
Exploring the Scope of Tandem Palladium and Isothiourea Relay Catalysis for the Synthesis of α-Amino Acid Derivatives



Abstract
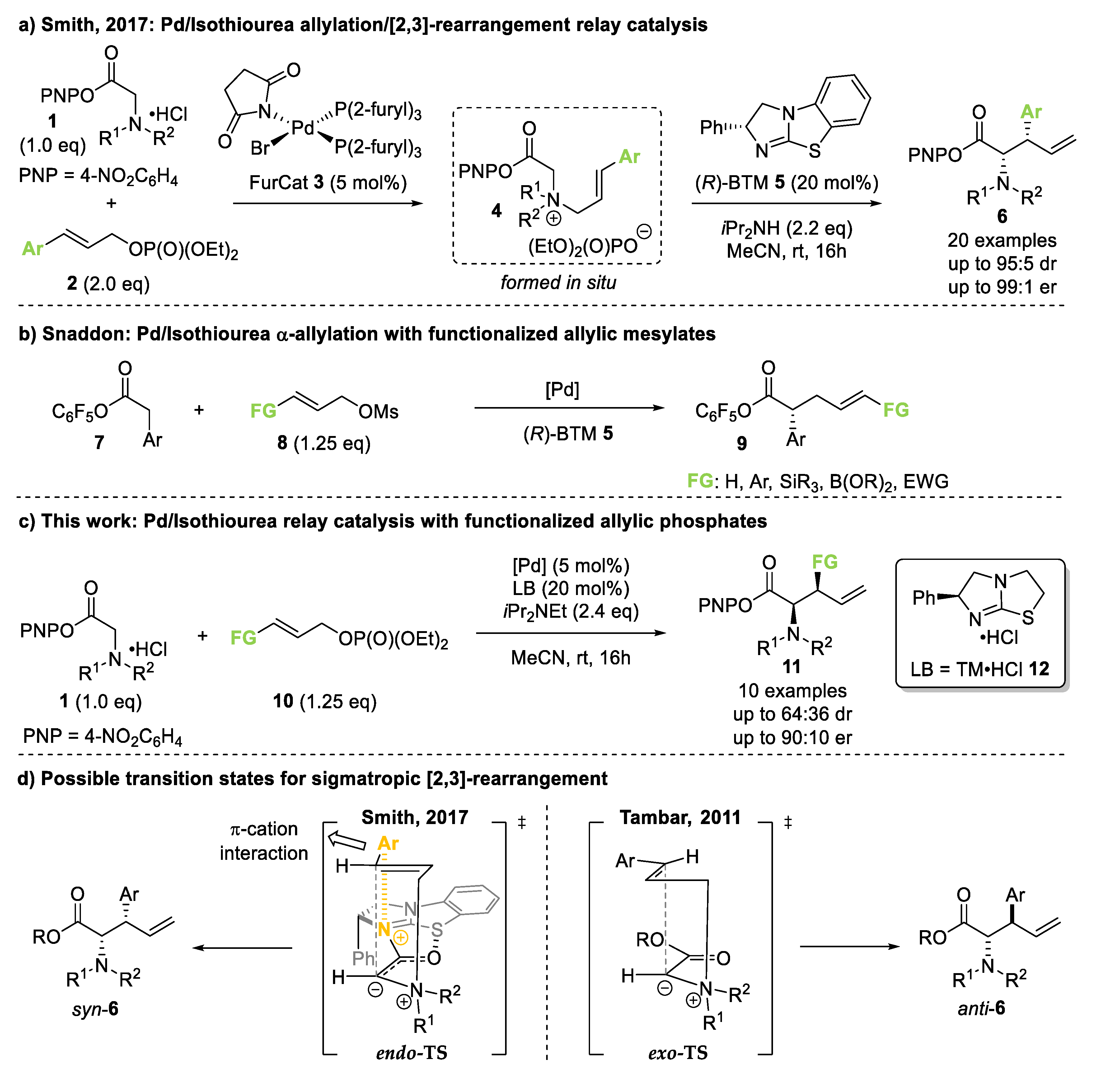
:1. Introduction
2. Results and Discussion
2.1. Initial Functional Group Assessment
2.2. Reaction Optimization for Ethyl Ester Containing Allylic Phosphate
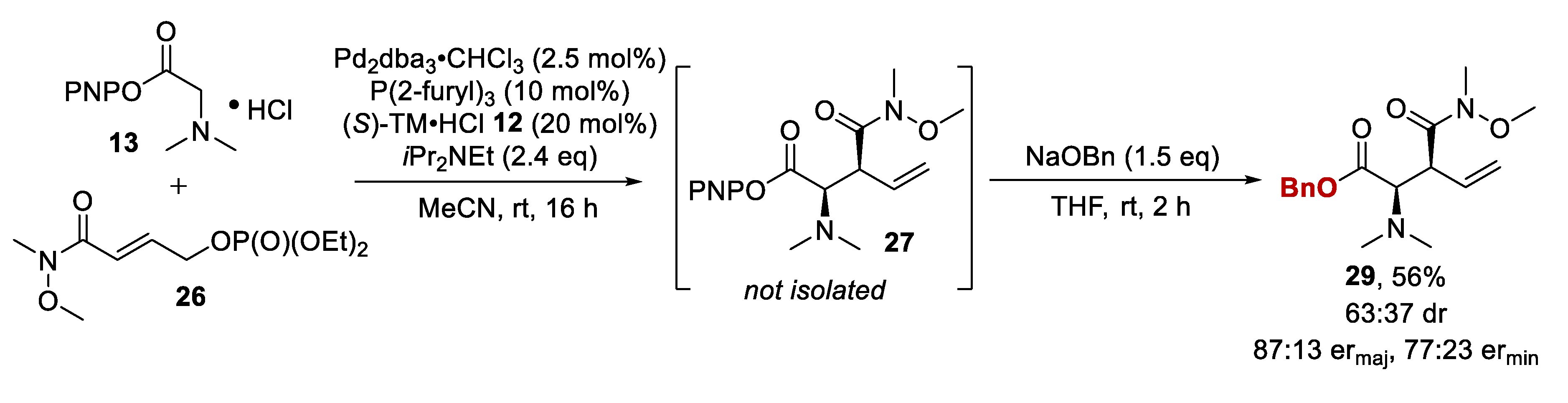
2.3. Amide Containing Allylic Phosphates
2.3.1. Initial Assessment
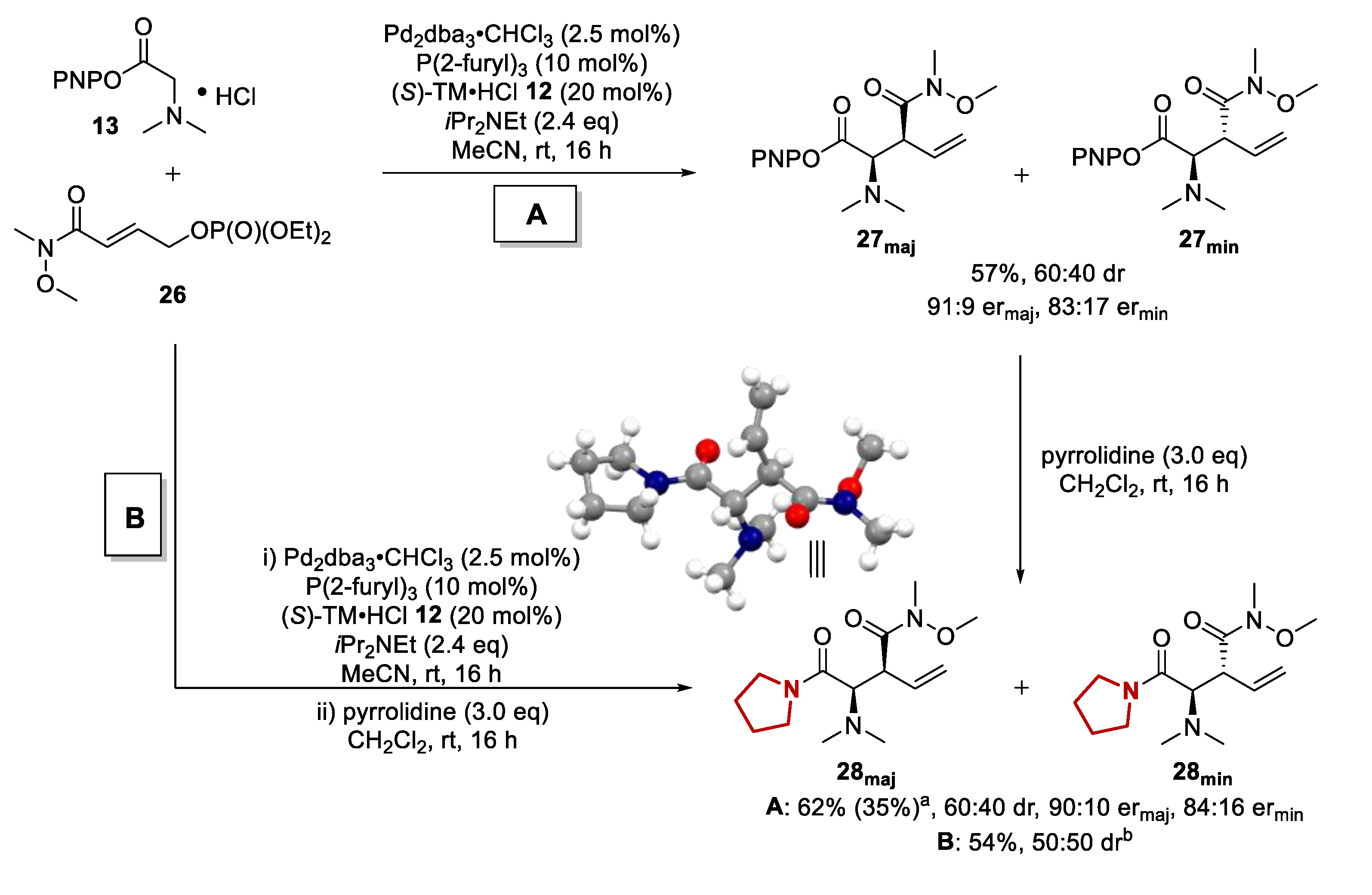
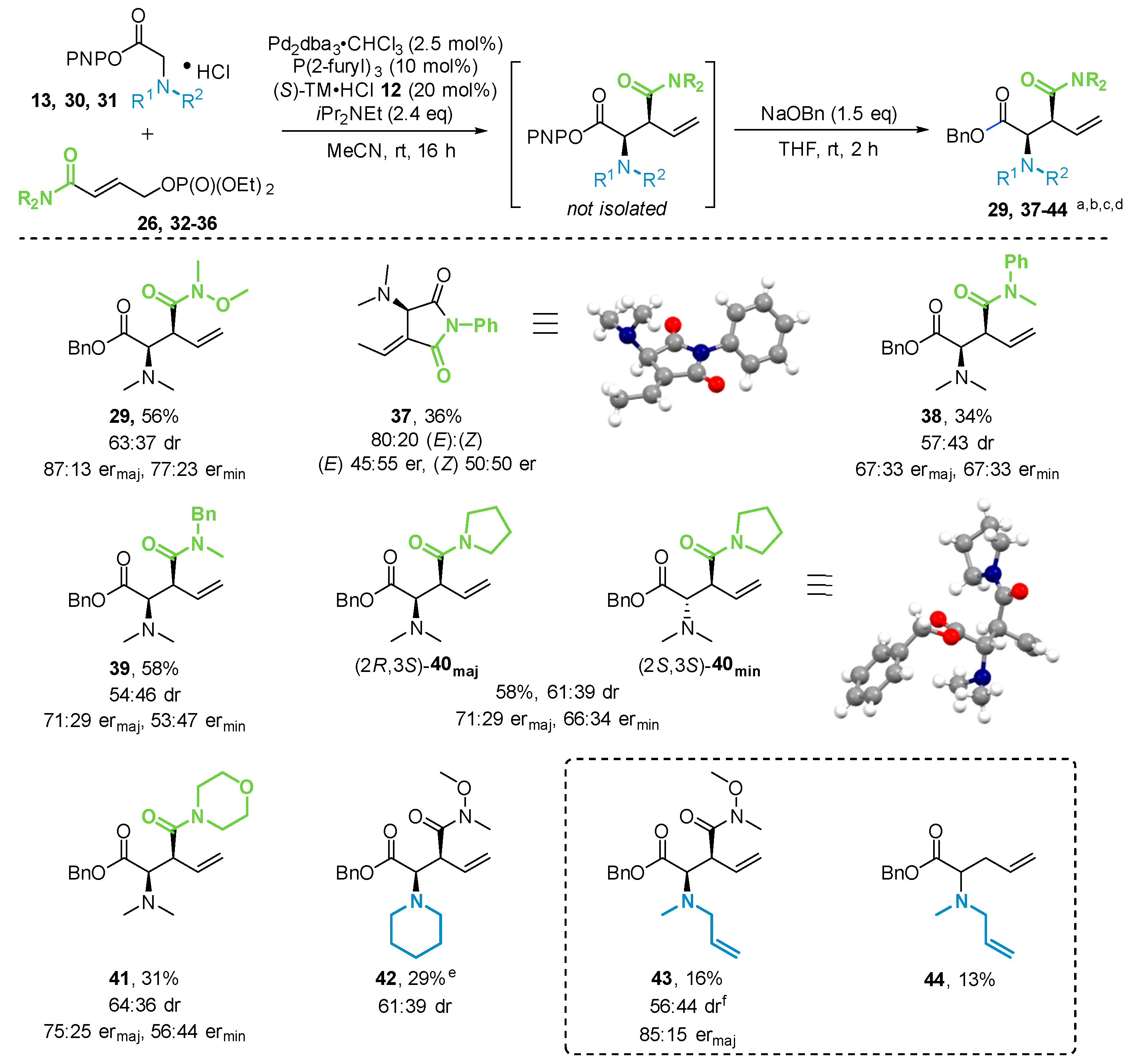
2.3.2. Scope of Amide Containing Allylic Phosphates
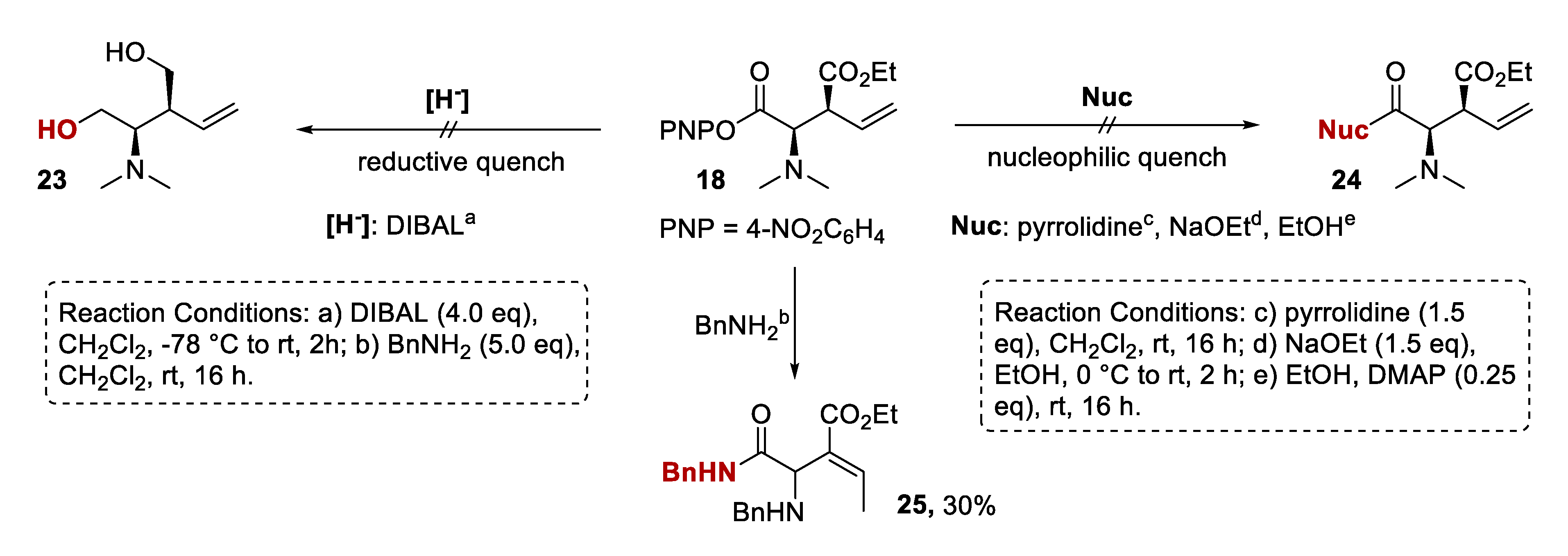
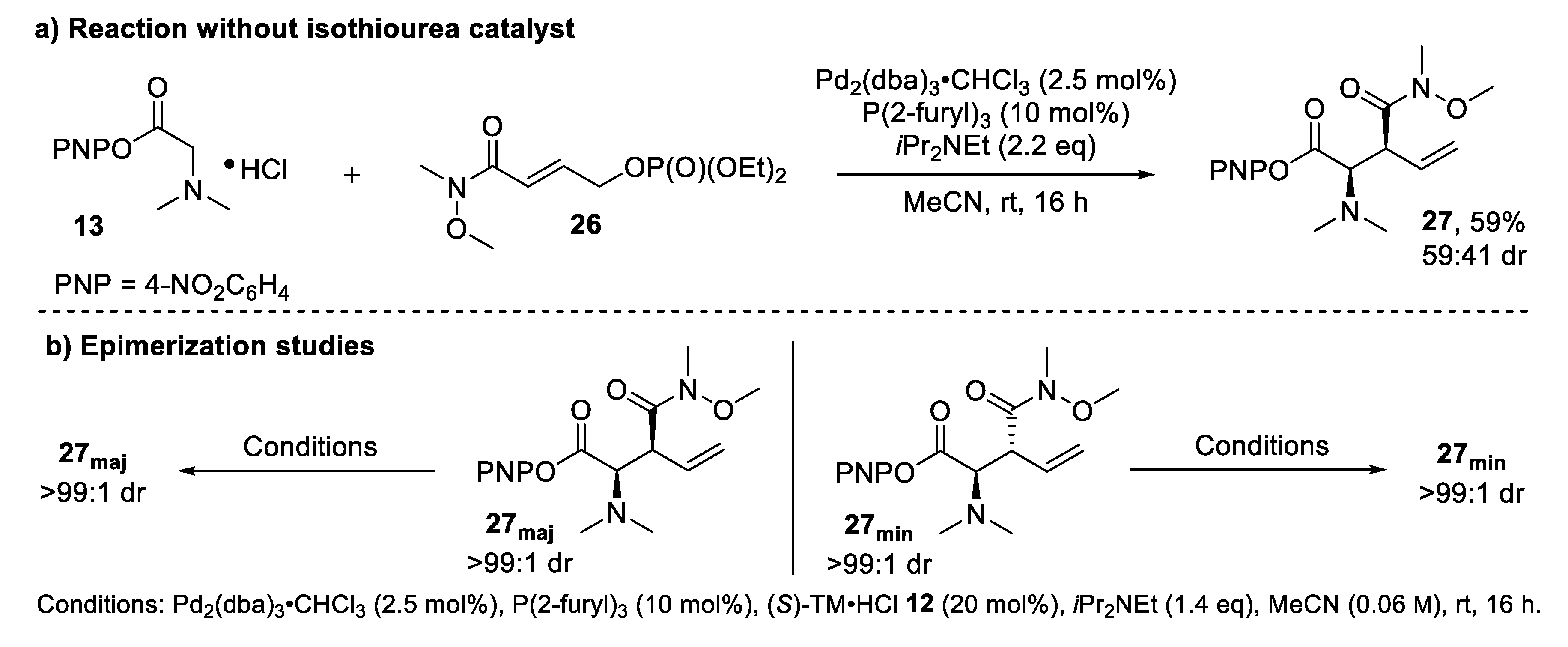
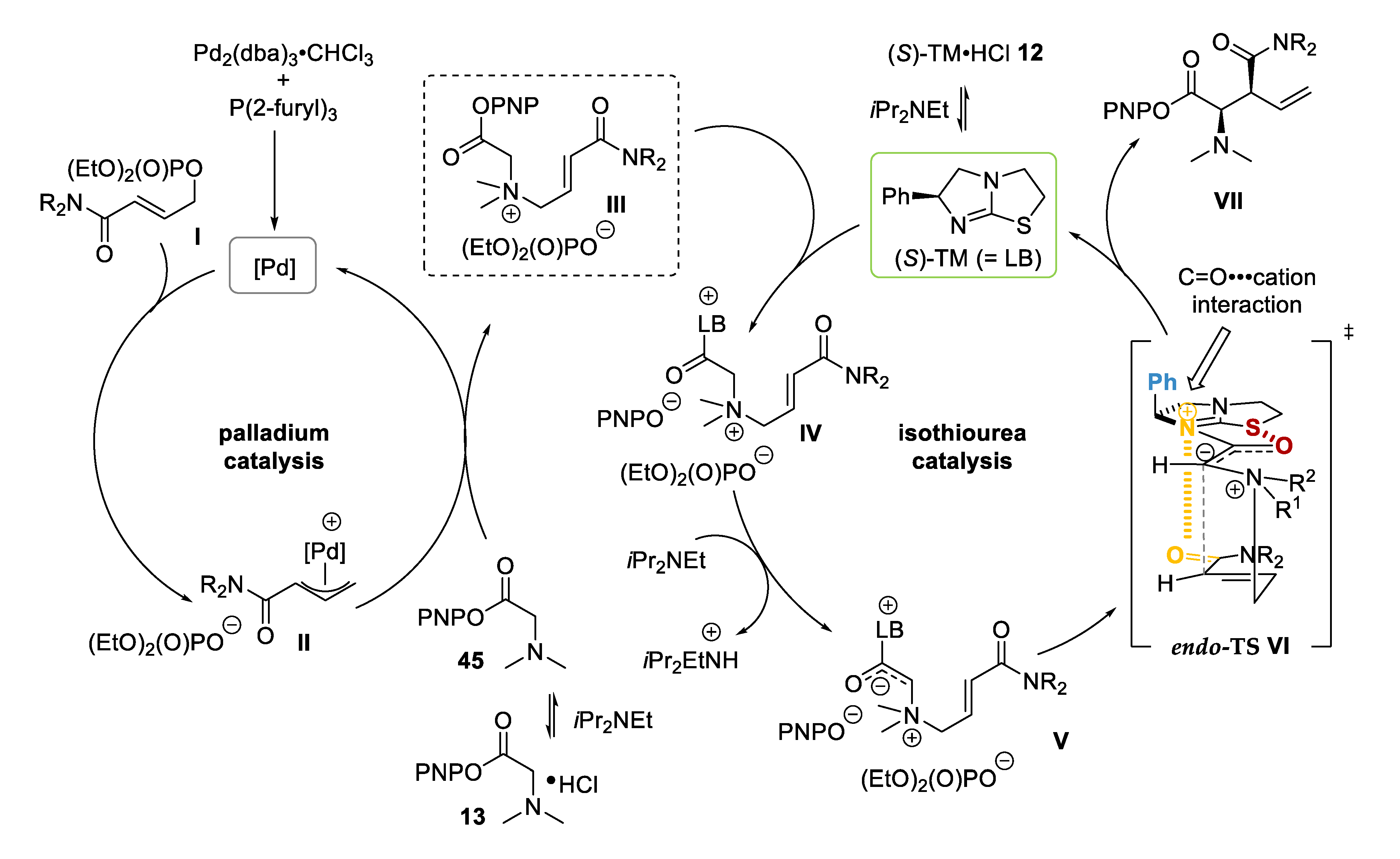
2.3.3. Mechanistic Control Experiments
3. Materials and Methods
3.1. General Procedure for the Tandem Palladium and Isothiourea Relay Catalysis
3.2. Representative Synthesis and Characterization of Compound 29
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Grayson, I.; Kessler, C. Modern applications of amino acids and dipeptides. Chim. Oggi–Chem. Today 2015, 33, 46–51. [Google Scholar]
