Application of a Substrate-Mediated Selection with c-Src Tyrosine Kinase to a DNA-Encoded Chemical Library

 , and
, and
Abstract
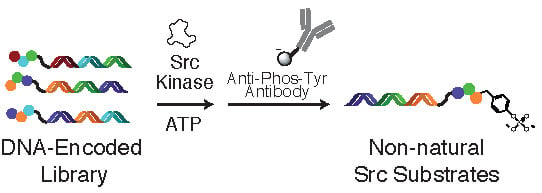
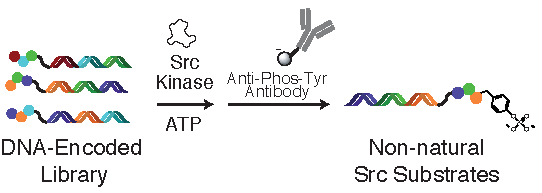
:
1. Introduction
2. Results
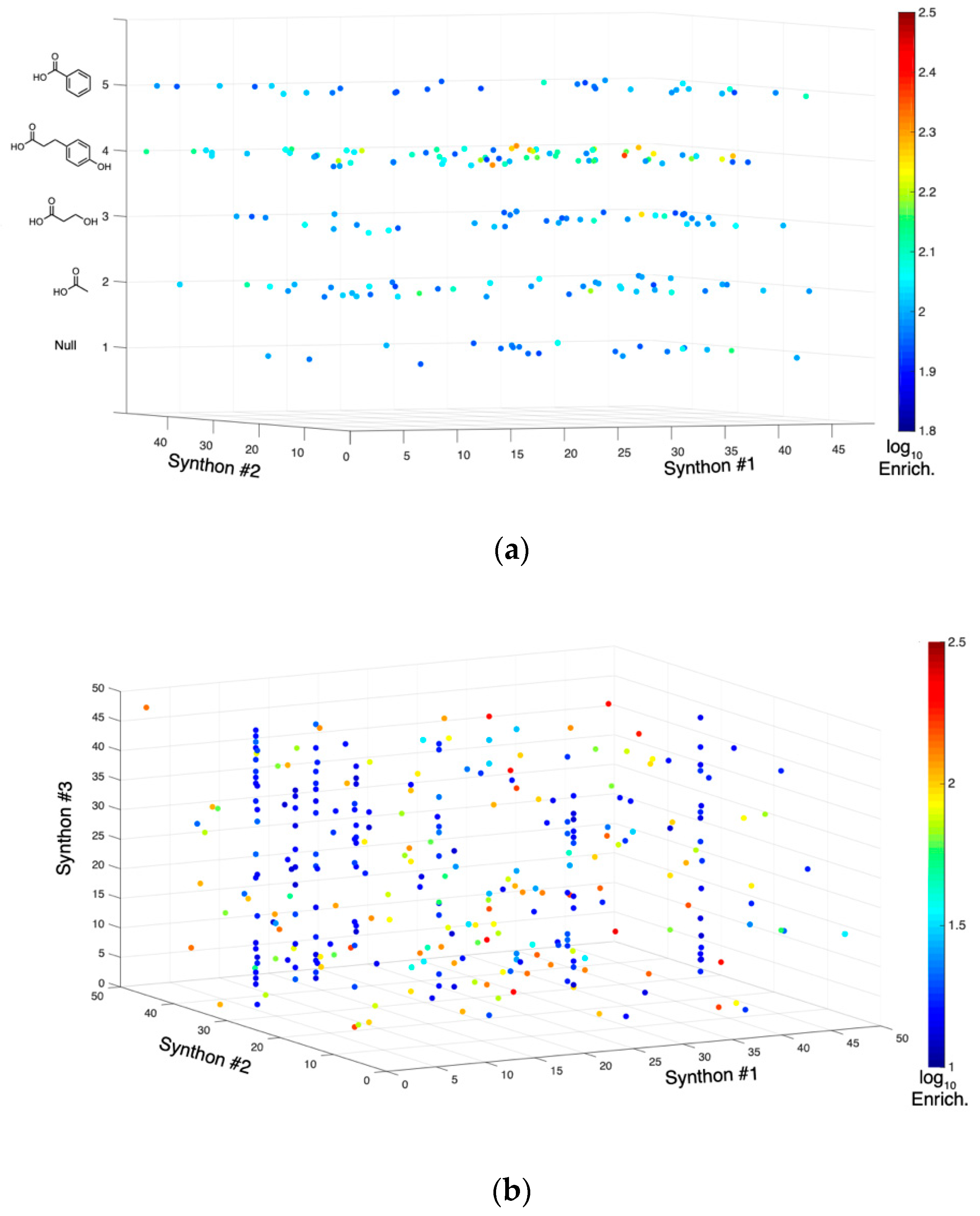
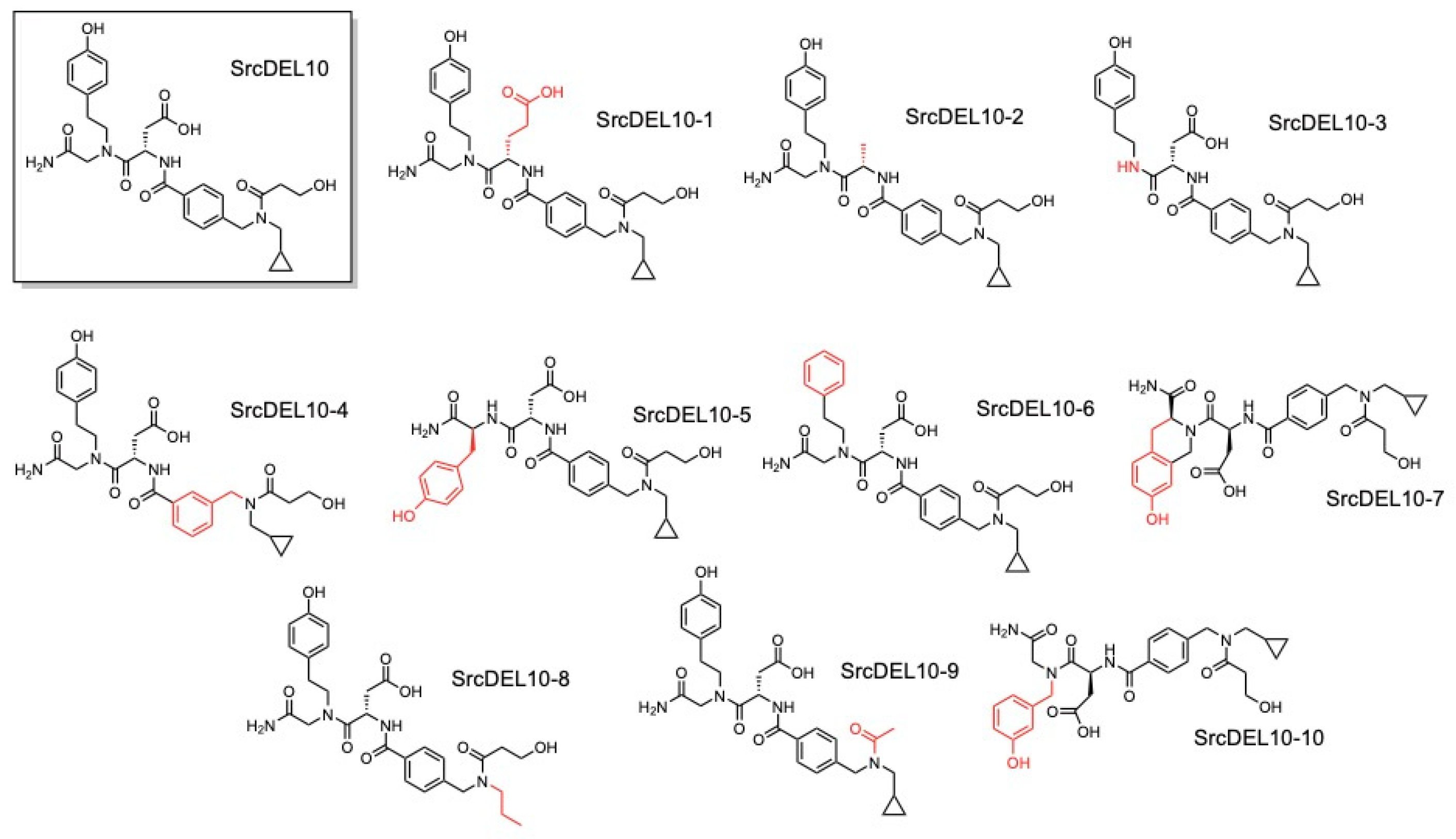
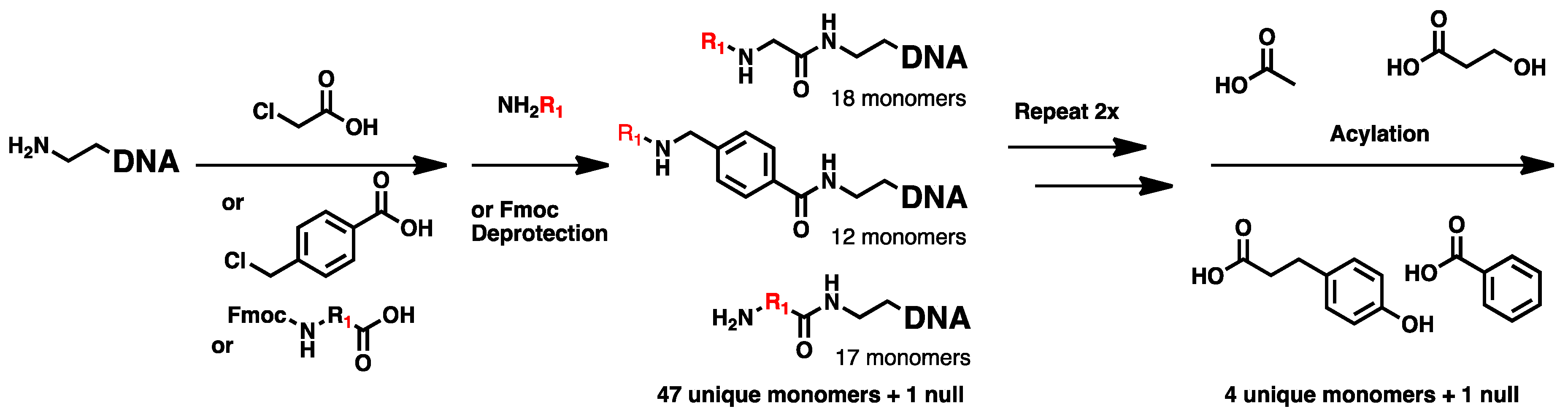
2.1. DNA-Encoded Chemical Library
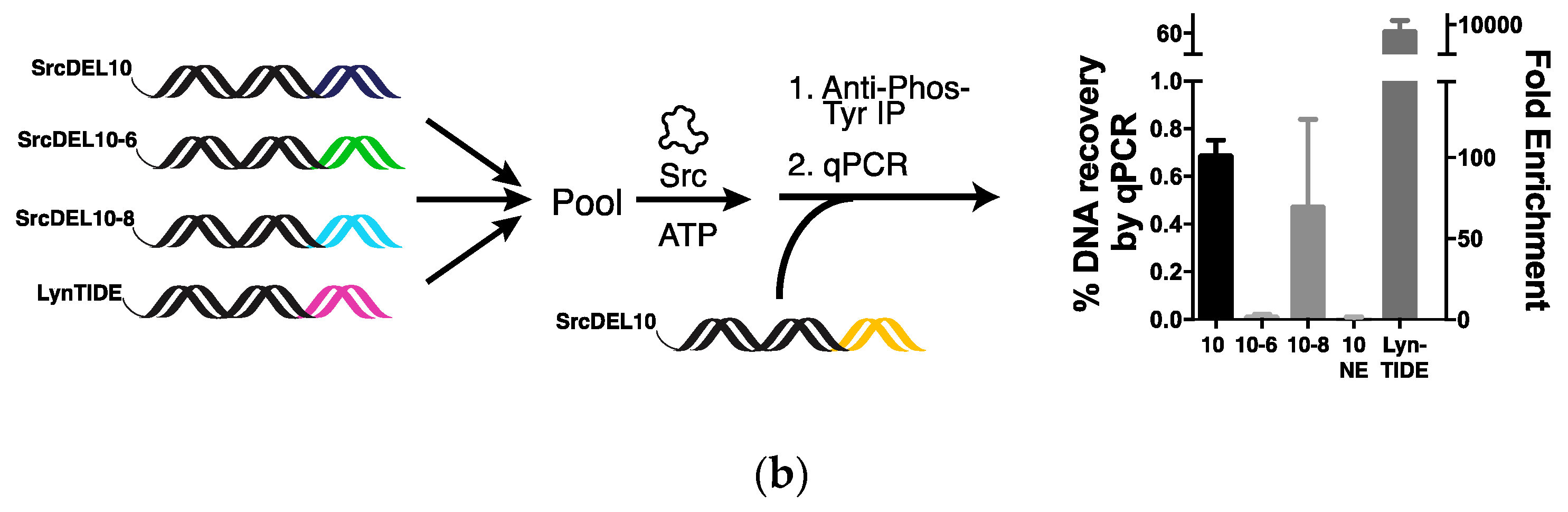
2.2. Tyrosine Kinase Substrate-Mediated Selection
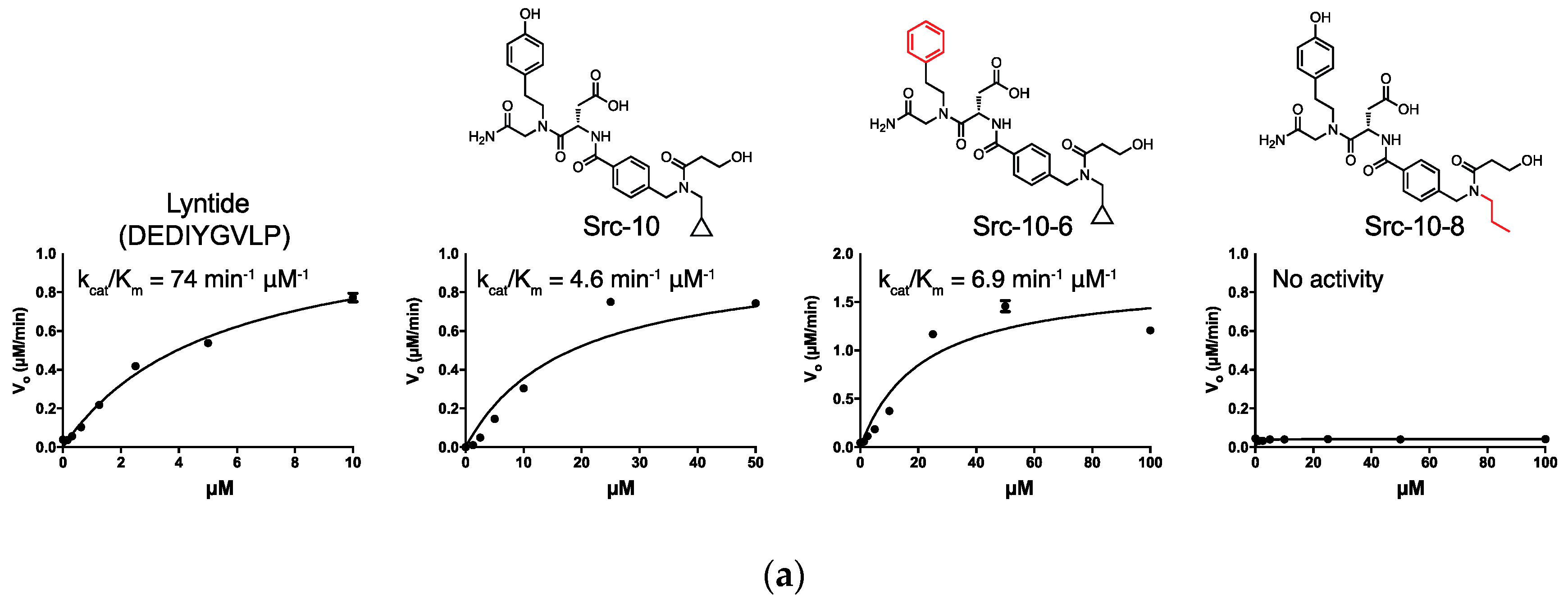
2.3. c-Src Activity and Binding Assays
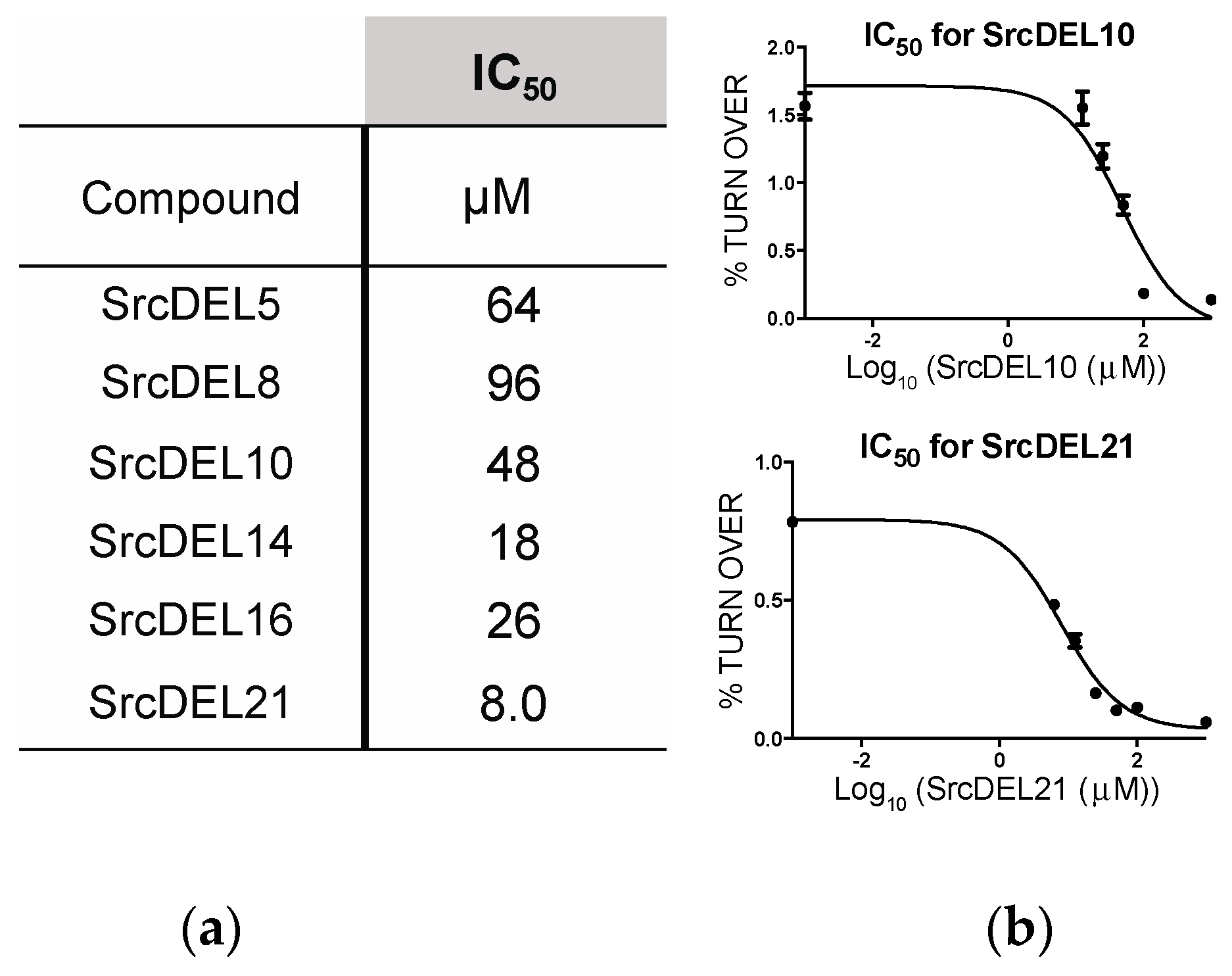
2.4. Src Activity Inhibition
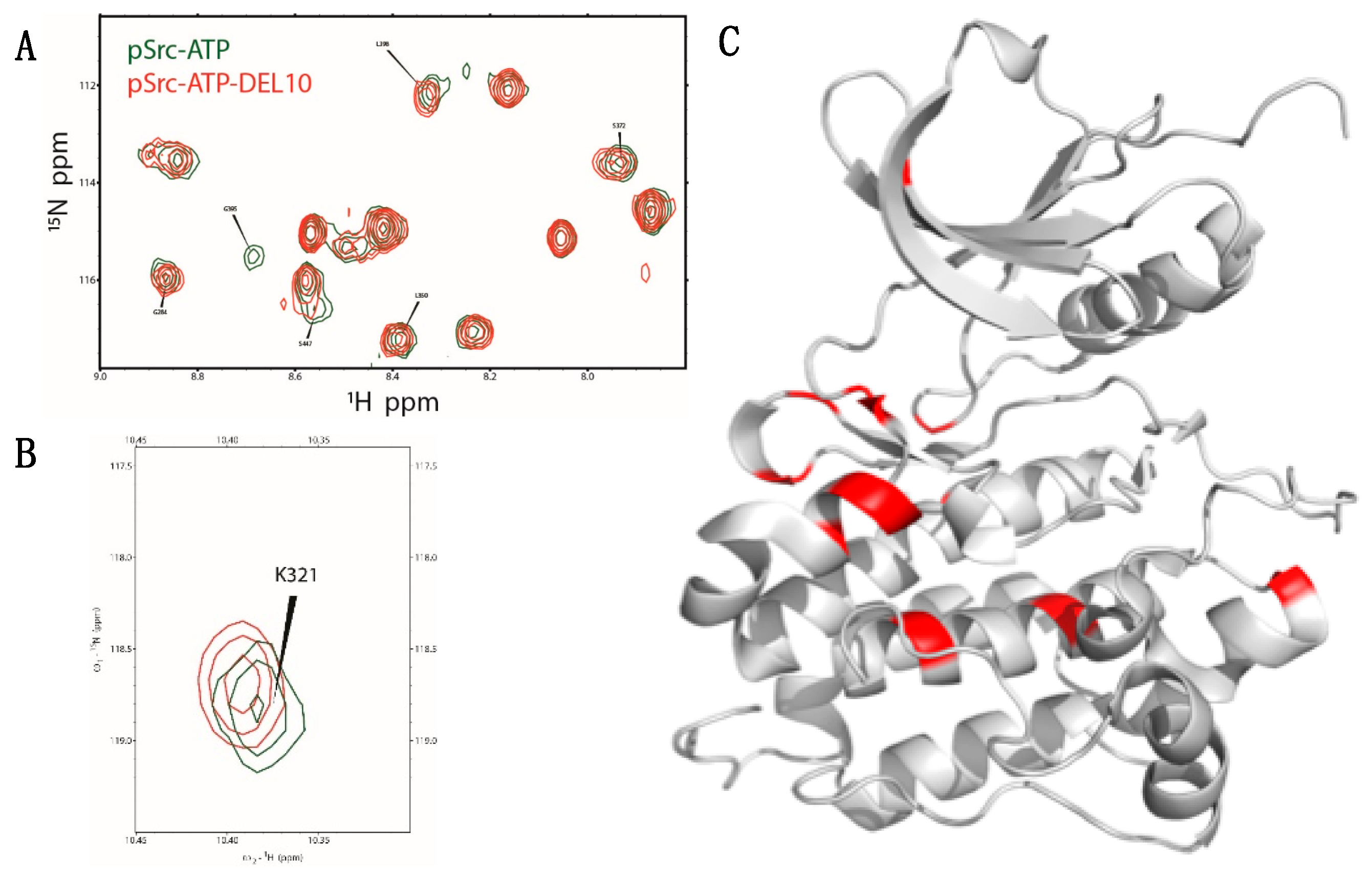
2.5. NMR Analysis
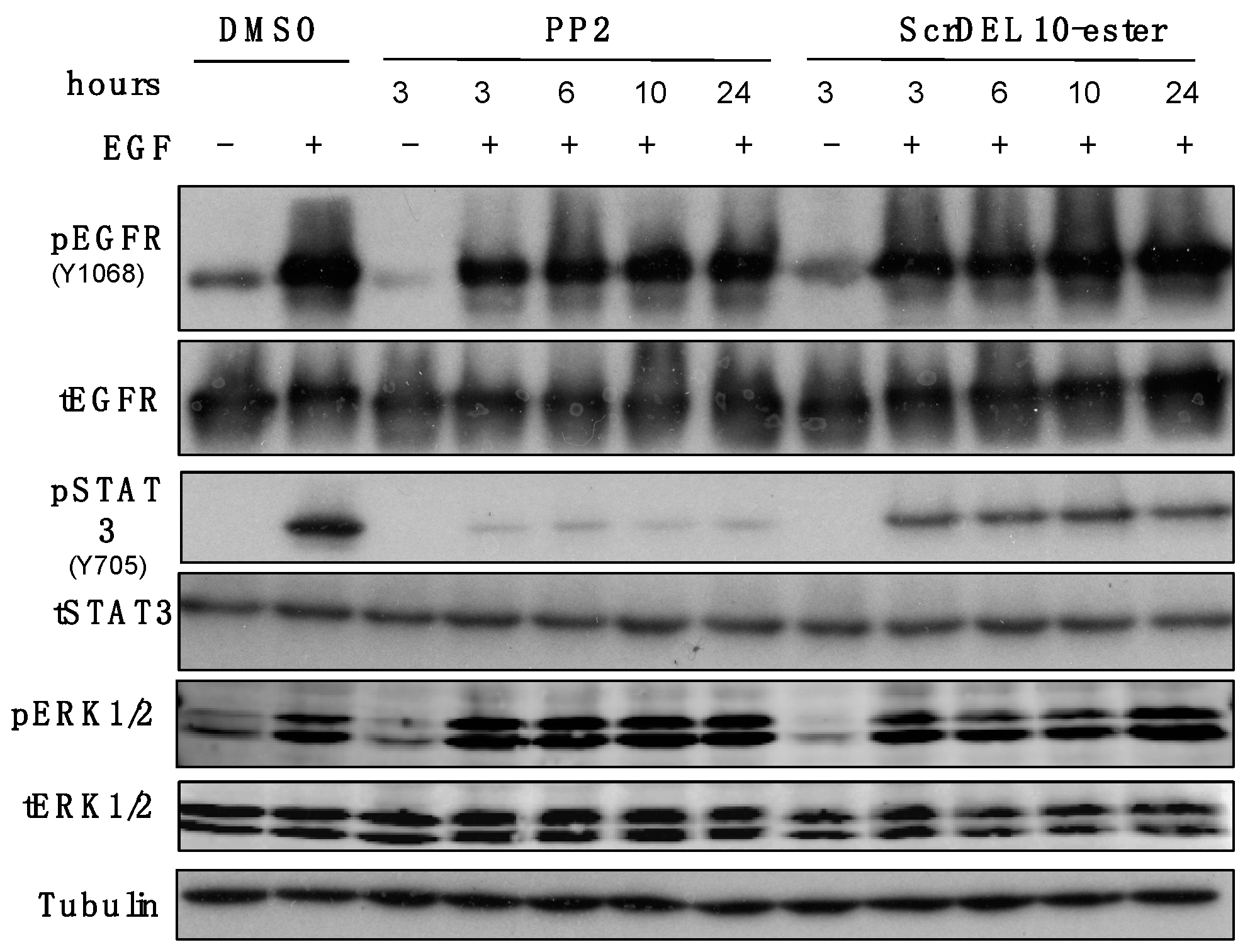
2.6. Activity in Cell Culture
3. Summary Discussion and Conclusion
4. Materials and Methods
4.1. DNA Library Preparation
4.2. Chemical Translation
4.3. Substrate-Mediated Selection for Tyrosine Kinases
4.4. qPCR Assay
4.5. ADP-Glo Assay
4.6. NADH Coupled Assay
4.7. Radiolabeled Assay
4.8. Cell Signaling Assays
4.9. Expression, Purification, and Phosphorylation of c-Src Catalytic Domain
4.10. NMR Experiments
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Goodnow, R.A.; Dumelin, C.E.; Keefe, A.D. DNA-encoded chemistry: Enabling the deeper sampling of chemical space. Nat. Rev. Drug Discov. 2016. [Google Scholar] [CrossRef]
- Neri, D.; Lerner, R.A. DNA-Encoded Chemical Libraries: A Selection System Based on Endowing Organic Compounds with Amplifiable Information. Annu. Rev. Biochem. 2018, 87, 479–502. [Google Scholar] [CrossRef]
- Zimmermann, G.; Neri, D. DNA-encoded chemical libraries: Foundations and applications in lead discovery. Drug Discov. Today 2016, 21, 1828–1834. [Google Scholar] [CrossRef]
- Decurtins, W.; Wichert, M.; Franzini, R.M.; Buller, F.; Stravs, M.A.; Zhang, Y.; Neri, D.; Scheuermann, J. Automated screening for small organic ligands using DNA-encoded chemical libraries. Nat. Protoc. 2016, 11, 764–780. [Google Scholar] [CrossRef]
- Cuozzo, J.W.; Centrella, P.A.; Gikunju, D.; Habeshian, S.; Hupp, C.D.; Keefe, A.D.; Sigel, E.A.; Soutter, H.H.; Thomson, H.A.; Zhang, Y.; et al. Discovery of a Potent BTK Inhibitor with a Novel Binding Mode by Using Parallel Selections with a DNA-Encoded Chemical Library. ChemBioChem 2017, 18, 864–871. [Google Scholar] [CrossRef] [Green Version]
- Cochrane, W.G.; Malone, M.L.; Dang, V.Q.; Cavett, V.; Satz, A.L.; Paegel, B.M. Activity-Based DNA-Encoded Library Screening. ACS Comb. Sci. 2019, 21, 425–435. [Google Scholar] [CrossRef]
- Jetson, R.R.; Krusemark, C.J. Sensing Enzymatic Activity by Exposure and Selection of DNA-Encoded Probes. Angew. Chem. Int. Ed. Engl. 2016, 55, 9562–9566. [Google Scholar] [CrossRef]
- Ratnikov, B.; Cieplak, P.; Smith, J.W. High throughput substrate phage display for protease profiling. Methods Mol. Biol. 2009, 539, 93–114. [Google Scholar]
- Valencia, C.A.; Cotten, S.W.; Dong, B.; Liu, R. mRNA-display-based selections for proteins with desired functions: A protease-substrate case study. Biotechnol. Prog. 2008, 24, 561–569. [Google Scholar] [CrossRef]
- Krusemark, C.J.; Tilmans, N.P.; Brown, P.O.; Harbury, P.B. Directed Chemical Evolution with an Outsized Genetic Code. PLoS ONE 2016, 11, 1–16. [Google Scholar] [CrossRef]
- Barouch-Bentov, R.; Sauer, K. Mechanisms of Drug-Resistance in Kinases. Expert Opin. Investig. Drugs 2011, 20, 153–208. [Google Scholar] [CrossRef] [PubMed]
- Bhullar, K.S.; Lagarón, N.O.; McGowan, E.M.; Parmar, I.; Jha, A.; Hubbard, B.P.; Rupasinghe, H.P.V. Kinase-targeted cancer therapies: Progress, challenges and future directions. Mol. Cancer 2018, 17, 48. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.I.; Hunt, J.P.; Herrgard, S.; Ciceri, P.; Wodicka, L.M.; Pallares, G.; Hocker, M.; Treiber, D.K.; Zarrinkar, P.P. Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2011, 29, 1046–1051. [Google Scholar] [CrossRef] [PubMed]
- Scapin, G. Protein kinase inhibition: Different approaches to selective inhibitor design. Curr. Drug Targets 2006, 7, 1443–1454. [Google Scholar] [CrossRef] [PubMed]
- Breen, M.E.; Soellner, M.B. Small molecule substrate phosphorylation site inhibitors of protein kinases: Approaches and challenges. ACS Chem. Biol. 2015, 10, 175–189. [Google Scholar] [CrossRef]
- Turk, B.E.; Hutti, J.E.; Cantley, L.C. Determining protein kinase substrate specificity by parallel solution-phase assay of large numbers of peptide substrates. Nat. Protoc. 2006, 1, 375–379. [Google Scholar] [CrossRef]
- Brooijmans, N.; Chang, Y.-W.; Mobilio, D.; Denny, R.A.; Humblet, C. An enriched structural kinase database to enable kinome-wide structure-based analyses and drug discovery. Protein Sci. 2010, 19, 763–774. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.; Alicea-Velazquez, N.L.; Bannwarth, L.; Lehtonen, S.I.; Boggon, T.J.; Cheng, H.-C.; Hytonen, V.P.; Turk, B.E. Global analysis of human nonreceptor tyrosine kinase specificity using high-density peptide microarrays. J. Proteome Res. 2014, 13, 4339–4346. [Google Scholar] [CrossRef]
- Al-Obeidi, F.A.; Wu, J.J.; Lam, K.S. Protein tyrosine kinases: Structure, substrate specificity, and drug discovery. Pept. Sci. 1998, 47, 197–223. [Google Scholar] [CrossRef]
- Lou, Q.; Leftwich, M.E.; McKay, R.T.; Salmon, S.E.; Rychetsky, L.; Lam, K.S. Potent Pseudosubstrate-based Peptide Inhibitors for p60c-src Protein Tyrosine Kinase. Cancer Res. 1997, 57, 1877–1881. [Google Scholar]
- Alfaro-Lopez, J.; Yuan, W.; Phan, B.C.; Kamath, J.; Lou, Q.; Lam, K.S.; Hruby, V.J. Discovery of a Novel Series of Potent and Selective Substrate-Based Inhibitors of p60c-src Protein Tyrosine Kinase: Conformational and Topographical Constraints in Peptide Design. J. Med. Chem. 1998, 41, 2252–2260. [Google Scholar] [CrossRef] [PubMed]
- Bogoyevitch, M.A.; Barr, R.K.; Ketterman, A.J. Peptide inhibitors of protein kinases-discovery, characterisation and use. Biochim. Biophys. Acta 2005, 1754, 79–99. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Nandy, S.K.; Lawrence, D.S. A highly potent and selective PKCalpha inhibitor generated via combinatorial modification of a peptide scaffold. J. Am. Chem. Soc. 2004, 126, 3394–3395. [Google Scholar] [CrossRef] [PubMed]
- Breen, M.E.; Steffey, M.E.; Lachacz, E.J.; Kwarcinski, F.E.; Fox, C.C.; Soellner, M.B. Substrate activity screening with kinases: Discovery of small-molecule substrate-competitive c-Src inhibitors. Angew. Chem. Int. Ed. Engl. 2014, 53, 7010–7013. [Google Scholar] [CrossRef] [PubMed]
- Georghiou, G.; Kleiner, R.E.; Pulkoski-Gross, M.; Liu, D.R.; Seeliger, M.A. Highly specific, bisubstrate-competitive Src inhibitors from DNA-templated macrocycles. Nat. Chem. Biol. 2012, 8, 366. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.K.; Wood, E.R. Cell-based assays for kinase drug discovery. Drug Discov. Today. Technol. 2010, 7, e1–e94. [Google Scholar] [CrossRef]
- Folkvord, S.; Flatmark, K.; Dueland, S.; de Wijn, R.; Groholt, K.K.; Hole, K.H.; Nesland, J.M.; Ruijtenbeek, R.; Boender, P.J.; Johansen, M.; et al. Prediction of response to preoperative chemoradiotherapy in rectal cancer by multiplex kinase activity profiling. Int. J. Radiat. Oncol. Biol. Phys. 2010, 78, 555–562. [Google Scholar] [CrossRef]
- Reimer, U.; Reineke, U.; Schneider-Mergener, J. Peptide arrays: From macro to micro. Curr. Opin. Biotechnol. 2002, 13, 315–320. [Google Scholar] [CrossRef]
- Placzek, E.A.; Plebanek, M.P.; Lipchik, A.M.; Kidd, S.R.; Parker, L.L. A peptide biosensor for detecting intracellular Abl kinase activity using matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Anal. Biochem. 2010, 397, 73–78. [Google Scholar] [CrossRef] [Green Version]
- Peterson, L.B.; Imperiali, B. Real-Time and Continuous Sensors of Protein Kinase Activity Utilizing Chelation-Enhanced Fluorescence. Concepts Case Stud. Chem. Biol. 2014, 1–16. [Google Scholar]
- Turner, A.H.; Lebhar, M.S.; Proctor, A.; Wang, Q.; Lawrence, D.S.; Allbritton, N.L. Rational Design of a Dephosphorylation-Resistant Reporter Enables Single-Cell Measurement of Tyrosine Kinase Activity. ACS Chem. Biol. 2016, 11, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Bantscheff, M.; Eberhard, D.; Abraham, Y.; Bastuck, S.; Boesche, M.; Hobson, S.; Mathieson, T.; Perrin, J.; Raida, M.; Rau, C.; et al. Quantitative chemical proteomics reveals mechanisms of action of clinical ABL kinase inhibitors. Nat. Biotechnol. 2007, 25, 1035–1044. [Google Scholar] [CrossRef] [PubMed]
- Patricelli, M.P.; Szardenings, A.K.; Liyanage, M.; Nomanbhoy, T.K.; Wu, M.; Weissig, H.; Aban, A.; Chun, D.; Tanner, S.; Kozarich, J.W. Functional interrogation of the kinome using nucleotide acyl phosphates. Biochemistry 2007, 46, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Halpin, D.R.; Harbury, P.B. DNA display I. Sequence-encoded routing of DNA populations. PLoS Biol. 2004, 2, E173. [Google Scholar] [CrossRef] [PubMed]
- Halpin, D.R.; Harbury, P.B. DNA display II. Genetic manipulation of combinatorial chemistry libraries for small-molecule evolution. PLoS Biol. 2004, 2, E174. [Google Scholar] [CrossRef]
- Simon, R.J.; Kania, R.S.; Zuckermann, R.N.; Huebner, V.D.; Jewell, D.A.; Banville, S.; