neutral Rhodium(III)-Catalyzed C-H Activation for the Synthesis of N-Heterocycles. Angew. Chem. Int. Ed. 2014, 53, 2754–2758. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; Tiwari, D.P.; Bolm, C. 1,2-Benzothiazines from Sulfoximines and Allyl Methyl Carbonate by Rhodium-Catalyzed Cross-Coupling and Oxidative Cyclization. Org. Lett. 2017, 19, 1706–1709. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Lan, J.; Gui, J.; Chen, F.; Jiang, H.; Zeng, W.; Jiang, H. Ru (II)-Catalyzed Coupling-Cyclization of Sulfoximines with alpha -Carbonyl Sulfoxonium Ylides as an Approach to 1,2-Benzothiazines. Adv. Synth. Catal. 2018, 360, 3534–3543. [Google Scholar] [CrossRef]
- Dong, W.; Wang, L.; Parthasarathy, K.; Pan, F.; Bolm, C. Rhodium-Catalyzed Oxidative Annulation of Sulfoximines and Alkynes as an Approach to 1,2-Benzothiazines. Angew. Chem. Int. Ed. 2013, 52, 11573–11576. [Google Scholar] [CrossRef]
- Cheng, Y.; Bolm, C. Regioselective Syntheses of 1,2-Benzothiazines by Rhodium-Catalyzed Annulation Reactions. Angew. Chem. Int. Ed. 2015, 54, 12349–12352. [Google Scholar] [CrossRef]
- Jeon, W.H.; Son, J.-Y.; Kim, J.E.; Lee, P.H. ChemInform Abstract: Synthesis of 1,2-Benzothiazines by a Rhodium-Catalyzed Domino C-H Activation/Cyclization/Elimination Process from S-Aryl Sulfoximines and Pyridotriazoles. Chemin 2016, 47, 3498–3501. [Google Scholar] [CrossRef]
- Liu, Y.-Z.; Hu, Y.; Lv, G.-H.; Nie, R.-F.; Peng, Y.; Zhang, C.; Lv, S.-Y.; Hai, L.; Wang, H.-J.; Wu, Y. Synthesis of 1,2-Benzothiazines via C–H Activation/Cyclization in a Recyclable, Mild System. ACS Sustain. Chem. Eng. 2019, 7, 13425–13429. [Google Scholar] [CrossRef]
- Ko, G.H.; Son, J.-Y.; Kim, H.; Maeng, C.; Baek, Y.; Seo, B.; Um, K.; Lee, P.H. Synthesis of Indolo-1,2-Benzothiazines from Sulfoximines and 3-Diazoindolin-2-imines. Adv. Synth. Catal. 2017, 359, 3362–3370. [Google Scholar] [CrossRef]
- Wu, X.; Wang, B.; Zhou, S.; Zhou, Y.; Liu, H. Ruthenium-Catalyzed Redox-Neutral [4 + 1] Annulation of Benzamides and Propargyl Alcohols via C–H Bond Activation. ACS Catal. 2017, 7, 2494–2499. [Google Scholar] [CrossRef]
- Xie, Y.; Wu, X.; Cai, J.; Wang, J.; Li, J.; Liu, H. Ruthenium(II)-Catalyzed Redox-Neutral [3+2] Annulation of Indoles with Internal Alkynes via C–H Bond Activation: Accessing a Pyrroloindolone Scaffold. J. Org. Chem. 2017, 82, 5263–5273. [Google Scholar] [CrossRef]
- Han, X.; Gao, F.; Li, C.; Fang, D.; Xie, X.; Zhou, Y.; Liu, H. Synthesis of Highly Fused Pyrano[2,3-b]pyridines via Rh(III)-Catalyzed C–H Activation and Intramolecular Cascade Annulation under Room Temperature. J. Org. Chem. 2020, 85, 6281–6294. [Google Scholar] [CrossRef] [PubMed]
- Back, T.G. Organosulfur Chemistry in Asymmetric Synthesis. Angew. Chem. 2009, 121, 2112–2114. [Google Scholar] [CrossRef]
- Otocka, S.; Kwiatkowska, M.; Madalińska, L.; Kiełbasiński, P. Chiral Organosulfur Ligands/Catalysts with a Stereogenic Sulfur Atom: Applications in Asymmetric Synthesis. Chem. Rev. 2017, 117, 4147–4181. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Wan, B.; Li, X. Enantiodivergent Desymmetrization in the Rhodium(III)-Catalyzed Annulation of Sulfoximines with Diazo Compounds. Angew. Chem. Int. Ed. 2018, 57, 15534–15538. [Google Scholar] [CrossRef]
- Sun, Y.; Cramer, N. Enantioselective Synthesis of Chiral-at-Sulfur 1,2-Benzothiazines by CpXRhIII-Catalyzed C-H Functionalization of Sulfoximines. Angew. Chem. Int. Ed. 2018, 57, 15539–15543. [Google Scholar] [CrossRef] [Green Version]
- Brauns, M.; Cramer, N. Efficient Kinetic Resolution of Sulfur-Stereogenic Sulfoximines by Exploiting CpXRhIII -Catalyzed C-H Functionalization. Angew. Chem. Int. Ed. 2019, 58, 8902–8906. [Google Scholar] [CrossRef]
- Simmons, E.M.; Hartwig, J.F. On the Interpretation of Deuterium Kinetic Isotope Effects in C?H Bond Functionalizations by Transition-Metal Complexes. Angew. Chem. Int. Ed. 2012, 51, 3066–3072. [Google Scholar] [CrossRef]
- Wang, H.; Li, L.; Yu, S.; Li, Y.; Li, X. ChemInform Abstract: Rh(III)-Catalyzed C-C/C-N Coupling of Imidates with α-Diazo Imidamide: Synthesis of Isoquinoline-Fused Indoles. Chemin 2016, 47, 2914–2917. [Google Scholar] [CrossRef]
- Zhu, R.-Y.; He, J.; Wang, X.-C.; Yu, J.-Q. Ligand-Promoted Alkylation of C(sp3)–H and C(sp2)–H Bonds. J. Am. Chem. Soc. 2014, 136, 13194–13197. [Google Scholar] [CrossRef] [Green Version]
Sample Availability: Samples of the compounds are not available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Catalyst (mol%) | Additive (mol%) | Solvent | Temp (°C) | Yield of 3aa (%) b |
|---|---|---|---|---|---|
| 1 | [Cp*RhCl2]2 (10) | AgSbF6 (40) | DCE | 100 | ND |
| 2 | [Cp*RhCl2]2 (10) | AgSbF6 (40) | DCE | 80 | 16 |
| 3 | [Cp*RhCl2]2 (10) | AgSbF6 (40) | HFIP | 80 | trace |
| 4 | [Cp*RhCl2]2 (10) | AgSbF6 (40) | DCE | rt | trace |
| 5 | [Cp*RhCl2]2 (10) | AgSbF6 (40) | HFIP | rt | 47 |
| 6 | [Cp*RhCl2]2 (10) | AgSbF6 (40) | TFE | rt | 75 |
| 7 | [Cp*RhCl2]2 (10) | AgSbF6 (40) | EtOH | rt | 57 |
| 8 | [Cp*RhCl2]2 (10) | AgSbF6 (40) | THF | rt | ND |
| 9 | [Cp*RhCl2]2 (10) | AgSbF6 (40) | DME | rt | trace |
| 10 | [Cp*RhCl2]2 (10) | AgOAc (40) | TFE | rt | 69 |
| 11 | [Cp*RhCl2]2 (10) | AgNTf2 (40) | TFE | rt | 27 |
| 12 | [Cp*RhCl2]2 (10) | AgBF4 (40) | TFE | rt | ND |
| 13 | [Cp*RhCl2]2 (10) | AgOPiv (40) | TFE | rt | 99 (92) c |
| 14 | [Cp*RhCl2]2 (20) | AgOPiv (40) | TFE | rt | 47 |
| 15 | [Cp*RhCl2]2 (5) | AgOPiv (40) | TFE | rt | 77 |
| 16 | [Cp*RhCl2]2 (5) | AgOPiv (20) | TFE | rt | 97 (93) c |
| 17 | [Cp*RhCl2]2 (2.5) | AgOPiv (10) | TFE | rt | 87 |
| 18 d | [Cp*RhCl2]2 (10) | AgOPiv (40) | TFE | rt | 90 (89) c |
| 19 e | [Cp*RhCl2]2 (10) | AgOPiv (40) | TFE | rt | 82 |
| 20 | [Cp*RuCl2]2 (10) | AgOPiv (40) | TFE | rt | trace |
| 21 | [Cp*lrCl2]2 (10) | AgOPiv (40) | TFE | rt | 85 |
| 22 | - | AgOPiv (40) | TFE | rt | ND |
 |
 |
 |
 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, B.; Han, X.; Li, J.; Li, C.; Liu, H. Sulfoximines-Assisted Rh(III)-Catalyzed C–H Activation and Intramolecular Annulation for the Synthesis of Fused Isochromeno-1,2-Benzothiazines Scaffolds under Room Temperature. Molecules 2020, 25, 2515. https://doi.org/10.3390/molecules25112515
Wang B, Han X, Li J, Li C, Liu H. Sulfoximines-Assisted Rh(III)-Catalyzed C–H Activation and Intramolecular Annulation for the Synthesis of Fused Isochromeno-1,2-Benzothiazines Scaffolds under Room Temperature. Molecules. 2020; 25(11):2515. https://doi.org/10.3390/molecules25112515
Chicago/Turabian StyleWang, Bao, Xu Han, Jian Li, Chunpu Li, and Hong Liu. 2020. "Sulfoximines-Assisted Rh(III)-Catalyzed C–H Activation and Intramolecular Annulation for the Synthesis of Fused Isochromeno-1,2-Benzothiazines Scaffolds under Room Temperature" Molecules 25, no. 11: 2515. https://doi.org/10.3390/molecules25112515