- Hedges, J.B.; Ryan, K.S. Biosynthetic Pathways to Nonproteinogenic α-Amino Acids. Chem. Rev. 2020, 120, 3161–3209. [Google Scholar] [CrossRef] [PubMed]
- Paek, S.-M.; Jeong, M.; Jo, J.; Heo, Y.; Han, Y.; Yun, H. Recent Advances in Substrate-Controlled Asymmetric Induction Derived from Chiral Pool α-Amino Acids for Natural Product Synthesis. Molecules 2016, 21, 951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blaser, H.U. The chiral pool as a source of enantioselective catalysts and auxiliaries. Chem. Rev. 1992, 92, 935–952. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Izawa, K. (Eds.) Asymmetric Synthesis and Application of α-Amino Acids; American Chemical Society: Washington, DC, USA, 2009; Volume 1009, ISBN 9780841269743. [Google Scholar]
- Ager, D.J. Synthesis of Unnatural/Nonproteinogenic α-Amino Acids. In Amino Acids, Peptides and Proteins in Organic Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2010; Volume 1, pp. 495–526. ISBN 9783527320967. [Google Scholar]
- Nájera, C.; Sansano, J.M. Catalytic Asymmetric Synthesis of α-Amino Acids. Chem. Rev. 2007, 107, 4584–4671. [Google Scholar] [CrossRef] [PubMed]
- West, T.H.; Daniels, D.S.B.; Slawin, A.M.Z.; Smith, A.D. An Isothiourea-Catalyzed Asymmetric [2,3]-Rearrangement of Allylic Ammonium Ylides. J. Am. Chem. Soc. 2014, 136, 4476–4479. [Google Scholar] [CrossRef] [Green Version]
- Spoehrle, S.S.M.; West, T.H.; Taylor, J.E.; Slawin, A.M.Z.; Smith, A.D. Tandem Palladium and Isothiourea Relay Catalysis: Enantioselective Synthesis of α-Amino Acid Derivatives via Allylic Amination and [2,3]-Sigmatropic Rearrangement. J. Am. Chem. Soc. 2017, 139, 11895–11902. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, K.J.; Amos, J.L.; Klein, J.C.; Do, D.T.; Snaddon, T.N. Uniting C1-Ammonium Enolates and Transition Metal Electrophiles via Cooperative Catalysis: The Direct Asymmetric α-Allylation of Aryl Acetic Acid Esters. J. Am. Chem. Soc. 2016, 138, 5214–5217. [Google Scholar] [CrossRef]
- Scaggs, W.R.; Snaddon, T.N. Enantioselective α-Allylation of Acyclic Esters Using B(pin)-Substituted Electrophiles: Independent Regulation of Stereocontrol Elements through Cooperative Pd/Lewis Base Catalysis. Chemistry 2018, 24, 14378–14381. [Google Scholar] [CrossRef]
- Fyfe, J.W.B.; Kabia, O.M.; Pearson, C.M.; Snaddon, T.N. Si-directed regiocontrol in asymmetric Pd-catalyzed allylic alkylations using C1-ammonium enolate nucleophiles. Tetrahedron 2018, 74, 5383–5391. [Google Scholar] [CrossRef]
- Hutchings-Goetz, L.; Yang, C.; Snaddon, T.N. Enantioselective α-Allylation of Aryl Acetic Acid Esters via C1-Ammonium Enolate Nucleophiles: Identification of a Broadly Effective Palladium Catalyst for Electron-Deficient Electrophiles. ACS Catal. 2018, 8, 10537–10544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, T.H.; Walden, D.M.; Taylor, J.E.; Brueckner, A.C.; Johnston, R.C.; Cheong, P.H.Y.; Lloyd-Jones, G.C.; Smith, A.D. Catalytic Enantioselective [2,3]-Rearrangements of Allylic Ammonium Ylides: A Mechanistic and Computational Study. J. Am. Chem. Soc. 2017, 139, 4366–4375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenhalgh, M.D.; Smith, S.M.; Walden, D.M.; Taylor, J.E.; Brice, Z.; Robinson, E.R.T.; Fallan, C.; Cordes, D.B.; Slawin, A.M.Z.; Richardson, H.C.; et al. A C=O⋅⋅⋅Isothiouronium Interaction Dictates Enantiodiscrimination in Acylative Kinetic Resolutions of Tertiary Heterocyclic Alcohols. Angew. Chem. Int. Ed. 2018, 57, 3200–3206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiina, I.; Nakata, K.; Ono, K.; Sugimoto, M.; Sekiguchi, A. Kinetic Resolution of the Racemic 2-Hydroxyalkanoates Using the Enantioselective Mixed-Anhydride Method with Pivalic Anhydride and a Chiral Acyl-Transfer Catalyst. Chem.—A Eur. J. 2010, 16, 167–172. [Google Scholar] [CrossRef]
- Nakata, K.; Gotoh, K.; Ono, K.; Futami, K.; Shiina, I. Kinetic Resolution of Racemic 2-Hydroxy-γ-butyrolactones by Asymmetric Esterification Using Diphenylacetic Acid with Pivalic Anhydride and a Chiral Acyl-Transfer Catalyst. Org. Lett. 2013, 15, 1170–1173. [Google Scholar] [CrossRef]
- Soheili, A.; Tambar, U.K. Tandem catalytic allylic amination and [2,3]-stevens rearrangement of tertiary amines. J. Am. Chem. Soc. 2011, 133, 12956–12959. [Google Scholar] [CrossRef]
- Schwarz, K.J.; Pearson, C.M.; Cintron-Rosado, G.A.; Liu, P.; Snaddon, T.N. Traversing Steric Limitations by Cooperative Lewis Base/Palladium Catalysis: An Enantioselective Synthesis of α-Branched Esters Using 2-Substituted Allyl Electrophiles. Angew. Chem. Int. Ed. 2018, 57, 7800–7803. [Google Scholar] [CrossRef]
- Soheili, A.; Tambar, U.K. Synthesis of (±)-amathaspiramide F and discovery of an unusual stereocontrolling element for the [2,3]-stevens rearrangement. Org. Lett. 2013, 15, 5138–5141. [Google Scholar] [CrossRef]
- Liu, P.; Yang, X.; Birman, V.B.; Houk, K.N. Origin of Enantioselectivity in Benzotetramisole-Catalyzed Dynamic Kinetic Resolution of Azlactones. Org. Lett. 2012, 14, 3288–3291. [Google Scholar] [CrossRef]
- Abbasov, M.E.; Hudson, B.M.; Tantillo, D.J.; Romo, D. Acylammonium Salts as Dienophiles in Diels–Alder/Lactonization Organocascades. J. Am. Chem. Soc. 2014, 136, 4492–4495. [Google Scholar] [CrossRef]
- Robinson, E.R.T.; Walden, D.M.; Fallan, C.; Greenhalgh, M.D.; Cheong, P.H.-Y.; Smith, A.D. Non-bonding 1,5-S⋯O interactions govern chemo- and enantioselectivity in isothiourea-catalyzed annulations of benzazoles. Chem. Sci. 2016, 7, 6919–6927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, C.M.; Elmi, A.; Pascoe, D.J.; Morris, R.K.; McLaughlin, C.; Woods, A.M.; Frost, A.B.; Houpliere, A.; Ling, K.B.; Smith, T.K.; et al. The Importance of 1,5-Oxygen⋅⋅⋅Chalcogen Interactions in Enantioselective Isochalcogenourea Catalysis. Angew. Chem. Int. Ed. 2020, 59, 3705–3710. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry a | Allyl Precursor | Product | Yield (%) b | dr c |
|---|---|---|---|---|
| 1 |  |  | 70 (61) | 60:40 |
| 2 |  |  | - | - |
| 3 |  |  | 33 (30) | 55:45 |
| 4 |  |  | 32 (-) | - |

| Entry a | LB catalyst | Base | Phosphate | Yield (%) b | dr c |
|---|---|---|---|---|---|
| 1 | (±)-BTM | iPr2NH (2.2 eq) | 2.0 eq | 70 | 60:40 |
| 2 | (±)-BTM | iPr2NEt (2.2 eq) | 2.0 eq | 65 (17) | 56:44 |
| 3 | (±)-TM·HCl | iPr2NEt (2.4 eq) | 2.0 eq | 87 (56) | 67:33 |
| 4 | (±)-TM·HCl | iPr2NEt (2.4 eq) | 1.25 eq | (62) | 55:45 |
| 5 | (S)-TM·HCl | iPr2NEt (2.4 eq) | 1.25 eq | (70) | 60:40 d |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bitai, J.; Slawin, A.M.Z.; Cordes, D.B.; Smith, A.D. Exploring the Scope of Tandem Palladium and Isothiourea Relay Catalysis for the Synthesis of α-Amino Acid Derivatives. Molecules 2020, 25, 2463. https://doi.org/10.3390/molecules25102463
Bitai J, Slawin AMZ, Cordes DB, Smith AD. Exploring the Scope of Tandem Palladium and Isothiourea Relay Catalysis for the Synthesis of α-Amino Acid Derivatives. Molecules. 2020; 25(10):2463. https://doi.org/10.3390/molecules25102463
Chicago/Turabian StyleBitai, Jacqueline, Alexandra M. Z. Slawin, David B. Cordes, and Andrew D. Smith. 2020. "Exploring the Scope of Tandem Palladium and Isothiourea Relay Catalysis for the Synthesis of α-Amino Acid Derivatives" Molecules 25, no. 10: 2463. https://doi.org/10.3390/molecules25102463