Efficient and Divergent Enantioselective Syntheses of DHPVs and Anti-Inflammatory Effect on IEC-6 Cells

 ,
,
Abstract
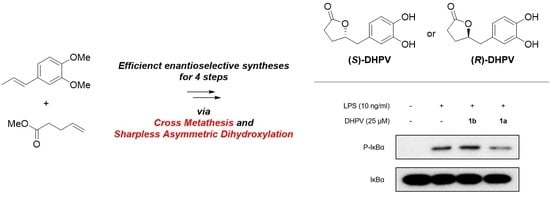
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
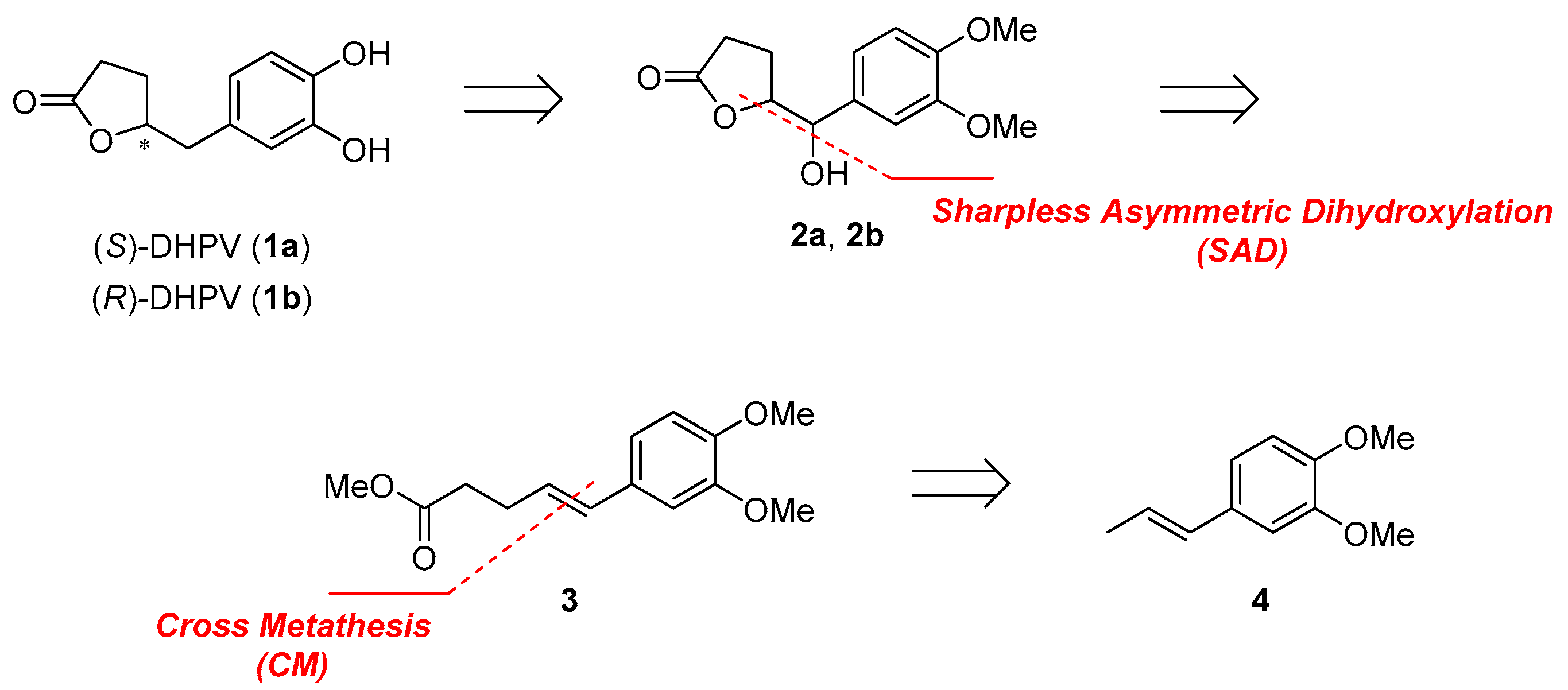
2.1. Retrosynthetic Analysis of 5-(3′,4′-Dihydroxyphenyl)-γ-Valerolactone (DHPV)
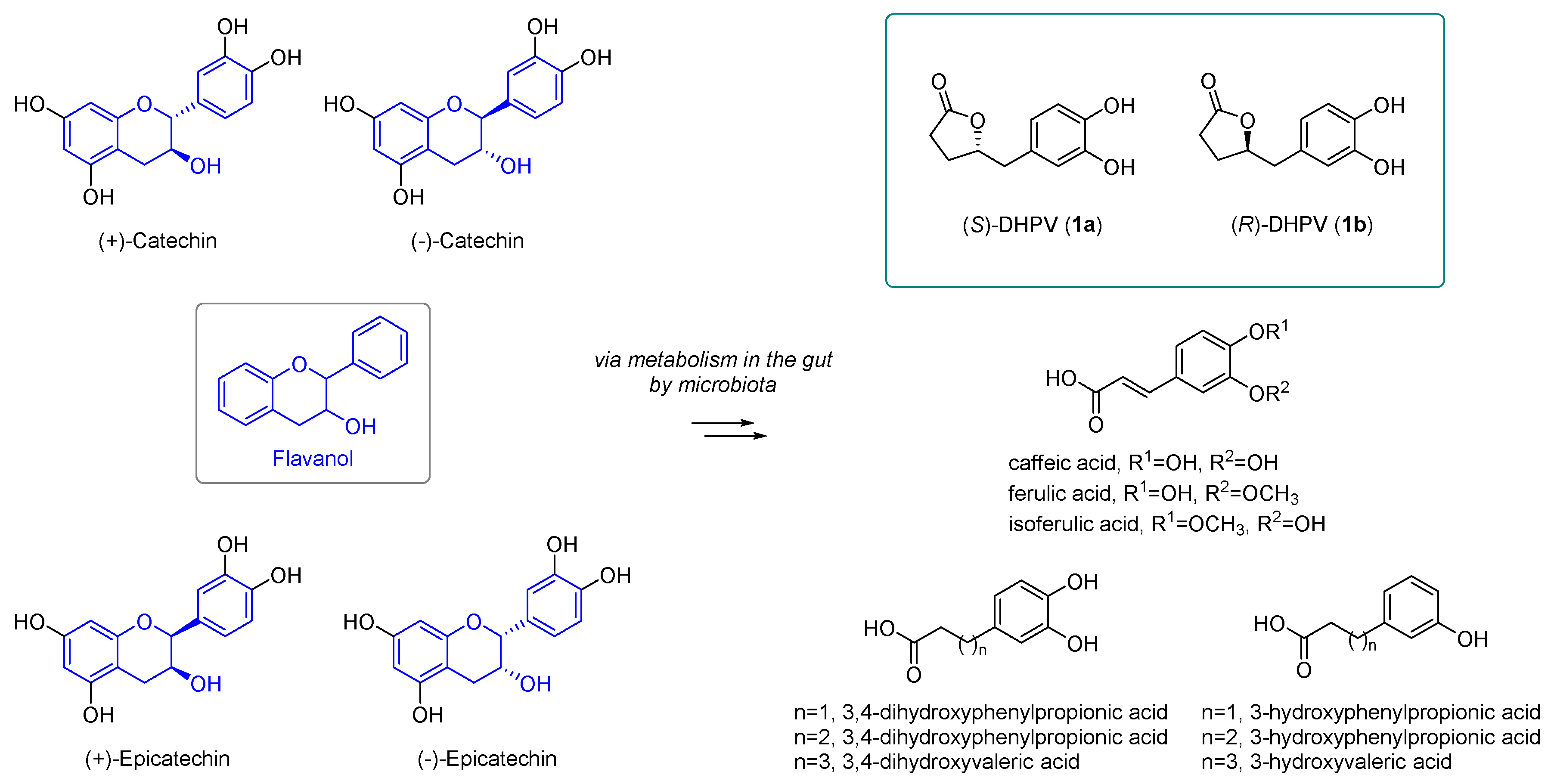
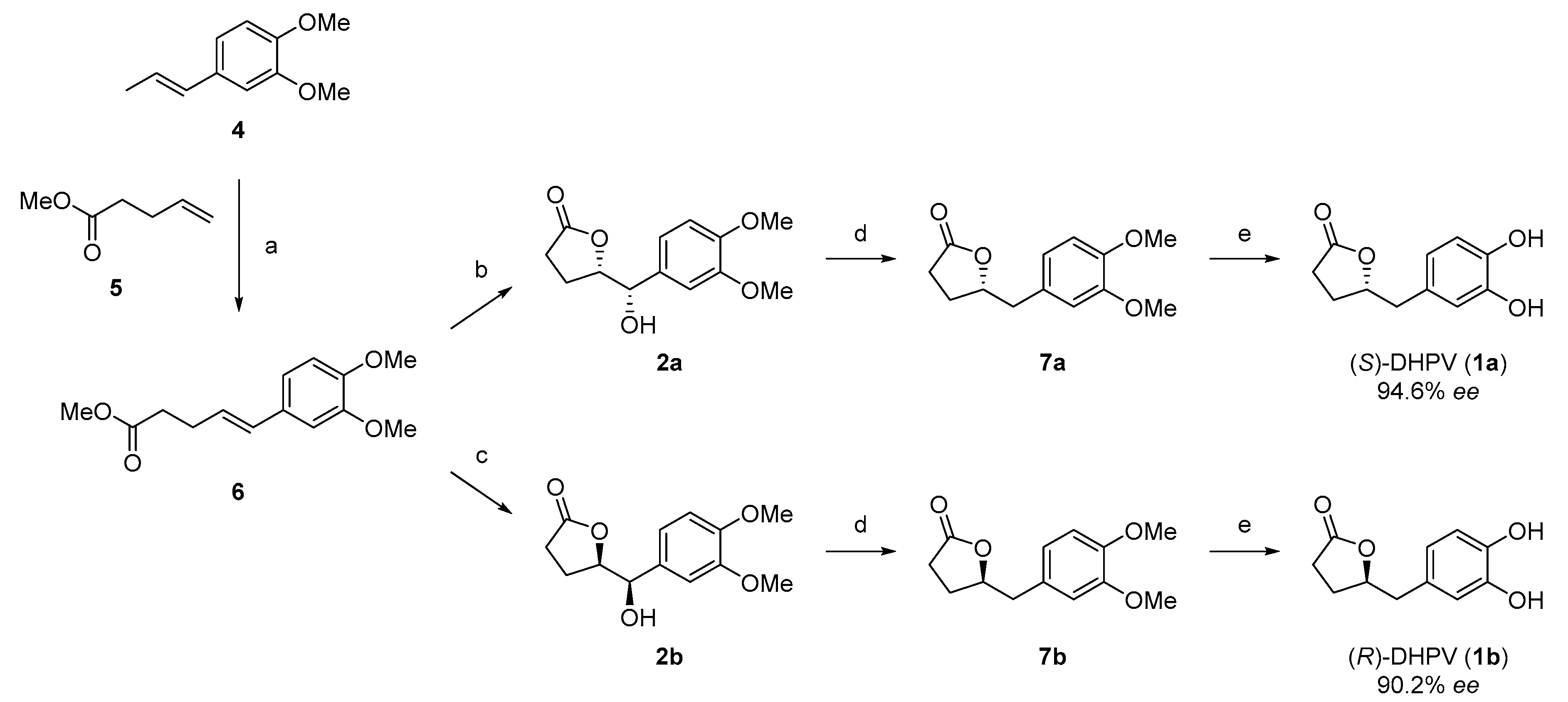
2.2. Divergent Synthesis of DHPV
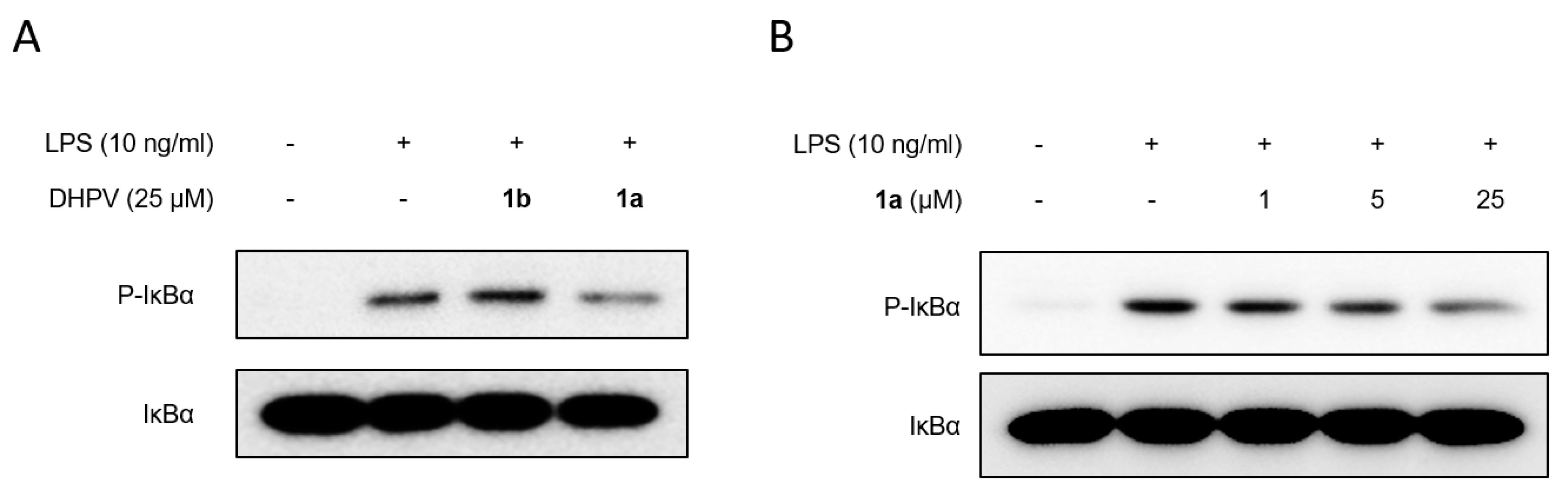
2.3. Anti-Inflammatory Activity of (R)- and (S)-DHPV on IEC-6 Cells
3. Materials and Methods
3.1. General Information
3.1.1. General Methods and Materials
3.1.2. Materials
3.1.3. Instrumentation
3.2. Experimental Part
3.3. Cell Culture and Treatments
3.4. Cell Lysates and Western Blot Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Kaser, A.; Lee, A.-H.; Franke, A.; Glickman, J.N.; Zeissig, S.; Tilg, H.; Nieuwenhuis, E.E.; Higgins, D.E.; Schreiber, S.; Glimcher, L.H. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell 2008, 134, 743–756. [Google Scholar] [CrossRef] [Green Version]
- Halpin, S.J.; Ford, A.C. Prevalence of symptoms meeting criteria for irritable bowel syndrome in inflammatory bowel disease: Systematic review and meta-analysis. Am. J. Gastroenterol. 2012, 107, 1474–1482. [Google Scholar] [CrossRef] [PubMed]
- Molodecky, N.A.; Soon, S.; Rabi, D.M.; Ghali, W.A.; Ferris, M.; Chernoff, G.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Barkema, H.W. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology 2012, 142, 46–54.e42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, E.R.; Chang, D.K. Colorectal cancer in inflammatory bowel disease: The risk, pathogenesis, prevention and diagnosis. World J. Gastroenterol. 2014, 20, 9872–9881. [Google Scholar] [CrossRef] [PubMed]
- Baumgart, D.C.; Sandborn, W.J. Crohn’s disease. Lancet 2012, 380, 1590–1605. [Google Scholar] [CrossRef] [Green Version]
- Roda, G.; Narula, N.; Pinotti, R.; Skamnelos, A.; Katsanos, K.; Ungaro, R.; Burisch, J.; Torres, J.; Colombel, J.F. Systematic review with meta-analysis: Proximal disease extension in limited ulcerative colitis. Aliment. Pharm. Ther. 2017, 45, 1481–1492. [Google Scholar] [CrossRef] [PubMed]
- Neish, A.S. Microbes in gastrointestinal health and disease. Gastroenterology 2009, 136, 65–80. [Google Scholar] [CrossRef] [Green Version]
- McDaniel, D.K.; Eden, K.; Ringel, V.M.; Allen, I.C. Emerging roles for noncanonical NF-κB signaling in the modulation of inflammatory bowel disease pathobiology. Inflamm. Bowel Dis. 2016, 22, 2265–2279. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Wang, H.; Liang, X.; Roberts, M. Hepatic Metabolism in Liver Health and Disease. In Liver Pathophysiology; Elsevier: Amsterdam, The Netherlands, 2017; pp. 391–400. [Google Scholar]
- Rinaldo, D.; Batista Jr, J.M.; Rodrigues, J.; Benfatti, A.C.; Rodrigues, C.M.; Dos Santos, L.C.; Furlan, M.; Vilegas, W. Determination of catechin diastereomers from the leaves of Byrsonima species using chiral HPLC-PAD-CD. Chirality 2010, 22, 726–733. [Google Scholar]
- Monagas, M.; Urpi-Sarda, M.; Sanchez-Patan, F.; Llorach, R.; Garrido, I.; Gomez-Cordoves, C.; Andres-Lacueva, C.; Bartolome, B. Insights into the metabolism and microbial biotransformation of dietary flavan-3-ols and the bioactivity of their metabolites. Food Funct. 2010, 1, 233–253. [Google Scholar] [CrossRef] [Green Version]
- H Brooks, W.; C Guida, W.; G Daniel, K. The significance of chirality in drug design and development. Curr. Top. Med. Chem. 2011, 11, 760–770. [Google Scholar] [CrossRef] [PubMed]
- Saha, D.; Kharbanda, A.; Yan, W.; Lakkaniga, N.R.; Frett, B.; Li, H.-Y. The Exploration of Chirality for Improved Druggability within the Human Kinome. J. Med. Chem. 2020, 63, 441–469. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H. The Chemical Structure of the Intermediate Metabolites of Catechin I–IV: Chemical Properties of the Intermediate Metabolites (G and H) and their Derivatives Oxidative Decomposition of the Intermediate Metabolites (G and H) Synthesis of the Intermediate Metabolites (G and H) Structure of the Intermediate Metabolite (F). J. Agric. Chem. Soc. Jpn. 1959, 23, 257–271. [Google Scholar]
- Lambert, J.D.; Rice, J.E.; Hong, J.; Hou, Z.; Yang, C.S. Synthesis and biological activity of the tea catechin metabolites, M4 and M6 and their methoxy-derivatives. Bioorg. Med. Chem. Lett. 2005, 15, 873–876. [Google Scholar] [CrossRef]
- Nakano, S.; Hamada, M.; Kishimoto, T.; Nakajima, N. Synthesis of γ-valerolactones as the tea catechin metabolites. Heterocycles 2008, 76, 1001–1005. [Google Scholar]
- Hamada, M.; Furuno, A.; Nakano, S.; Kishimoto, T.; Nakajima, N. Synthesis of optically pure lactone metabolites of tea catechins. Synthesis 2010, 1512–1520. [Google Scholar]
- Curti, C.; Brindani, N.; Battistini, L.; Sartori, A.; Pelosi, G.; Mena, P.; Brighenti, F.; Zanardi, F.; Del Rio, D. Catalytic, Enantioselective Vinylogous Mukaiyama Aldol Reaction of Furan-Based Dienoxy Silanes: A Chemodivergent Approach to γ-Valerolactone Flavan-3-ol Metabolites and δ-Lactone Analogues. Adv. Synth. Catal. 2015, 357, 4082–4092. [Google Scholar] [CrossRef]
- Wu, B.; Gao, X.; Yan, Z.; Chen, M.-W.; Zhou, Y.-G. C–H Oxidation/Michael Addition/Cyclization Cascade for Enantioselective Synthesis of Functionalized 2-Amino-4H-chromenes. Org. Lett. 2015, 17, 6134–6137. [Google Scholar] [CrossRef]
- Shopsowitz, K.E.; Edwards, D.; Gallant, A.J.; MacLachlan, M.J. Highly substituted Schiff base macrocycles via hexasubstituted benzene: A convenient double Duff formylation of catechol derivatives. Tetrahedron 2009, 65, 8113–8119. [Google Scholar] [CrossRef]
- Kolb, H.C.; VanNieuwenhze, M.S.; Sharpless, K.B. Catalytic asymmetric dihydroxylation. Chem. Rev. 1994, 94, 2483–2547. [Google Scholar] [CrossRef]
- Chatterjee, A.K.; Choi, T.-L.; Sanders, D.P.; Grubbs, R.H. A general model for selectivity in olefin cross metathesis. J. Am. Chem. Soc. 2003, 125, 11360–11370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hur, J.; Kim, A.-R.; Kim, H.S.; Lim, C.; Kim, T.; Kim, T.-A.; Sim, J.; Suh, Y.-G. Concise Synthesis of Catechin Metabolites 5-(3′,4′-Dihydroxyphenyl)-γ-valerolactones (DHPV) in Optically Pure Form and Their Stereochemical Effects on Skin Wrinkle-Reducing Activities. Molecules 2020, 25, 1970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neish, A.S. The gut microflora and intestinal epithelial cells: A continuing dialogue. Microb. Infect. 2002, 4, 309–317. [Google Scholar] [CrossRef]
- Atreya, I.; Atreya, R.; Neurath, M.F. NF-κB in inflammatory bowel disease. J. Intern. Med. 2008, 263, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Neurath, M.F.; Pettersson, S.; Meyer zum Büschenfelde, K.H.; Strober, W. Local administration of antisense phosphorothiate olignucleotides to the p65 subunit of NF–κB abrogates. Nat. Med. 1996, 2, 998–1004. [Google Scholar] [CrossRef] [PubMed]
- Rogler, G.; Brand, K.; Vogl, D.; Page, S.; Hofmeister, R.; Andus, T.; Knuechel, R.; Baeuerle, P.A.; Schölmerich, J.; Gross, V. Nuclear factor κB is activated in macrophages and epithelial cells of inflamed intestinal mucosa. Gastroenterology 1998, 115, 357–369. [Google Scholar] [CrossRef]
- Harikumar, K.B.; Kunnumakkara, A.B.; Ahn, K.S.; Anand, P.; Krishnan, S.; Guha, S.; Aggarwal, B.B. Modification of the cysteine residues in IκBα kinase and NF-κB (p65) by xanthohumol leads to suppression of NF-κB–regulated gene products and potentiation of apoptosis in leukemia cells. Blood 2009, 113, 2003–2013. [Google Scholar] [CrossRef] [Green Version]
- Escobar, J.; Pereda, J.; Arduini, A.; Sandoval, J.; Sabater, L.; Aparisi, L.; López-Rodas, G.; Sastre, J. Cross-talk between oxidative stress and pro-inflammatory cytokines in acute pancreatitis: A key role for protein phosphatases. Curr. Pharm. Des. 2009, 15, 3027–3042. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 6, 2a, 2b, 7a, 7b, 1a and 1b are available from the authors. |




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, H.S.; Chung, S.; Song, M.-Y.; Lim, C.; Shin, H.; Hur, J.; Kwon, H.; Suh, Y.-G.; Kim, E.-H.; Shin, D.; et al. Efficient and Divergent Enantioselective Syntheses of DHPVs and Anti-Inflammatory Effect on IEC-6 Cells. Molecules 2020, 25, 2215. https://doi.org/10.3390/molecules25092215
Kim HS, Chung S, Song M-Y, Lim C, Shin H, Hur J, Kwon H, Suh Y-G, Kim E-H, Shin D, et al. Efficient and Divergent Enantioselective Syntheses of DHPVs and Anti-Inflammatory Effect on IEC-6 Cells. Molecules. 2020; 25(9):2215. https://doi.org/10.3390/molecules25092215
Chicago/Turabian StyleKim, Hyun Su, Sungkyun Chung, Moon-Young Song, Changjin Lim, Hyeyoung Shin, Joonseong Hur, Hyuk Kwon, Young-Ger Suh, Eun-Hee Kim, Dongyun Shin, and et al. 2020. "Efficient and Divergent Enantioselective Syntheses of DHPVs and Anti-Inflammatory Effect on IEC-6 Cells" Molecules 25, no. 9: 2215. https://doi.org/10.3390/molecules25092215