
Total Syntheses of Cathepsin D Inhibitory Izenamides A, B, and C and Structural Confirmation of Izenamide B

College of Pharmacy, CHA University, 120 Haeryong-ro, Pocheon 11160, Gyeonggi-do, Korea
Molecules 2019, 24(19), 3424; https://doi.org/10.3390/molecules24193424
Submission received: 29 August 2019
/
Revised: 17 September 2019
/
Accepted: 19 September 2019
/
Published: 20 September 2019
(This article belongs to the Section Organic Chemistry)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:The first total syntheses of izenamides A, B, and C, which are depsipeptides inhibitor of cathepsin D, were accomplished. In addition, the stereochemistry of izenamide B was confirmed by our syntheses. The key features of our synthetic route involve the avoidance of critical 2,5-diketopiperazine (DKP) formation and the minimization of epimerization during the coupling of amino acids for the target peptides.
1. Introduction
Cathepsin D is an aspartic protease and has attracted considerable attention from both biologists and chemists due to its interesting biological functions [1,2]. This aspartic protease, which is involved in protein catabolism and required in certain epithelial cells for tissue remodeling and renewal, regulates lysosomal proteolysis and endogenous fibrinolysis [3,4,5,6]. Cathepsin D also maintains hormone and antigen processing [7,8,9]. In particular, cathepsin D is involved in various diseases, including breast cancer metastasis [10,11,12], atherosclerosis [8,13], Alzheimer’s disease [14,15,16] and neuronal ceroid lipofuscinosis (NCL) [6,17,18,19]. Therefore, therapeutic modulators of the proteolytic activity of cathepsin D have been extensively investigated for the development of new drugs although there have been few reports regarding inhibitors of cathepsin D [20,21,22,23,24].
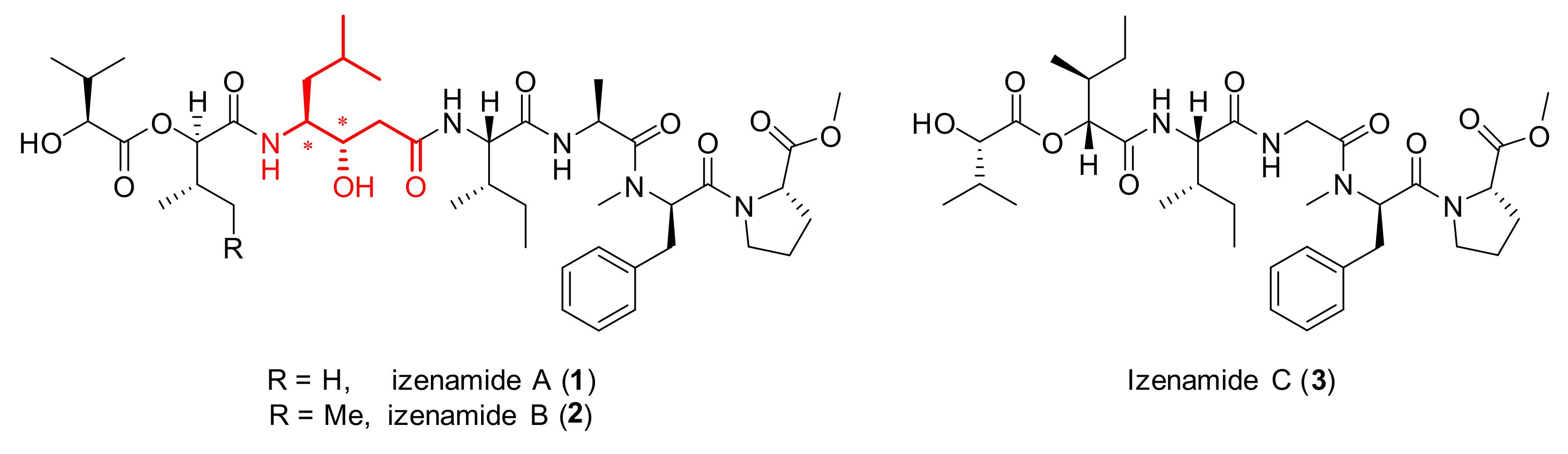
Recently, three new depsipeptides, namely, izenamides A (1), B (2), and C (3), were isolated from the marine cyanobacterium 1605-5 by Suenaga and coworkers (Figure 1) [25]. Interestingly, izenamides A (1) and B (2) were shown to inhibit cathepsin D in vitro. These two depsipeptides consist of seven monomers, including four amino acids, two hydroxy acids, and a γ-amino-β-hydroxy acid called statine. Izenamide C (3), which lacks a statine moiety and possesses a glycine unit instead of the alanine seen in izenamides A (1) and B (2), has no inhibitory activity against cathepsin D. The absolute configuration of the statine moiety in izenamide B (2) was indirectly determined by numerous efforts by Suenaga and coworkers [25].
In our pursuit of developing novel cathepsin D inhibitors, we recently became interested in establishing an efficient synthetic route towards izenamides A, B, and C. We were also interested in confirming the structure of izenamide B to ensure the stereochemistry of the statine moiety before we started medicinal chemistry studies. We herein report the first total syntheses of izenamides A, B, and C as well as the structural confirmation of izenamide B.
2. Results and Discussion
2.1. Synthetic Strategy for Izenamides A (1), B (2), and C (3)
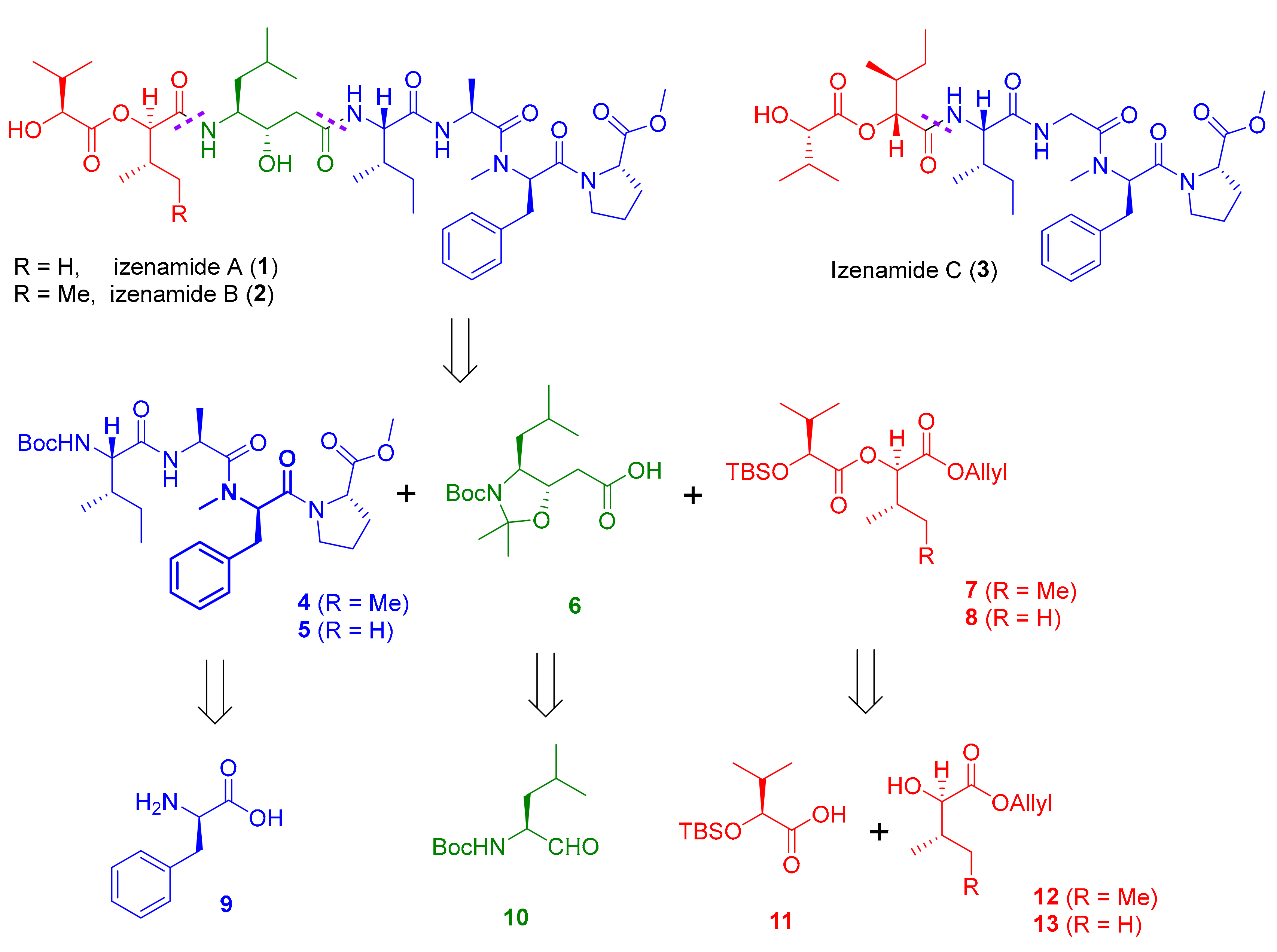
Our retrosynthetic analysis is outlined in Scheme 1. We envisioned that the target izenamides could be accessed via a versatile convergent approach. Izenamides A (1) and B (2) would be synthesized by assembling three fragments, tetrapeptide 4, statine 6, and esters 7 or 8. Considering that izenamide C (3) has a very similar structure except for the statine moiety, it could also be convergently synthesized by the amide coupling of tetrapeptide 5 and ester 7. Tetrapeptides 4 and 5 were anticipated to be derived from commercially available amino acids and NMe-d-Phe, which could be obtained from d-Phe (9). Protected statine 6 could be prepared from Boc-l-leucinal (10) through the known procedure [26,27]. Esters 7 and 8 can be conveniently obtained by the esterification of protected hydroxy acids 11 and 12 (or 13).
2.2. Syntheses of Fragments
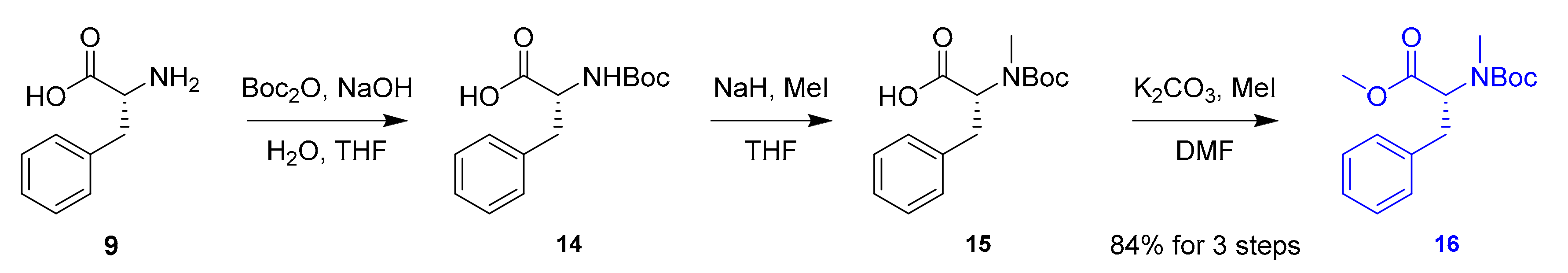
The synthesis of izenamides commenced with synthesizing tetrapeptides 4 and 5. Initially, NMe-d-Phe 16 was prepared from commercially available d-Phe (9) as shown in Scheme 2. Amine protection of d-Phe 9 with Boc anhydride and N-methylation of resulting carbamate 14 afforded acid 15, which was esterified to produce desired monomer 16 in 84% yield over three steps.
With monomer 16 in hand, we turned our attention to the synthesis of tetrapeptides 4 and 5 (Scheme 3). To avoid facile DKP formation upon sequential elongation from the C-terminal NMe-Phe and Pro residue, we synthesized tetrapeptides 4 and 5 according to the optimized sequence of fragment couplings. Amidation of l-alanine methyl ester hydrochloride (17) and glycine methyl ester hydrochloride (18) with Boc-Ile-OH followed by ester hydrolysis with LiOH afforded dipeptides 21 and 22 in high yields. Deprotection of 16 and coupling of resulting free amine 23 with acids 21 and 22 produced tripeptides 24 and 25 in 77 and 87% yields over two steps, respectively. Unfortunately, epimerization was observed during the amidation to form 24. After thorough optimization of the coupling conditions, amidation in the presence of DEPBT in CH2Cl2 at 0 °C provided the best results in terms of high chemical yield and minimal epimerization (2.8:1). Tripeptides 24 and 25 were subjected to hydrolysis and then amidation with l-proline methyl ester hydrochloride to afford tetrapeptides 4 and 5 in 84 and 81% yield over two steps, respectively. In general, the N-Me peptide backbone near the C-terminal is prone to α-epimerization upon carboxy activation due to steric hindrance and electronic effects [28]. However, we could obtain the desired tetrapeptides without epimerization under the optimized conditions (DEPBT, CH2Cl2, 0 °C).
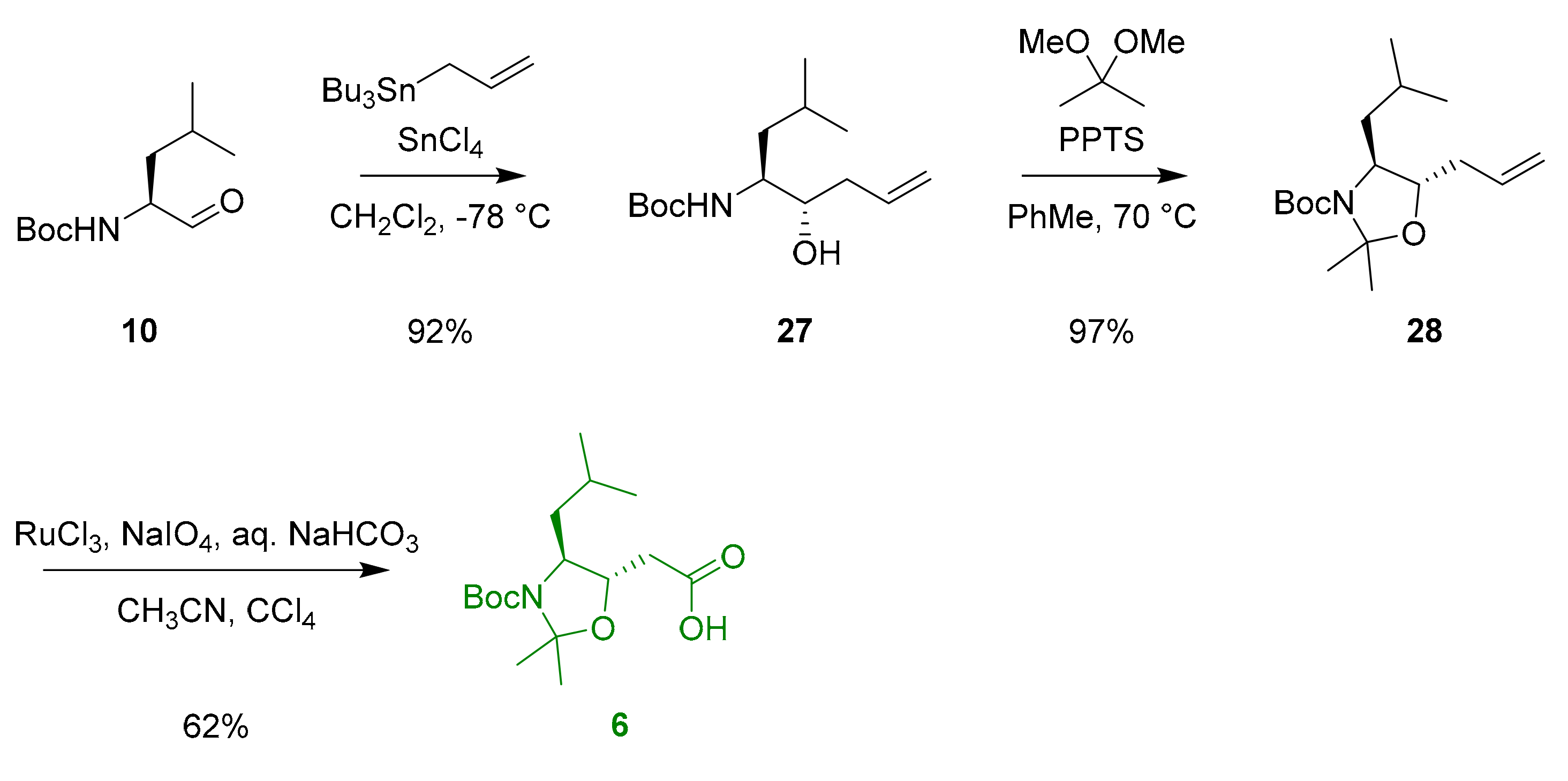
Optically pure statine 6 was prepared by the known procedure [26,27] (Scheme 4). An elegant highly diastereoselective allyl addition to N-Boc-l-leucinal in the presence of SnCl4 [26] provided desired threo isomer 27. The reaction of homoallylic alcohol 27 with 2,2-dimethoxypropane in the presence of catalytic PPTS followed by RuO4-mediated oxidative degradation [27] afforded monomer acid 6.
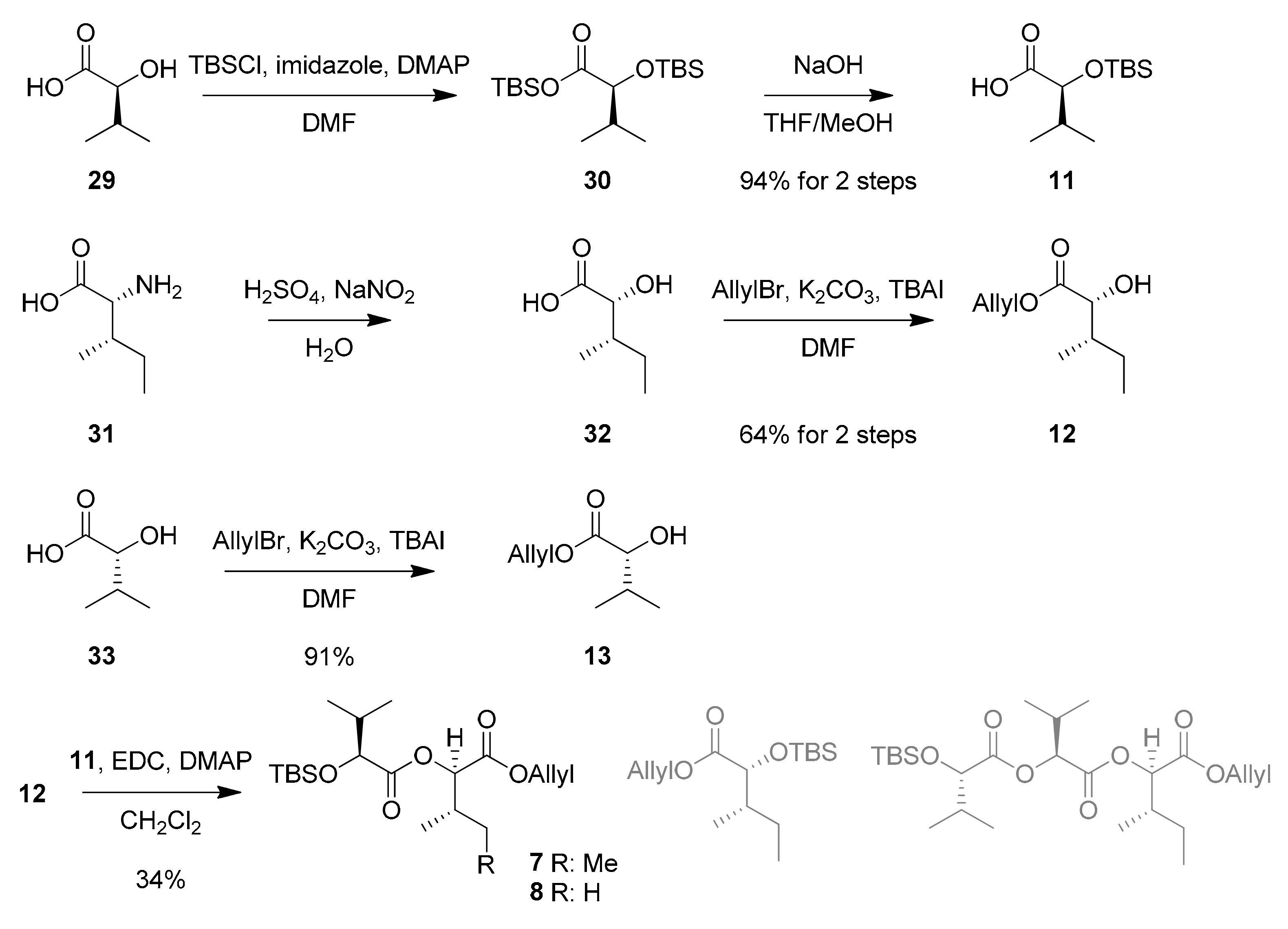
The synthesis of esters 7 and 8, which are the C-terminal fragments of izenamides, began with the preparation of silyl-protected valic acid 11 (Scheme 5). Global TBS-protection of commercially available l-valic acid (29) and hydrolysis of resulting silyl ester 30 produced C-terminal acid 11 in 94% yield over 2 steps. d-Allo-isoleucic acid 32 [29], which was prepared through the diazotization of commercially available d-allo-Ile (31), and d-valic acid (33) were converted to corresponding allyl esters 12 (64% for 2 steps) and 13 (91%), respectively. The EDC-mediated esterification of acid 11 with alcohol 12 afforded ester 7. Unfortunately, a satisfactory yield for this esterification was not achieved due the production of undesired esters by inevitable silyl transfer between hydroxyl groups.
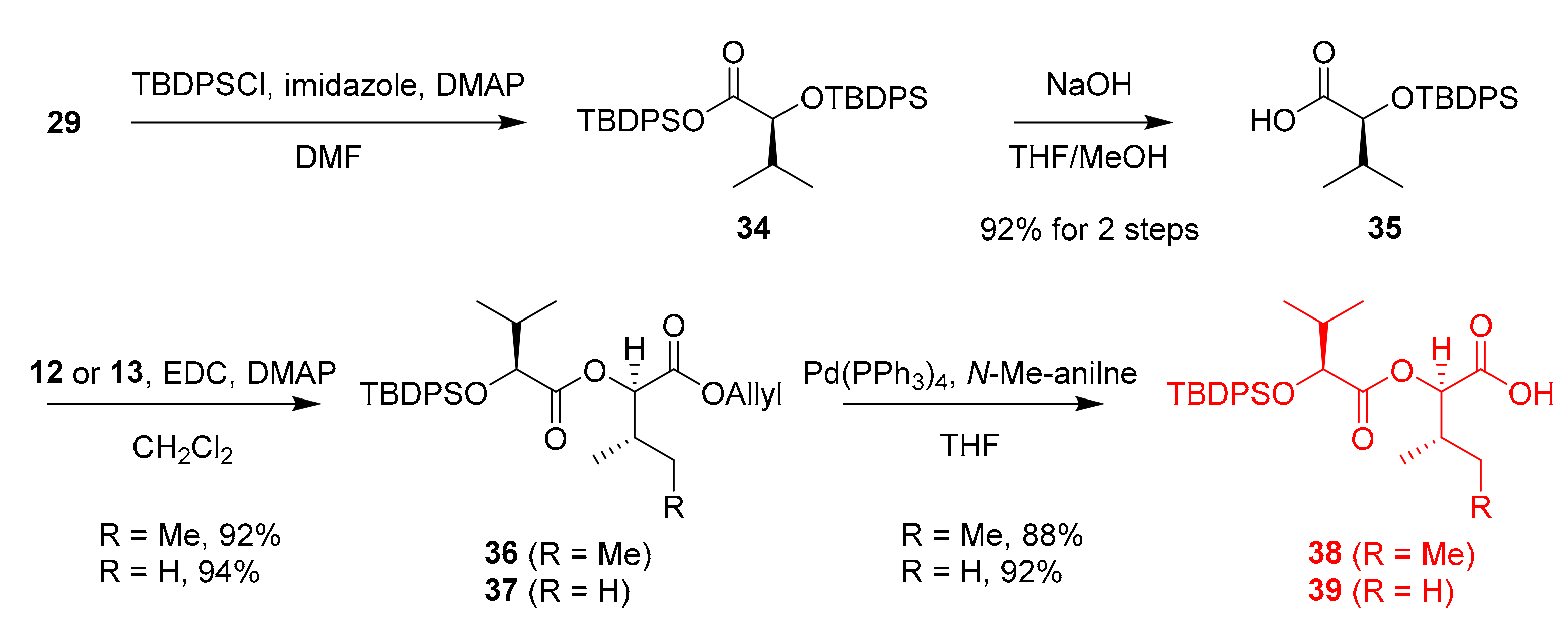
Therefore, we decided to change the TBS protecting group to a bulkier TBDPS group, which was anticipated to minimize silyl transfer (Scheme 6). To our delight, the silyl transfer was minimized in the coupling reactions, and fragments 36 and 37 were obtained in 92% and 94% yields, respectively. Finally, allyl deprotection of esters 36 and 37 with N-Me-aniline in the presence of Pd(0) produced acids 38 in 88% and 39 in 92% yield, respectively.
2.3. Completion of the Syntheses
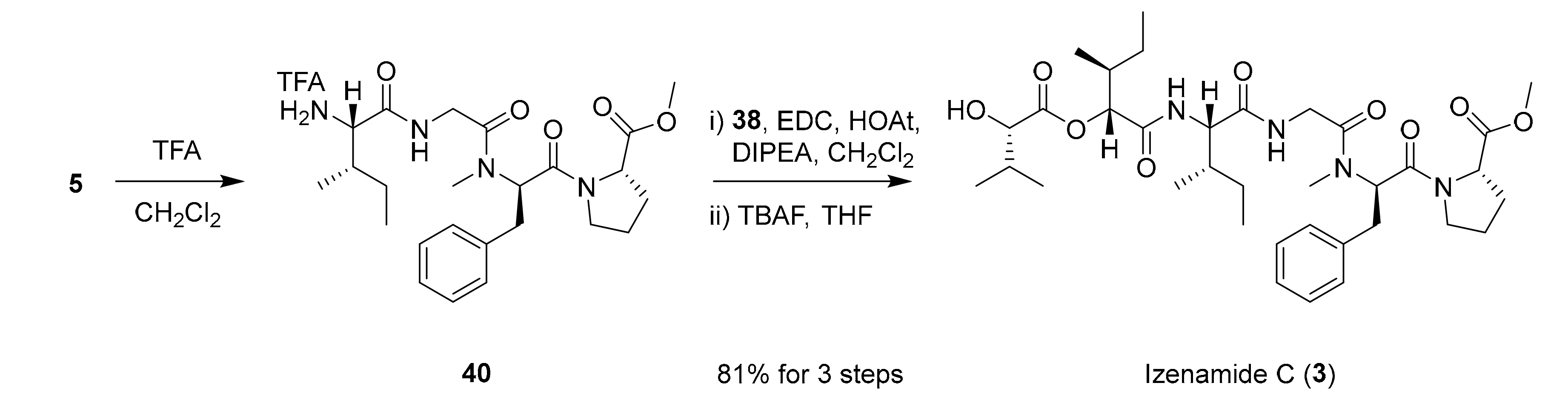
We finally assembled the prepared fragments of izenamide C as shown in Scheme 7. The EDC-mediated amide coupling of amine 40, which was prepared from tetrapeptide 5, with acid 38 followed by silyl-deprotection produced izenamide C (3) in 81% over 3 steps.
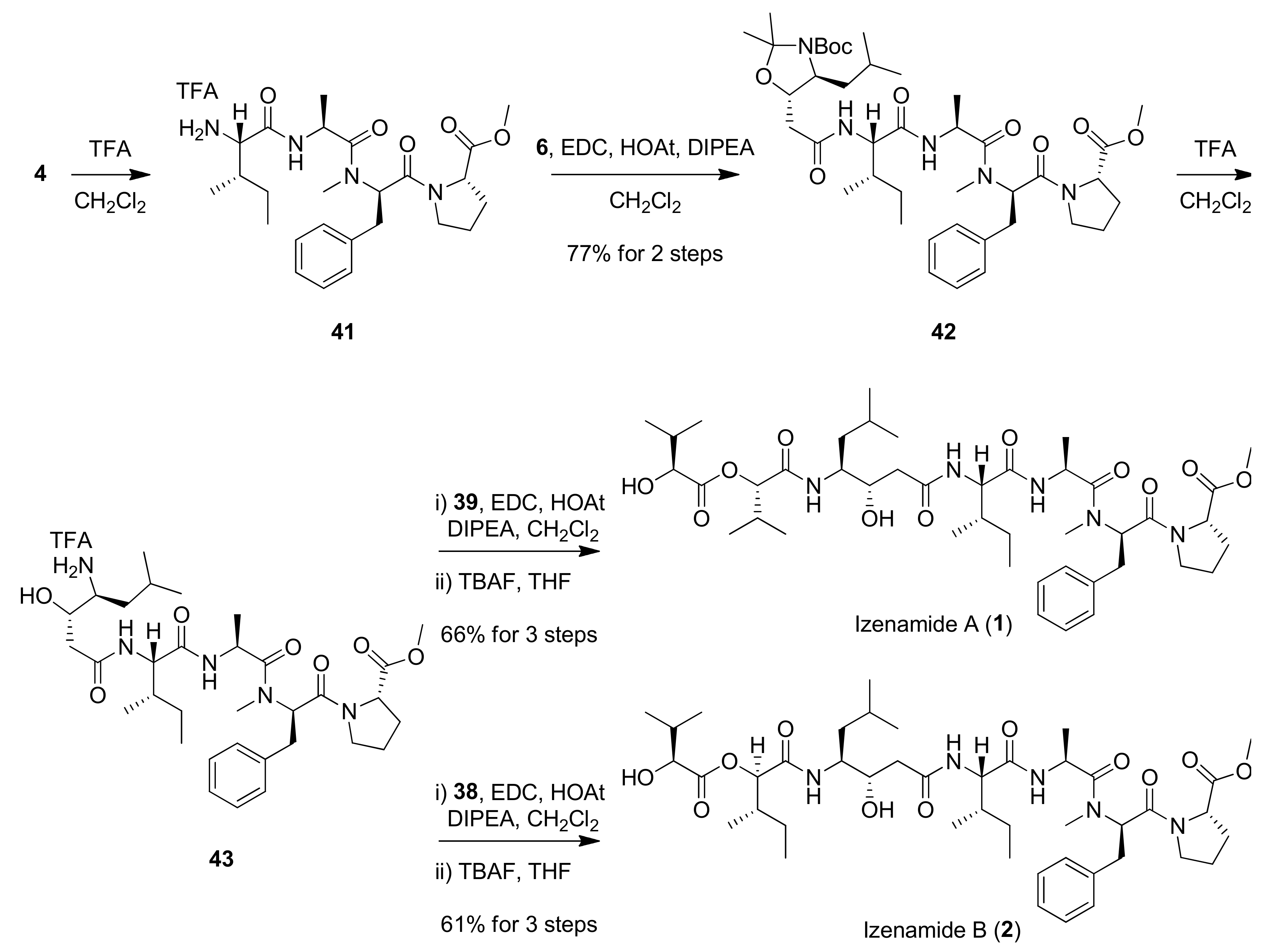
The completed synthesis of izenamides A (1) and B (2) is illustrated in Scheme 8. Boc-deprotection of tetrapeptide 4 with TFA and amide coupling with acid 6 afforded pentapeptide 42 in 77% yield over two steps. Global deprotection under acidic conditions followed by amidation of the resulting amine with acids 39 or 38 produced the corresponding heptapeptides. Finally, desilylation of heptapeptides afforded izenamides A (1) and B (2) in 66% and 61% over 3 steps, respectively. Spectral data of the synthesized depsipeptides 1, 2, and 3 were all identical with the reported data of natural 1, 2 and 3 [25]. Moreover, the absolute configuration of the statine moiety in 2 was identical to that of 1. The 1H and 13C NMR spectra of some compounds are in the Supplementary Materials.
3. Materials and Methods
3.1. General Information
Unless noted otherwise, all starting materials and reagents were obtained from commercial suppliers (Sigma-Aldrich, St. Louis, MO, USA; TCI, Tokyo, Japan; Combi-Blocks, San Diego, CA, USA) and were used without further purification. Tetrahydrofuran and Et2O were distilled from sodium benzophenone ketyl. Dichloromethane, chloroform and acetonitrile were freshly distilled from calcium hydride. All solvents used for routine isolation of products and chromatography were reagent grade and glass distilled. Reaction flasks were dried at 100 °C. Air and moisture sensitive reactions were performed under argon atmosphere. Flash column chromatography was performed using silica gel 60 (230–400 mesh, Merck, Kenilworth, NJ, USA) with the indicated solvents. Thin-layer chromatography was performed using 0.25 mm silica gel plates (Merck, Kenilworth, NJ, USA). Optical rotations were measured with JASCO P-2000 digital polarimeter (Tokyo, Japan) at ambient temperature using cylindrical cell of 10 mm or 100 mm pathlength. Infrared spectra were recorded on a JASCO FT-IR-4200 spectrometer (Tokyo, Japan). High resolution mass spectra were obtained with JEOL JMS-700 (Tokyo, Japan) and Agilent Q TOF 6530 (Santa Clara, CA, USA) instruments. 1H and 13C NMR spectra were recorded using BRUKER AVANCE-800 (Billerica, MA, USA). Chemical shifts are expressed in parts per million (ppm, δ) downfield from tetramethylsilane and are referenced to the deuterated solvent (CHCl3, 1H δ 7.24, 13C δ 77.0; MeOH-d4, 1H δ 3.30, 13C δ 49.00). 1H-NMR data were reported in the order of chemical shift, multiplicity (s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet and/or multiple resonances; br, broad signal), coupling constant in hertz (Hz) and number of protons.
3.2. Experimental Part
Boc-NMe-d-Phe-OMe (16). To a solution of d-Phe 9 (1.7 g, 10.3 mmol) and Boc2O (3.5 mL, 15.4 mmol) in a mixture THF and H2O (1:1, 50 mL) was added NaOH (0.6 g, 15.4 mmol) at room temperature. After stirring overnight, the reaction mixture was quenched with 1N HCl and extracted with EtOAc. The combined organic layer was washed with brine, dried over MgSO4, and concentrated in vacuo. To a solution of above crude Boc-d-Phe-OH 14 (10.3 mmol) in dry THF (20 mL) was added NaH (60% dispersion in mineral oil, 2.1 g, 51.5 mmol) at room temperature. After stirring for 1 h, iodomethane (3.2 mL, 51.5 mmol) was added to the reaction mixture. The reaction mixture was stirred for 12 h, quenched with 1N HCl, and extracted with EtOAc. The combined organic layer was washed with brine, dried over MgSO4, and concentrated in vacuo. The residue was used in the next step without further purification. To a solution of crude acid 15 (10.3 mmol) in dry DMF (20 mL) were added iodomethane (1.3 mL, 20.6 mmol) and K2CO3 (2.8 g, 20.6 mmol) at room temperature. After stirring overnight, the reaction mixture was quenched with 1N HCl and extracted with Et2O. The combined organic layer was washed with brine, dried over MgSO4, and concentrated in vacuo. The residue was purified by flash column chromatography (EtOAc/Hexane = 1:20) to give 2.5 g (84% for 3 steps) of ester 16 as a colorless oil. [α]D20 = +109.84 (c 1.00, CHCl3); 1H-NMR (800 MHz, CDCl3, 3:2 mixture of two rotamers). Major rotamer δ 7.28–7.20 (m, 2H), 7.19–7.07 (m, 3H), 4.50 (dd, J = 10.4, 3.8 Hz, 1H), 3.70 (s, 3H), 3.23 (dd, J = 14.2, 4.4 Hz, 1H), 3.01–2.94 (m, 1H), 2.68 (s, 3H), 1.29 (s, 9H), minor rotamer δ 7.28–7.20 (m, 2H), 7.19–7.07 (m, 3H), 4.89 (dd, J = 10.6, 5.2 Hz, 1H), 3.68 (s, 3H), 3.27 (dd, J = 14.4, 5.1 Hz, 1H), 3.01–2.94 (m, 1H), 2.66 (s, 3H), 1.33 (s, 9H); 13C-NMR (200 MHz, CDCl3, 3:2 mixture of two rotamers). Major rotamer δ 171.4, 154.8, 137.5, 128.9, 128.4, 126.5, 80.1, 61.5, 52.0, 35.4, 32.4, 28.0, minor rotamer δ 171.7, 155.6, 137.2, 128.8, 128.2, 126.3, 79.8, 59.4, 52.0, 34.9, 31.8, 28.1; IR (thin film, neat) νmax 2977, 1746, 1698, 1393, 1332, 1227, 1145, 751 cm−1; LR-MS (ESI+) m/z 316 (M + Na+); HR-MS (ESI+) calcd for C16H23NNaO4 (M + Na+) 316.1519; found 316.1523.
Boc-L-Ile-L-Ala-OH (21). To a solution of l-alanine methyl ester hydrochloride 17 (1.7 g, 12.2 mmol), Boc-l-Ile-OH (2.3 g, 9.9 mmol), DIPEA (5.2 mL, 29.8 mmol), and HOAt (1.4 g, 10.4 mmol) in CH2Cl2 (40 mL) was added EDC∙HCl (3.8 g, 19.9 mmol) at room temperature. After stirring for 4 h, the reaction mixture was quenched with 1N HCl, extracted with CH2Cl2. The combined organic layer was washed with aqueous NaHCO3, dried over MgSO4, and concentrated in vacuo. The residue was purified by flash column chromatography (EtOAc/Hexane = 1:2 to 1:1) to give 2.8 g (88%) of dipeptide 19 as white solid. [α]D20 = +16.54 (c 1.00, CHCl3); 1H NMR (800 MHz, CDCl3) δ 6.40 (s, 1H), 5.03 (d, J = 6.7 Hz, 1H), 4.56 (dq, J = 7.2, 7.1 Hz, 1H), 3.94 (t, J = 6.2 Hz, 1H), 3.72 (s, 3H), 1.88–1.81 (m, 1H), 1.53–1.44 (m, 1H), 1.42 (s, 9H), 1.39 (d, J = 7.2 Hz, 3H), 1.17–1.09 (m, 1H), 0.92 (d, J = 6.8 Hz, 3H), 0.89 (t, J = 7.4 Hz, 3H); 13C NMR (200 MHz, CDCl3) δ 173.1, 171.1, 155.7, 79.9, 59.1, 52.4, 48.0, 37.3, 28.3, 24.7, 18.3, 15.4, 11.4; IR (thin film, neat) νmax 3313, 2977, 1751, 1682, 1651, 1528, 1367, 1252, 870 cm−1; LR-MS (ESI+) m/z 317 (M + H+); HR-MS (ESI+) calcd for C15H29N2O5 (M + H+) 317.2071; found 317.2072. To a solution of Boc-l-Ile-l-Ala-OMe 19 (2.0 g, 6.3 mmol) in a mixture of THF, MeOH and H2O (1:1:1, 10 mL) was added LiOH∙H2O (0.6 g, 14.3 mmol) at room temperature. After stirring for 2 h, the reaction mixture was quenched with 1N HCl and extracted with EtOAc. The combined organic layer was dried over MgSO4 and concentrated in vacuo to afford 1.9 g (99%) of Boc-l-Ile-l-Ala-OH 21 as a white solid. The free acid 21 was used in the next step without further purification.
Boc-L-Ile-Gly-OH (22). To a solution of glycine methyl ester hydrochloride 18 (1.5 g, 11.9 mmol), Boc-l-Ile-OH (2.3 g, 9.9 mmol), DIPEA (5.2 mL, 29.8 mmol), and HOAt (1.4 g, 10.4 mmol) in CH2Cl2 (40 mL) was added EDC∙HCl (3.8 g, 19.9 mmol) at room temperature. After stirring for 5 h, the reaction mixture was quenched with 1N HCl and extracted with CH2Cl2. The combined organic layer was washed with aqueous NaHCO3, dried over MgSO4, and concentrated in vacuo. The residue was purified by flash column chromatography (EtOAc/Hexane = 1:2 to 1:1) to give 2.7 g (91%) of dipeptide 20 as a white solid. [α]D20 = −5.85 (c 1.00, CHCl3); 1H-NMR (800 MHz, CDCl3) δ 6.81 (s, 1H), 5.17 (d, J = 7.5 Hz, 1H), 4.08–3.91 (m, 3H), 3.70 (s, 3H), 1.84 (s, 1H), 1.51–1.45 (m, 1H), 1.39 (s, 9H), 1.13–1.06 (m, 1H), 0.91 (d, J = 6.9 Hz, 3H), 0.86 (t, J = 7.4 Hz, 3H); 13C-NMR (200 MHz, CDCl3) δ 172.1, 170.1, 155.8, 79.9, 59.0, 52.2, 41.0, 37.2, 28.2, 24.6, 15.4, 11.3; IR (thin film, neat) νmax 3324, 2968, 1759, 1687, 1661, 1527, 1367, 1173, 862 cm−1; LR-MS (ESI+) m/z 303 (M + H+); HR-MS (ESI+) calcd for C14H27N2O5 (M + H+) 303.1914; found 303.1923. To a solution of Boc-l-Ile-Gly-OMe 20 (2.0 g, 6.6 mmol) in a mixture of THF, MeOH, and H2O (1:1:1, 33 mL) was added LiOH∙H2O (0.6 g, 14.3 mmol) at room temperature. After stirring for 2 h, the reaction mixture was quenched with 1N HCl and extracted with EtOAc. The combined organic layer was dried over MgSO4 and concentrated in vacuo to afford 1.9 g (99%) of Boc-l-Ile-Gly-OH 22 as white solid. The free acid 22 was used in the next step without further purification.
Boc-l-Ile-l-Ala-NMe-d-Phe-OMe (24). To a solution of Boc-NMe-d-Phe-OMe 16 (440 mg, 1.5 mmol) in CH2Cl2 (6.0 mL) was added TFA (1.5 mL) dropwise at room temperature. After stirring for 1 h, the reaction mixture was concentrated in vacuo. The residue was used in the next step without further purification. To a solution of above amine salt 23 (1.5 mmol), Boc-l-Ile-l-Ala-OH 21 (544 mg, 1.8 mmol), and DIPEA (0.8 mL, 4.6 mmol) in CH2Cl2 (7.5 mL) was added DEPBT (898 mg, 3.0 mmol) at 0 °C. After stirring for overnight at 0 °C, the reaction mixture was quenched with 1N HCl, extracted with CH2Cl2. The combined organic layer was washed with aqueous NaHCO3, dried over MgSO4, and concentrated in vacuo. The residue was purified by flash column chromatography (EtOAc/Hexane = 1:1) to give 407 mg (57% for 2 steps) of tripeptide 24 as white solid and 145 mg (20% for 2 steps) of its epimer as white solid. [α]D20 = +61.58 (c 1.00, CHCl3); 1H-NMR (800 MHz, CDCl3, a mixture of rotamers) Major rotamer δ 7.24 (t, J = 7.4 Hz, 2H), 7.18 (t, J = 7.4 Hz, 1H), 7.13 (d, J = 7.3 Hz, 2H), 6.81 (d, J = 7.1 Hz, 1H), 5.29 (d, J = 7.1 Hz, 1H), 4.98 (d, J = 8.3 Hz, 1H), 4.69 (dq, J = 6.9, 6.8 Hz, 1H ), 3.95 (dd, J = 7.3, 6.1 Hz, 1H), 3.73 (s, 3H), 3.39 (dd, J = 14.8, 5.0 Hz, 1H), 3.01 (dd, J = 14.7, 11.8 Hz, 1H), 2.83 (s, 3H), 1.87–1.79 (m, 1H), 1.74–1.64 (m, 1H), 1.40 (s, 9H), 1.12–1.02 (m, 1H), 0.88 (d, J = 6.8 Hz, 3H), 0.87 (t, J = 7.4 Hz, 3H), 0.84 (d, J = 6.8 Hz, 3H).; 13C-NMR (200 MHz, CDCl3, a mixture of rotamers) Major rotamer δ 172.9, 170.7, 170.5, 155.6, 136.4, 128.7, 128.6, 126.9, 79.8, 59.2, 58.3, 52.4, 45.5, 37.6, 34.7, 32.5, 28.3, 24.6, 17.9, 15.6, 11.5; IR (thin film, neat) νmax 3319, 2970, 1743, 1710, 1637, 1498, 1366, 1173, 1017, 871 cm−1; LR-MS (ESI+) m/z 478 (M + H+); HR-MS (ESI+) calcd for C25H40N3O6 (M + H+) 478.2912; found 478.2915.
Boc-l-Ile-Gly-NMe-d-Phe-OMe (25). To a solution of Boc-NMe-d-Phe-OMe 16 (426 mg, 1.5 mmol) in CH2Cl2 (6.0 mL) was added TFA (1.5 mL) dropwise at room temperature. After stirring for 1 h, the reaction mixture was concentrated in vacuo. The residue was used in the next step without further purification. To a solution of above amine salt 23 (1.5 mmol), Boc-l-Ile-Gly-OH 22 (502 mg, 1.7 mmol) and DIPEA (0.8 mL, 4.4 mmol) in CH2Cl2 (7.3 mL) was added DEPBT (869 mg, 2.9 mmol) at 0 °C. After stirring overnight at 0 °C, the reaction mixture was quenched with 1N HCl and extracted with CH2Cl2. The combined organic layer was washed with aqueous NaHCO3, dried over MgSO4, and concentrated in vacuo. The residue was purified by flash column chromatography (EtOAc/Hexane = 1:1) to give 586 mg (87% for 2 steps) of tripeptide 25 as white solid. [α]D20 = +38.96 (c 1.00, CHCl3); 1H-NMR (800 MHz, CDCl3, a mixture of rotamers) Major rotamer δ 7.21 (t, J = 7.5 Hz, 2H), 7.15 (t, J = 7.4 Hz, 1H), 7.09 (d, J = 7.3 Hz, 2H), 6.84 (s, 1H), 5.19 (dd, J = 10.7, 5.3 Hz, 1H), 5.10 (d, J = 8.4 Hz, 1H), 4.04–4.01 (m, 1H), 3.95 (dd, J = 17.9, 3.7 Hz, 1H), 3.81 (dd, J = 17.7, 3.1 Hz, 1H), 3.67 (s, 3H), 3.32 (dd, J = 14.6, 5.4 Hz, 1H), 2.98 (dd, J = 14.6, 10.9 Hz, 1H), 2.74 (s, 3H), 1.81 (s, 1H), 1.44–1.36 (m, 1H), 1.37 (s, 9H), 1.07–1.02 (m, 1H), 0.85 (d, J = 6.9 Hz, 3H), 0.83 (t, J = 7.4 Hz, 3H); 13C-NMR (200 MHz, CDCl3, a mixture of rotamers) Major rotamer δ 171.3, 170.6, 168.4, 155.5, 136.4, 128.5, 128.5, 126.8, 79.5, 58.9, 58.7, 52.3, 41.2, 37.5, 34.4, 31.7, 28.1, 24.5, 15.4, 11.4; IR (thin film, neat) νmax 3328, 2967, 1743, 1712, 1643, 1497, 1366, 1172, 1016, 867 cm−1; LR-MS (ESI+) m/z 464 (M + H+); HR-MS (ESI+) calcd for C24H38N3O6 (M + H+) 464.2755; found 464.2751.
Boc-l-Ile-l-Ala-NMe-d-Phe-l-Pro-OMe (4). To a solution of Boc-l-Ile-l-Ala-NMe-d-Phe-OMe 24 (491 mg, 1.0 mmol) in a mixture of THF, MeOH, and H2O (1:1:1, 10 mL) was added LiOH∙H2O (86 mg, 2.1 mmol) at room temperature. After stirring for 2 h, the reaction mixture was quenched with 1N HCl and extracted with EtOAc. The combined organic layer was dried over MgSO4 and concentrated in vacuo. The free acid 26 was used in the next step without further purification. To a solution of above acid 26 (1.0 mmol), l-proline methyl ester hydrochloride (203 mg, 1.3 mmol), and DIPEA (0.6 mL, 3.4 mmol) in CH2Cl2 (10 mL) was added DEPBT (615 mg, 2.1 mmol) at 0 °C. After stirring for overnight at 0 °C, the reaction mixture was quenched with 1N HCl and extracted with CH2Cl2. The combined organic layer was washed with aqueous NaHCO3, dried over MgSO4, and concentrated in vacuo. The residue was purified by flash column chromatography (EtOAc/Hexane = 1:1 to 2:1) to give 496 mg (84% for 2 steps) of tetrapeptide 4 as white solid. [α]D20 = +45.90 (c 1.00, CHCl3); 1H-NMR (800 MHz, CDCl3, a mixture of rotamers) Major rotamer δ 7.23–7.13 (m, 5H), 6.64 (d, J = 7.2 Hz, 1H), 5.66 (dd, J = 9.5, 6.4 Hz, 1H), 4.98 (d, J = 8.2 Hz, 1H), 4.74 (dq, J = 7.1, 6.9 Hz, 1H), 4.42 (t, J = 7.5 Hz, 1H), 3.90 (t, J = 6.4 Hz, 1H), 3.70 (s, 3H), 3.47–3.42 (m, 1H), 3.22–3.15 (m, 1H), 2.98 (s, 3H), 2.94 (dd, J = 14.4, 9.6 Hz, 1H), 2.22–2.17 (m, 1H), 1.97–1.76 (m, 6H), 1.39 (s, 9H), 1.08–1.01 (m, 1H), 0.84 (t, J = 7.4 Hz, 3H), 0.83 (d, J = 6.8 Hz, 3H), 0.82 (d, J = 6.9 Hz, 3H); 13C-NMR (200 MHz, CDCl3, a mixture of rotamers) Major rotamer δ 172.3, 172.2, 170.5, 168.1, 155.5, 136.7, 129.5, 128.2, 126.6, 79.7, 59.2, 59.0, 55.5, 52.2, 46.9, 45.1, 37.6, 34.6, 30.2, 28.8, 28.2, 25.3, 24.6, 17.7, 15.4, 11.5; IR (thin film, neat) νmax 3316, 2971, 1748, 1709, 1643, 1497, 1365, 1245, 1174, 1045, 870 cm−1; LR-MS (ESI+) m/z 575 (M + H+); HR-MS (ESI+) calcd for C30H47N4O7 (M + H+) 575.3439; found 575.3442.
Boc-l-Ile-Gly-NMe-d-Phe-l-Pro-OMe (5). To a solution of Boc-l-Ile-Gly-NMe-d-Phe-OMe 25 (511 mg, 1.0 mmol) in a mixture of THF, MeOH, and H2O (1:1:1, 11 mL) was added LiOH∙H2O (92 mg, 2.2 mmol) at room temperature. After stirring for 2 h, the reaction mixture was quenched with 1N HCl and extracted with EtOAc. The combined organic layer was dried over MgSO4 and concentrated in vacuo. The free acid 27 was used in the next step without further purification. To a solution of above acid 27 (1.1 mmol), l-proline methyl ester hydrochloride (216 mg, 1.4 mmol) and DIPEA (0.6 mL, 3.4 mmol) in CH2Cl2 (11 mL) was added DEPBT (660 mg, 2.2 mmol) at 0 °C. After stirring overnight at 0 °C, the reaction mixture was quenched with 1N HCl, extracted with CH2Cl2. The combined organic layer was washed with aqueous NaHCO3, dried over MgSO4, and concentrated in vacuo. The residue was purified by flash column chromatography (EtOAc/Hexane = 1:1 to 2:1) to give 501 mg (81% for 2 steps) of tetrapeptide 5 as a white solid. [α]D20 = +69.20 (c 1.00, CHCl3); 1H-NMR (800 MHz, CDCl3, a mixture of rotamers) Major rotamer δ 7.23 (t, J = 7.4 Hz, 2H), 7.20 (d, J = 6.9 Hz, 2H), 7.17 (t, J = 7.2 Hz, 1H), 6.77 (s, 1H), 5.56 (t, J = 7.6 Hz, 1H), 5.00 (d, J = 8.0 Hz, 1H), 4.40 (dd, J = 8.5, 5.7 Hz, 1H), 4.09–4.01 (m, 1H), 3.92–3.85 (m, 1H), 3.70 (s, 3H), 3.38–3.34 (m, 1H), 3.33–3.29 (m, 1H), 3.25 (dd, J = 13.7, 8.0 Hz, 1H), 2.96 (s, J = 6.5 Hz, 3H), 2.81 (dd, J = 13.7, 7.1 Hz, 1H), 2.16–2.11 (m, 1H), 1.95–1.74 (m, 6H), 1.42 (s, 9H), 1.12–1.02 (m, 1H), 0.90 (d, J = 6.8 Hz, 3H), 0.88 (t, J = 7.4 Hz, 3H).; 13C-NMR (200 MHz, CDCl3, a mixture of rotamers) Major rotamer δ 172.4, 171.5, 167.9, 167.9, 155.7, 136.9, 129.4, 128.4, 126.7, 79.9, 59.2, 58.9, 56.2, 52.2, 46.8, 41.1, 37.5, 34.9, 29.7, 28.8, 28.3, 25.0, 24.5, 15.6, 11.6; IR (thin film, neat) νmax 3328, 2968, 1748, 1710, 1640, 1497, 1365, 1246, 1174, 1044, 869 cm−1; LR-MS (ESI+) m/z 561 (M + H+); HR-MS (ESI+) calcd for C29H45N4O7 (M + H+) 561.3283; found 561.3290.
Allyl (2R,3S)-2-hydroxy-3-methylpentanoate (12). To a solution of d-allo-Ile 31 (1.4 g, 10.7 mmol) in dilute sulfuric acid (0.8N in H2O, 26.7 mL, 21.3 mmol) was added NaNO2 (2.2 g, 32.0 mmol) slowly at 0°C. After stirring for 6 h at the same temperature, the reaction mixture was diluted with Et2O and extracted with Et2O. The combined organic layer was dried over MgSO4 and concentrated in vacuo. The residue was used in the next step without further purification. To a solution of crude acid 32 (10.7 mmol) and TBAI (0.79 g, 2.13 mmol) in dry DMF (500 mL) were added allyl bromide (1.8 mL, 21.3 mmol) and K2CO3 (3.0 g, 21.3 mmol) at room temperature. After stirring for overnight, the reaction mixture was quenched with 1N HCl and extracted with Et2O. The combined organic layer was washed with brine, dried over MgSO4, and concentrated in vacuo. The residue was purified by flash column chromatography (EtOAc/Hexane = 1:10) to give 1.2 g (65% for 2 steps) of ester 12 as a colorless oil. [α]D20 = +45.94 (c 0.50, CHCl3); 1H-NMR (800 MHz, CDCl3) δ 5.89 (ddt, J = 17.1, 10.5, 5.9 Hz, 1H), 5.31 (ddd, J = 17.2, 2.9, 1.5 Hz, 1H), 5.24 (dd, J = 10.4, 2.3, 1.2 Hz, 1H), 4.67 (ddt, J = 13.0, 5.9, 1.2 Hz, 1H), 4.63 (ddt, J = 13.1, 5.9, 1.3 Hz, 1H), 4.17 (d, J = 3.0 Hz, 1H), 2.61 (s, 1H), 1.83–1.75 (m, 1H), 1.50 (ddq, J = 13.8, 7.4, 7.2 Hz, 1H), 1.28 (ddq, J = 13.7, 7.5, 7.4 Hz, 1H), 0.91 (t, J = 7.5 Hz, 3H), 0.78 (d, J = 7.0 Hz, 3H); 13C-NMR (200 MHz, CDCl3) δ 175.0, 131.5, 119.1, 72.9, 66.0, 38.5, 25.9, 13.1, 11.8; IR (thin film, neat) νmax 3524, 2966, 1742, 1461, 1384, 1200, 1138, 934 cm−1; LR-MS (ESI+) m/z 173 (M + H+); HR-MS (ESI+) calcd for C29H45N4O7 (M + H+) 173.1172; found 173.1163.
Allyl (R)-2-hydroxy-3-methylbutanoate (13). To a solution of d-valic acid 33 (1.2 g, 10.2 mmol) and TBAI (0.75 g, 2.0 mmol) in dry DMF (25 mL) were added allyl bromide (1.8 mL, 20.3 mmol) and K2CO3 (2.8 g, 20.3 mmol) at room temperature. After stirring overnight, the reaction mixture was quenched with 1N HCl and extracted with Et2O. The combined organic layer was washed with brine, dried over MgSO4 and concentrated in vacuo. The residue was purified by flash column chromatography (EtOAc/Hexane = 1:20) to give 1.5 g (93%) of ester 13 as a colorless oil. [α]D20 = +24.77 (c 1.00, CHCl3); 1H-NMR (800 MHz, CDCl3) δ 5.84 (ddt, J = 17.1, 10.6, 5.9 Hz, 1H), 5.26 (dd, J = 17.2, 1.3 Hz, 1H), 5.18 (dd, J = 10.4, 1.1 Hz, 1H), 4.61 (dd, J = 13.1, 5.9 Hz, 1H), 4.57 (dd, J = 13.2, 5.9 Hz, 1H), 3.98 (d, J = 3.9 Hz, 1H), 2.87 (s, 1H), 2.01 (dtd, J = 13.9, 6.9, 3.7 Hz, 1H), 0.94 (d, J = 7.3 Hz, 3H), 0.79 (d, J = 7.2 Hz, 3H); 13C-NMR (200 MHz, CDCl3) δ 174.4, 131.4, 118.9, 74.9, 65.8, 32.0, 18.6, 15.8; IR (thin film, neat) νmax 3516, 2968, 1743, 1468, 1372, 1197, 1130, 933 cm−1; LR-MS (ESI+) m/z 181 (M + Na+); HR-MS (ESI+) calcd for C8H14NaO3 (M + Na+) 181.0835; found 181.0834.
(S)-2-((tert-Butyldiphenylsilyl)oxy)-3-methylbutanoic acid (35). To a solution of l-valic acid (1.2 g, 10.2 mmol) and DMAP (0.12 g, 1.0 mmol) in dry DMF (5.0 mL) were added imidazole (3.5 g, 50.8 mmol) and TBDPSCl (7.9 mL, 30.5 mmol) at room temperature. After stirring overnight, the reaction mixture was quenched with 1N HCl and extracted with Et2O. The combined organic layer was washed with brine, dried over MgSO4, and concentrated in vacuo. The residue was used in the next step without further purification. To a solution of crude silyl ether 34 (10.2 mmol) in a mixture of MeOH and H2O (3:1, 40 mL) was added K2CO3 (2.1 g, 15.2 mmol) at room temperature. After stirring for 1 h, the reaction mixture was quenched with 1N HCl and extracted with Et2O. The combined organic layer was washed with brine, dried over MgSO4 and concentrated in vacuo. The residue was purified by flash column chromatography (EtOAc/Hexane = 1:10) to give 1.1 g (92% for 2 steps) of acid 35 as a colorless oil. [α]D20 = −12.98 (c 1.00, CHCl3); 1H-NMR (800 MHz, CDCl3) δ 7.64 (d, J = 6.9 Hz, 2H), 7.60 (d, J = 6.9 Hz, 2H), 7.42 (dt, J = 13.4, 7.4 Hz, 2H), 7.39–7.33 (m, 4H), 4.12 (d, J = 3.8 Hz, 1H), 1.95–1.85 (m, 1H), 1.11 (s, 9H), 0.88 (d, J = 7.0 Hz, 3H), 0.86 (d, J = 6.9 Hz, 3H); 13C-NMR (200 MHz, CDCl3) δ 174.4, 135.9, 135.8, 132.7, 132.2, 130.2, 130.1, 127.8, 127.8, 77.4, 33.2, 27.0, 19.4, 17.6, 17.3; IR (thin film, neat) νmax 2962, 1720, 1471, 1428, 1183, 1113, 1071, 939 cm−1; LR-MS (ESI+) m/z 355 (M - H+); HR-MS (ESI+) calcd for C21H27OSi (M - H+) 355.1735; found 355.1742.
Allyl (2R,3S)-2-(((S)-2-((tert-Butyldiphenylsilyl)oxy)-3-methylbutanoyl)oxy)-3-methylpentanoate (36). To a solution of acid 35 (591 mg, 1.7 mmol), alcohol 12 (238 mg, 1.4 mmol), and DMAP (338 g, 2.8 mmol) in CH2Cl2 (14 mL) was added EDC∙HCl (530 g, 2.8 mmol) at room temperature. After stirring for 2 h, the reaction mixture was quenched with 1N HCl and extracted with CH2Cl2. The combined organic layer was washed with aqueous NaHCO3, dried over MgSO4, and concentrated in vacuo. The residue was purified by flash column chromatography (EtOAc/Hexane = 1:20) to give 649 mg (92%) of ester 36 as a colorless oil. [α]D20 = −23.21 (c 1.00, CHCl3); 1H NMR (800 MHz, CDCl3) δ 7.67–7.64 (m, 4H), 7.42–7.38 (m, 2H), 7.36–7.33 (m, 4H), 5.84 (ddt, J = 17.2, 10.4, 5.8 Hz, 1H), 5.29 (ddd, J = 17.2, 2.9, 1.4 Hz, 1H), 5.21 (ddd, J = 10.4, 2.3, 1.1 Hz, 1H), 4.82 (d, J = 3.5 Hz, 1H), 4.59 (ddt, J = 13.1, 5.7, 1.3 Hz, 1H), 4.52 (ddt, J = 13.1, 5.9, 1.3 Hz, 1H), 4.25 (d, J = 4.3 Hz, 1H), 2.06–1.96 (m, 1H), 1.90–1.85 (m, 1H), 1.36 (ddq, J = 13.8, 7.4, 7.3 Hz, 1H), 1.16 (ddq, J = 13.7, 7.6, 7.5 Hz, 1H), 1.14 (d, J = 7.5 Hz, 3H), 1.10 (s, 9H), 0.92 (d, J = 7.0 Hz, 3H), 0.89 (d, J = 6.9 Hz, 3H), 0.86 (t, J = 7.5 Hz, 3H); 13C-NMR (200 MHz, CDCl3) δ 172.0, 169.2, 136.0, 135.9, 133.5, 133.3, 131.7, 129.7, 129.6, 127.5, 127.4, 118.7, 77.6, 75.1, 65.5, 36.7, 33.3, 26.9, 25.7, 19.6, 18.1, 17.1, 14.4, 11.7; IR (thin film, neat) νmax 2965, 1756, 1463, 1428, 1192, 1113, 1008, 935, 822 cm−1; LR-MS (ESI+) m/z 528 (M + NH4+); HR-MS (ESI+) calcd for C30H46NO5Si (M + NH4+) 528.3140; found 528.3149.
Allyl (R)-2-(((S)-2-((tert-Butyldiphenylsilyl)oxy)-3-methylbutanoyl)oxy)-3-methylbutanoate (37). To a solution of acid 35 (741 g, 2.1 mmol), alcohol 13 (274 mg, 1.7 mmol) and DMAP (423 mg, 3.5 mmol) in CH2Cl2 was added EDC∙HCl (664 mg, 3.5 mmol) at room temperature. After stirring for 2 h, the reaction mixture was quenched with 1N HCl, extracted with CH2Cl2. The combined organic layer was washed with aqueous NaHCO3, dried over MgSO4 and concentrated in vacuo. The residue was purified by flash column chromatography (EtOAc/Hexane = 1:20) to give 809 mg (94%) of ester 37 as a colorless oil. [α]D20 = +2.49 (c 1.00, CHCl3); 1H-NMR (800 MHz, CDCl3) δ 7.64 (td, J = 8.4, 1.2 Hz, 4H), 7.40 (tdt, J = 7.7, 7.6, 1.3 Hz, 2H), 7.34 (dd, J = 7.2, 7.0 Hz, 4H), 5.83 (ddt, J = 17.2, 10.4, 5.8 Hz, 1H), 5.28 (dq, J = 17.2, 1.4 Hz, 1H), 5.20 (ddt, J = 10.4, 1.2, 1.0 Hz, 1H), 4.58 (dt, J = 13.1, 5.8, 1.3 Hz, 1H), 4.58 (d, J = 4.4 Hz, 1H), 4.51 (ddt, J = 13.2, 5.9, 1.2 Hz, 1H), 4.22 (d, J = 4.4 Hz, 1H), 2.12–2.06 (m, 1H), 2.05–1.98 (m, 1H), 1.09 (s, 9H), 0.92 (d, J = 6.9 Hz, 3H), 0.92 (d, J = 6.9 Hz, 3H), 0.90 (d, J = 6.9 Hz, 3H), 0.90 (d, J = 7.0 Hz, 3H); 13C-NMR (200 MHz, CDCl3) δ 172.0, 168.9, 136.0, 135.9, 133.4, 133.3, 131.6, 129.7, 129.6, 127.5, 127.4, 118.8, 77.6, 77.0, 65.5, 33.3, 30.1, 26.9, 19.6, 18.5, 18.3, 17.4, 17.2; IR (thin film, neat) νmax 2965, 1754, 1471, 1428, 1185, 1113, 990, 934, 822 cm−1; LR-MS (ESI+) m/z 519 (M + Na+); HR-MS (ESI+) calcd for C29H40NaO5Si (M + Na+) 519.2537; found 519.2543.
(2R,3S)-2-(((S)-2-((tert-Butyldiphenylsilyl)oxy)-3-methylbutanoyl)oxy)-3-methylpentanoic acid (38). To a solution of ester 36 (345 mg, 0.7 mmol) and N-methylaniline (0.15 mL, 1.4 mmol) in dry THF (6.8 mL) was added Pd(PPh3)4 (78 mg, 0.1 mmol) at room temperature. After stirring for 2 h, the reaction mixture was quenched with 1N HCl and extracted with EtOAc. The combined organic layer was washed with brine, dried over MgSO4, and concentrated in vacuo. The residue was purified by flash column chromatography through a short pad of silica gel (CH2Cl2/MeOH = 16:1) to afford 280 mg (88%) of the acid 38 as a yellow oil. [α]D20 = −15.06 (c 1.00, CHCl3); 1H-NMR (800 MHz, CDCl3) δ 7.64–7.62 (m, 4H), 7.42–7.36 (m, 2H), 7.35–7.29 (m, 4H), 4.84 (d, J = 3.3 Hz, 1H), 4.25 (d, J = 4.1 Hz, 1H), 2.03–1.97 (m, 1H), 1.89 (dtd, J = 7.5, 7.0, 3.3 Hz, 1H), 1.36 (ddq, J = 13.9, 7.3, 7.1 Hz, 1H), 1.18 (ddq, J = 13.8, 7.6, 7.4 Hz, 1H), 1.09 (s, 9H), 0.92 (d, J = 7.0 Hz, 3H), 0.91 (d, J = 6.9 Hz, 3H), 0.90 (d, J = 6.9 Hz, 3H), 0.86 (t, J = 7.4 Hz, 3H); 13C-NMR (200 MHz, CDCl3) δ 174.8, 172.0, 136.0, 135.9, 133.5, 133.3, 129.7, 129.7, 127.5, 127.5, 77.4, 74.4, 36.6, 33.3, 26.9, 25.8, 19.6, 18.4, 17.1, 14.3, 11.7; IR (thin film, neat) νmax 2965, 1760, 1730, 1463, 1428, 1182, 1113, 1000, 822 cm−1; LR-MS (ESI+) m/z 488 (M + NH4+); HR-MS (ESI+) calcd for C27H42NO5Si (M + NH4+) 488.2827; found 488.2829.
(R)-2-(((S)-2-((tert-Butyldiphenylsilyl)oxy)-3-methylbutanoyl)oxy)-3-methylbutanoic acid (39). To a solution of ester 37 (362 mg, 0.7 mmol) and N-methylaniline (0.2 mL, 1.5 mmol) in dry THF (7.3 mL) was added Pd(PPh3)4 (84 mg, 0.1 mmol) at room temperature. After stirring for 2 h, the reaction mixture was quenched with 1N HCl and extracted with EtOAc. The combined organic layer was washed with brine, dried over MgSO4, and concentrated in vacuo. The residue was purified by flash column chromatography through a short pad of silica gel (CH2Cl2/MeOH = 16:1) to afford 306 mg (92%) of the acid 39 as a yellow oil. [α]D20 = −11.79 (c 1.00, CHCl3); 1H-NMR (800 MHz, CDCl3) δ 7.63 (td, J = 8.0, 1.1 Hz, 4H), 7.41–7.36 (m, 2H), 7.32 (dt, J = 9.0, 7.6 Hz, 4H), 4.61 (d, J = 4.3 Hz, 1H), 4.24 (d, J = 4.2 Hz, 1H), 2.14–2.09 (m, 1H), 2.06–1.98 (m, 1H), 1.08 (s, 9H), 0.93 (d, J = 7.2 Hz, 3H), 0.92 (d, J = 7.0 Hz, 6H), 0.92 (d, J = 6.9 Hz, 3H); 13C-NMR (200 MHz, CDCl3) δ 173.6, 172.0, 136.0, 135.9, 133.4, 133.3, 129.7, 129.7, 127.5, 127.4, 77.4, 76.3, 33.3, 30.0, 26.9, 19.6, 18.6, 18.4, 17.2, 17.1; IR (thin film, neat) νmax 2965, 1760, 1726, 1470, 1428, 1181, 1113, 999, 822 cm−1; LR-MS (ESI+) m/z 474 (M + NH4+); HR-MS (ESI+) calcd for C26H40NO5Si (M + NH4+) 474.2670; found 474.2674.
Izenamide C (3). To a solution of Boc-l-Ile-Gly-NMe-d-Phe-l-Pro-OMe 5 (34 mg, 0.1 mmol) in CH2Cl2 (0.8 mL) was added TFA (0.2 mL) dropwise at room temperature. After stirring for 1 h, the reaction mixture was concentrated in vacuo. To a solution of above amine salt 40 (0.1 mmol), acid 38 (37 mg, 0.1 mmol), DIPEA (0.1 mL, 0.2 mmol), and HOAt (11 mg, 0.1 mmol) in CH2Cl2 (1.0 mL) was added EDC∙HCl (24 mg, 0.1 mmol) at room temperature. After stirring overnight, the reaction mixture was quenched with 1N HCl and extracted with CH2Cl2. The combined organic layer was washed with aqueous NaHCO3, dried over MgSO4, and concentrated in vacuo. The residue was used in the next step without further purification. To a solution of crude hexapeptide (0.1 mmol) in THF (1.0 mL) was added TBAF (1M in THF, 0.2 mL, 0.2 mmol) at room temperature. After stirring for 4 h, the reaction mixture was quenched with H2O and extracted with EtOAc. The combined organic layer was washed with brine, dried over MgSO4, and concentrated in vacuo. The residue was purified by flash column chromatography (Acetone/Hexane = 1:3) to give 33 mg (83% for 3 steps) of izenamide C (3) as white solid. [α]D20 = +21.50 (c 0.10, MeOH); 1H-NMR (800 MHz, CD3OD, a mixture of rotamers) Major rotamer δ 7.25–7.19 (m, 4H), 7.18–7.14 (m, 1H), 5.57 (t, J = 7.6 Hz, 1H), 5.06 (d, J = 4.5 Hz, 1H), 4.37 (dd, J = 8.3, 5.6 Hz, 1H), 4.33 (d, J = 7.0 Hz, 1H), 4.15 (d, J = 17.0 Hz, 1H), 4.11 (d, J = 4.4 Hz, 1H), 3.90 (d, J = 17.0 Hz, 1H), 3.70 (s, 3H), 3.44 (dt, J = 10.2, 6.9 Hz, 1H), 3.39 (dt, J = 10.2, 5.6 Hz, 1H), 3.19 (dd, J = 13.7, 7.8 Hz, 1H), 3.02 (s, 3H), 2.84 (dd, J = 13.7, 7.4 Hz, 1H), 2.21–2.17 (m, 1H), 2.11 (qqd, J = 6.9, 6.8, 4.5 Hz, 1H), 1.97–1.89 (m, 3H), 1.88–1.81 (m, 2H), 1.50–1.43 (m, 1H), 1.44 (ddq, J = 13.8, 7.4, 7.3 Hz, 1H), 1.28 (ddq, J = 13.7, 7.6, 7.5 Hz, 1H), 1.18–1.11 (m, 1H), 1.01 (d, J = 6.9 Hz, 3H), 0.95 (t, J = 6.7 Hz, 3H), 0.95 (d, J = 6.5 Hz, 3H), 0.93 (d, J = 6.9 Hz, 3H), 0.91 (d, J = 6.8 Hz, 3H), 0.89 (t, J = 7.4 Hz, 3H); 13C-NMR (200 MHz, CD3OD, a mixture of rotamers) Major rotamer δ 175.2, 174.0, 173.4, 172.2, 170.2, 170.1, 138.6, 130.5, 129.4, 127.6, 78.0, 76.5, 60.6, 58.9, 57.8, 52.7, 48.3, 41.9, 38.5, 37.9, 35.7, 33.3, 30.5, 29.9, 27.0, 26.0, 25.7, 19.4, 16.9, 16.1, 14.7, 11.8, 11.4; IR (thin film, neat) νmax 3317, 2960, 2877, 1744, 1637, 1519, 1447, 1199, 1176, 1137, 1030, 753 cm−1; LR-MS (ESI+) m/z 675 (M + H+); HR-MS (ESI+) calcd for C35H55N4O9 (M + H+) 675.3964; found 675.3949.
Pentapeptide (42). To a solution of Boc-l-Ile-l-Ala-NMe-d-Phe-l-Pro-OMe 4 (140 mg, 0.2 mmol) in CH2Cl2 (1.6 mL) was added TFA (0.4 mL) dropwise at room temperature. After stirring for 1 h, the reaction mixture was concentrated in vacuo. The residue was used in the next step without further purification. To a solution of above amine salt 41 (0.2 mmol), acid 6 (100 mg, 0.3 mmol), DIPEA (0.1 mL, 0.7 mmol), and HOAt (43 mg, 0.3 mmol) in CH2Cl2 (2.4 mL) was added EDC∙HCl (93 mg, 0.5 mmol) at room temperature. After stirring overnight, the reaction mixture was quenched with 1N HCl and extracted with CH2Cl2. The combined organic layer was washed with aqueous NaHCO3, dried over MgSO4, and concentrated in vacuo. The residue was purified by flash column chromatography (Acetone/Hexane = 1:3) to give 145 mg (77% for 2 steps) of pentapeptide 42 as white solid. [α]D20 = +67.88 (c 0.50, CHCl3); 1H-NMR (800 MHz, CDCl3, a mixture of rotamers) Major rotamer δ 7.23–7.18 (m, 4H), 7.16 (t, J = 7.2 Hz, 1H), 6.64 (s, 1H), 6.51 (d, J = 6.8 Hz, 1H), 5.66 (dd, J = 9.5, 6.5 Hz, 1H), 4.72 (dq, J = 7.2, 7.0 Hz, 1H), 4.44 (dd, J = 8.1, 6.7 Hz, 1H), 4.27–4.24 (m, 1H), 4.22–4.19 (m, 1H), 3.71 (s, 3H), 3.65–3.60 (m, 1H), 3.48–3.42 (m, 1H), 3.21 (dt, J = 10.6, 7.4 Hz, 1H), 3.19 (dd, J = 14.5, 6.4 Hz, 1H), 2.98 (s, 3H), 2.95 (dd, J = 14.4, 9.6 Hz, 1H), 2.58–2.51 (m, 1H), 2.48–2.40 (m, 1H), 2.23–2.18 (m, 1H), 1.93–1.87 (m, 1H), 1.87–1.75 (m, 4H), 1.61 (s, 3H), 1.52–1.46 (m, 5H), 1.46 (s, 9H), 1.46–1.42 (m, 1H), 1.12–1.02 (m, 1H), 0.90 (d, J = 6.2 Hz, 3H), 0.88 (d, J = 6.8 Hz, 3H), 0.85 (t, J = 7.4 Hz, 3H), 0.83 (d, J = 6.8 Hz, 3H), 0.82 (d, J = 6.9 Hz, 3H); 13C-NMR (200 MHz, CDCl3) δ 172.6, 172.4, 172.2, 169.8, 168.1, 156.3, 136.6, 129.5, 128.3, 126.7, 93.8, 79.3, 70.4, 59.3, 57.7, 55.6, 52.3, 52.2, 46.9, 45.2, 41.6, 40.1, 37.5, 34.7, 30.9, 30.3, 28.8, 28.4, 25.3, 24.9, 24.8, 24.8, 23.1, 22.1, 17.6, 15.3, 11.4; IR (thin film, neat) νmax 2961, 2875, 1751, 1700, 1632, 1532, 1455, 1366, 1257, 1175, 1047, 859 cm−1; LR-MS (ESI+) m/z 789 (M + NH4+); HR-MS (ESI+) calcd for C41H69N6O9 (M + NH4+) 789.5121; found 789.5134.
Izenamide A (1). To a solution of pentapeptide 42 (41 mg, 0.1 mmol) in CH2Cl2 (0.8 mL) was added TFA (0.2 mL) dropwise at room temperature. After stirring for 1 h, the reaction mixture was concentrated in vacuo. To a solution of above amine salt 43 (0.1 mmol), acid 39 (32 mg, 0.1 mmol), DIPEA (0.1 mL, 0.2 mmol), and HOAt (10 mg, 0.1 mmol) in CH2Cl2 (1.0 mL) was added EDC∙HCl (21 mg, 0.1 mmol) at room temperature. After stirring for 6 h, the reaction mixture was quenched with 1N HCl and extracted with CH2Cl2. The combined organic layer was washed with aqueous NaHCO3, dried over MgSO4 and concentrated in vacuo. The residue was used in the next step without further purification. To a solution of crude heptapeptide (0.1 mmol) in THF (1.0 mL) was added TBAF (1M in THF, 0.2 mL, 0.2 mmol) at room temperature. After stirring for 6 h, the reaction mixture was quenched with H2O and extracted with EtOAc. The combined organic layer was washed with brine, dried over MgSO4 and concentrated in vacuo. The residue was purified by flash column chromatography (Acetone/Hexane = 1:3 to 1:1) to give 29 mg (66% for 3 steps) of izenamide A (1) as white solid. [α]D20 = −17.15 (c 0.20, MeOH); 1H-NMR (800 MHz, CD3OD, a mixture of rotamers) Major rotamer δ 7.26–7.19 (m, 4H), 7.18–7.13 (m, 1H), 5.69 (dd, J = 10.0, 5.9 Hz, 1H), 4.71 (d, J = 5.9 Hz, 1H), 4.68 (q, J = 7.1 Hz, 1H), 4.41 (dd, J = 7.7, 7.4 Hz, 1H), 4.19 (d, J = 7.6 Hz, 1H), 4.11 (d, J = 4.4 Hz, 1H), 4.00–3.95 (m, 2H), 3.71 (s, 3H), 3.47–3.45 (m, 1H), 3.42–3.37 (m, 1H), 3.11 (dd, J = 14.3, 5.9 Hz, 1H), 3.06 (s, 3H), 2.93 (dd, J = 14.3, 10.0 Hz, 1H), 2.41–2.34 (m, 2H), 2.29–2.23 (m, 1H), 2.17–2.12 (m, 1H), 2.11–2.07 (m, 1H), 1.97–1.90 (m, 1H), 1.87–1.82 (m, 2H), 1.80–1.73 (m, 1H), 1.60–1.52 (m, 2H), 1.48 (ddd, J = 13.4, 7.6, 3.4 Hz, 1H), 1.31 (ddd, J = 9.3, 7.5, 4.3 Hz, 1H), 1.13 (ddd, J = 13.6, 9.5, 7.3 Hz, 1H), 1.01 (d, J = 6.8 Hz, 3H), 1.00 (d, J = 6.8 Hz, 6H), 0.92 (d, J = 6.8 Hz, 3H), 0.92 (d, J = 6.5 Hz, 3H), 0.89 (d, J = 6.3 Hz, 3H), 0.87 (t, J = 7.4 Hz, 3H), 0.86 (d, J = 6.7 Hz, 3H), 0.82 (d, J = 7.0 Hz, 3H); 13C-NMR (200 MHz, CD3OD, a mixture of rotamers) Major rotamer δ 175.8, 174.6, 173.9, 173.8, 173.0, 171.8, 170.4, 138.3, 130.8, 129.3, 127.6, 80.6, 76.4, 71.3, 60.9, 58.7, 57.2, 52.6, 52.2, 48.5, 46.6, 41.4, 41.4, 38.3, 35.6, 33.3, 31.7, 31.1, 30.0, 26.2, 26.0, 25.7, 23.8, 22.2, 19.3, 19.2, 18.1, 17.2, 16.6, 15.8, 11.4; IR (thin film, neat) νmax 3318, 2960, 2878, 1742, 1637, 1544, 1450, 1366, 1265, 1198, 1038, 753 cm−1; LR-MS (ESI+) m/z 854 (M + Na+); HR-MS (ESI+) calcd for C43H69N5NaO11 (M + Na+) 854.4886; found 854.4888.
Izenamide B (2). To a solution of pentapeptide 42 (46 mg, 0.1 mmol) in CH2Cl2 (0.8 mL) was added TFA (0.2 mL) dropwise at room temperature. After stirring for 1 h, the reaction mixture was concentrated in vacuo. To a solution of above amine salt 43 (0.1 mmol), acid 38 (36 mg, 0.1 mmol), DIPEA (0.1 mL, 0.2 mmol), and HOAt (11 mg, 0.1 mmol) in CH2Cl2 (1.0 mL) was added EDC∙HCl (23 mg, 0.1 mmol) at room temperature. After stirring for 6 h, the reaction mixture was quenched with 1N HCl and extracted with CH2Cl2. The combined organic layer was washed with aqueous NaHCO3, dried over MgSO4, and concentrated in vacuo. The residue was used in the next step without further purification. To a solution of crude heptapeptide (0.1 mmol) in THF (1.0 mL) was added TBAF (1M in THF, 0.2 mL, 0.2 mmol) at room temperature. After stirring for 6 h, the reaction mixture was quenched with H2O and extracted with EtOAc. The combined organic layer was washed with brine, dried over MgSO4, and concentrated in vacuo. The residue was purified by flash column chromatography (Acetone/Hexane = 1:3 to 1:1) to give 31 mg (61% from S5) of izenamide B (2) as white solid. [α]D20 = −11.62 (c 1.30, MeOH); 1H-NMR (800 MHz, CD3OD) δ 7.25–7.19 (m, 4H), 7.18–7.13 (m, 1H), 5.69 (dd, J = 10.0, 5.9 Hz, 1H), 4.93 (d, J = 4.4 Hz, 1H), 4.67 (q, J = 7.0 Hz, 1H), 4.41 (dd, J = 7.8, 7.3 Hz, 1H), 4.19 (d, J = 7.5 Hz, 1H), 4.13 (d, J = 4.3 Hz, 1H), 4.00–3.96 (m, 2H), 3.71 (s, 3H), 3.48–3.44 (m, 1H), 3.42–3.38 (m, 1H), 3.11 (dd, J = 14.3, 5.9 Hz, 1H), 3.06 (s, 3H), 2.93 (dd, J = 14.3, 10.0 Hz, 1H), 2.41–2.33 (m, 2H), 2.28–2.23 (m, 1H), 2.11–2.07 (m, 1H), 1.96–1.89 (m, 2H), 1.88–1.81 (m, 2H), 1.79–1.74 (m, 1H), 1.59–1.53 (m, 2H), 1.51–1.43 (m, 2H), 1.33–1.27 (m, 2H), 1.18–1.10 (m, 1H), 1.01 (d, J = 6.9 Hz, 3H), 0.97 (d, J = 6.9 Hz, 3H), 0.95 (t, J = 7.5 Hz, 3H), 0.92 (d, J = 6.4 Hz, 3H), 0.92 (d, J = 6.8 Hz, 3H), 0.89 (d, J = 6.2 Hz, 3H), 0.87 (t, J = 7.4 Hz, 3H), 0.86 (d, J = 6.8 Hz, 3H), 0.82 (d, J = 7.1 Hz, 3H); 13C-NMR (200 MHz, CD3OD, a mixture of rotamers) Major rotamer δ 175.7, 174.6, 173.9, 173.8, 173.0, 171.9, 170.3, 138.3, 130.8 129.3 127.6, 78.3, 76.4, 71.4, 60.9, 58.7, 57.2, 52.6, 52.3, 48.5, 46.6, 41.5, 41.4, 38.4, 38.3, 35.6, 33.3, 31.0, 30.0, 27.2, 26.2, 26.1, 25.7, 23.8, 22.2, 19.3, 17.1, 16.6, 15.8, 14.7, 11.9, 11.5; IR (thin film, neat) νmax 3308, 2960, 2877, 1743, 1636, 1533, 1448, 1361, 1265, 1195, 1038, 753 cm−1; LR-MS (ESI+) m/z 846 (M + H+); HR-MS (ESI+) calcd for C44H72N5O11 (M + H+) 846.5223; found 846.5229.
4. Conclusions
In conclusion, we have accomplished the first total syntheses of izenamides A (1), B (2), and C (3) and confirmed the stereochemistry of izenamide B, which was originally suggested by Suenaga and coworkers. The key features of the syntheses include the DKP formation-free amide coupling sequence and amidation with minimal epimerization. Our versatile and convergent synthesis of izenamides is expected to be widely utilized for the efficient synthesis of izenamides and their analogs for the development of novel cathepsin D inhibitors. Further studies on the structure-activity relationship of izenamides are currently underway and successful results will be reported in due course.
Supplementary Materials
The following are available online. The 1H and 13C NMR spectra of 16, 19, 20, 24, 25, 4, 5, 12, 13, 35, 36, 37, 38, 39, 3, 42, 1 and 2.
Funding
This research was funded by National Research Foundation of Korea, grant number 2019M3A9A8067070.
Acknowledgments
We thank Young-Ger Suh of Seoul National University for his helpful discussions in management of this work and preparation of manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Benes, P.; Vetvicka, V.; Fusek, M. Cathepsin D—many functions of one aspartic protease. Crit. Rev. Oncol. Hematol. 2008, 68, 12–28. [Google Scholar] [CrossRef] [PubMed]
- Vidoni, C.; Follo, C.; Savino, M.; Melone, M.A.; Isidoro, C. The role of cathepsin d in the pathogenesis of human neurodegenerative disorders. Med. Res. Rev. 2016, 36, 845–870. [Google Scholar] [CrossRef] [PubMed]
- Loscalzo, J. The Macrophage and Fibrinolysis; Seminars in thrombosis and hemostasis, 1996; Copyright© 1996 by Thieme Medical Publishers, Inc.: Stuttgart, Germany, 1996; pp. 503–506. [Google Scholar]
- Simon, D.I.; Ezratty, A.M.; Loscalzo, J. The fibrin (ogen) olytic properties of cathepsin d. Biochemistry 1994, 33, 6555–6563. [Google Scholar] [CrossRef] [PubMed]
- Koike, M.; Shibata, M.; Ohsawa, Y.; Nakanishi, H.; Koga, T.; Kametaka, S.; Waguri, S.; Momoi, T.; Kominami, E.; Peters, C. Involvement of two different cell death pathways in retinal atrophy of cathepsin D-deficient mice. Mol. Cell. Neurosci. 2003, 22, 146–161. [Google Scholar] [CrossRef]
- Saftig, P.; Hetman, M.; Schmahl, W.; Weber, K.; Heine, L.; Mossmann, H.; Köster, A.; Hess, B.; Evers, M.; von Figura, K. Mice deficient for the lysosomal proteinase cathepsin D exhibit progressive atrophy of the intestinal mucosa and profound destruction of lymphoid cells. EMBO J. 1995, 14, 3599–3608. [Google Scholar] [CrossRef] [PubMed]
- Baechle, D.; Flad, T.; Cansier, A.; Steffen, H.; Schittek, B.; Tolson, J.; Herrmann, T.; Dihazi, H.; Beck, A.; Mueller, G.A. Cathepsin D is present in human eccrine sweat and involved in the postsecretory processing of the antimicrobial peptide DCD-1L. J. Biol. Chem. 2006, 281, 5406–5415. [Google Scholar] [CrossRef]
- Hakala, J.K.; Oksjoki, R.; Laine, P.; Du, H.; Grabowski, G.A.; Kovanen, P.T.; Pentikäinen, M.O. Lysosomal enzymes are released from cultured human macrophages, hydrolyze LDL in vitro, and are present extracellularly in human atherosclerotic lesions. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1430–1436. [Google Scholar] [CrossRef] [PubMed]
- Bańkowska, A.; Gacko, M.; Chyczewska, E.; Worowska, A. Biological and diagnostic role of cathepsin D. Rocz. Akad. Med. Bialymst. 1997, 42, 79–85. [Google Scholar]
- Vetvicka, V.; Vetvickova, J.; Fusek, M. Anti-human procathepsin D activation peptide antibodies inhibit breast cancer development. Breast Cancer Res. Treat. 1999, 57, 261–269. [Google Scholar] [CrossRef]
- Vetvicka, V.; Vetvickova, J.; Hilgert, I.; Voburka, Z.; Fusek, M. Analysis of the interaction of procathepsin D activation peptide with breast cancer cells. Int. J. Cancer 1997, 73, 403–409. [Google Scholar] [CrossRef]
- Vignon, F.; Capony, F.; Ghambon, M.; Freiss, G.; Garcia, M.; Rochefort, H. Autocrine growth stimulation of the MCF 7 breast cancer cells by the estrogen-regulated 52 K protein. Endocrinology 1986, 118, 1537–1545. [Google Scholar] [CrossRef] [PubMed]
- Haidar, B.; Kiss, R.S.; Sarov-Blat, L.; Brunet, R.; Harder, C.; McPherson, R.; Marcel, Y.L. Cathepsin D, a lysosomal protease, regulates ABCA1-mediated lipid efflux. J. Biol. Chem. 2006, 281, 39971–39981. [Google Scholar] [CrossRef] [PubMed]
- Elliott, D.A.; Tsoi, K.; Holinkova, S.; Chan, S.L.; Kim, W.S.; Halliday, G.M.; Rye, K.-A.; Garner, B. Isoform-specific proteolysis of apolipoprotein-E in the brain. Neurobiol. Aging 2011, 32, 257–271. [Google Scholar] [CrossRef] [PubMed]
- Mutka, A.L.; Haapanen, A.; Käkelä, R.; Lindfors, M.; Wright, A.K.; Inkinen, T.; Hermansson, M.; Rokka, A.; Corthals, G.; Jauhiainen, M. Murine cathepsin D deficiency is associated with dysmyelination/myelin disruption and accumulation of cholesteryl esters in the brain. J. Neurochem. 2010, 112, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Scott, S.; Shelton, S.; Crutcher, K. Cathepsin D-mediated proteolysis of apolipoprotein E: Possible role in Alzheimer’s disease. Neuroscience 2006, 143, 689–701. [Google Scholar] [CrossRef] [PubMed]
- Myllykangas, L.; Tyynelä, J.; Page-McCaw, A.; Rubin, G.M.; Haltia, M.J.; Feany, M.B. Cathepsin D-deficient drosophila recapitulate the key features of neuronal ceroid lipofuscinoses. Neurobiol. Dis. 2005, 19, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Koike, M.; Nakanishi, H.; Saftig, P.; Ezaki, J.; Isahara, K.; Ohsawa, Y.; Schulz-Schaeffer, W.; Watanabe, T.; Waguri, S.; Kametaka, S. Cathepsin D deficiency induces lysosomal storage with ceroid lipofuscin in mouse CNS neurons. J. Neurosci. 2000, 20, 6898–6906. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Montealegre, D.; Rothberg, P.G.; Pearce, D.A. Another disorder finds its gene. Brain 2006, 129, 1353–1356. [Google Scholar] [CrossRef] [Green Version]
- Umezawa, H.; Aoyagi, T.; Morishima, H.; Matsuzaki, M.; Hamada, M.; Takeuchi, T. Pepstatin, a new pepsin inhibitor produced by agtinomygetes. J. Antibiot. 1970, 23, 259–262. [Google Scholar] [CrossRef]
- Kwan, J.C.; Eksioglu, E.A.; Liu, C.; Paul, V.J.; Luesch, H. Grassystatins A−C from marine cyanobacteria, potent cathepsin E inhibitors that reduce antigen presentation. J. Med. Chem. 2009, 52, 5732–5747. [Google Scholar] [CrossRef]
- Al-Awadhi, F.H.; Law, B.K.; Paul, V.J.; Luesch, H. Grassystatins D–F, potent aspartic protease inhibitors from marine cyanobacteria as potential antimetastatic agents targeting invasive breast cancer. J. Nat. Prod. 2017, 80, 2969–2986. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.G.; Yoshida, W.Y.; Moore, R.E.; Paul, V.J. The isolation and structure elucidation of tasiamide B, a 4-amino-3-hydroxy-5-phenylpentanoic acid containing peptide from the marine cyanobacterium symploca sp. J. Nat. Prod. 2003, 66, 1006–1009. [Google Scholar] [CrossRef] [PubMed]
- Al-Awadhi, F.H.; Ratnayake, R.; Paul, V.J.; Luesch, H. Tasiamide F, a potent inhibitor of cathepsins D and E from a marine cyanobacterium. Bioorgan. Med. Chem. 2016, 24, 3276–3282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanamori, Y.; Iwasaki, A.; Sumimoto, S.; Matsubara, T.; Sato, T.; Suenaga, K. Izenamides A and B, statine-containing depsipeptides, and an analogue from a marine cyanobacterium. J. Nat. Prod. 2018, 81, 1673–1681. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Bischoff, A.; Cappiello, J. Asymmetric total synthesis of the gastroprotective microbial agent AI-77-B. Eur. J. Org. Chem. 2003, 2003, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Veeresha, G.; Datta, A. Stereoselective synthesis of (−)-N-Boc-statine and (−)-N-Boc-norstatine. Tetrahedron Lett. 1997, 38, 5223–5224. [Google Scholar] [CrossRef]
- Fernández-Llamazares, A.I.; Spengler, J.; Albericio, F. Review backbone N-modified peptides: How to meet the challenge of secondary amine acylation. Biopolymers 2015, 104, 435–452. [Google Scholar] [CrossRef]
- Dai, L.; Chen, B.; Lei, H.; Wang, Z.; Liu, Y.; Xu, Z.; Ye, T. Total synthesis and stereochemical revision of lagunamide A. Chem. Commun. 2012, 48, 8697–8699. [Google Scholar] [CrossRef] [Green Version]
Sample Availability: Samples of the compounds 1–3, 4–6, 11–13, 16, 19–22, 24–27, 35–39, and 42 are available from the authors. |
Figure 1.
Structures of Izenamides.

Scheme 1.
Retrosynthetic analysis for the synthesis of izenamides 1, 2 and 3.

Scheme 2.
Preparation of NMe-d-Phe monomer 16.

Scheme 3.
Synthesis of tetrapeptides 4 and 5.

Scheme 4.
Synthesis of statine 6.

Scheme 5.
Synthesis of Ester 7.

Scheme 6.
Synthesis of fragments 38 and 39 with minimized silyl transfer.

Scheme 7.
Completion of the synthesis of izenamide C (3).

Scheme 8.
Completion of the syntheses of izenamides A (1) and B (2).

© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Lim, C. Total Syntheses of Cathepsin D Inhibitory Izenamides A, B, and C and Structural Confirmation of Izenamide B. Molecules 2019, 24, 3424. https://doi.org/10.3390/molecules24193424
AMA Style
Lim C. Total Syntheses of Cathepsin D Inhibitory Izenamides A, B, and C and Structural Confirmation of Izenamide B. Molecules. 2019; 24(19):3424. https://doi.org/10.3390/molecules24193424
Chicago/Turabian StyleLim, Changjin. 2019. "Total Syntheses of Cathepsin D Inhibitory Izenamides A, B, and C and Structural Confirmation of Izenamide B" Molecules 24, no. 19: 3424. https://doi.org/10.3390/molecules24193424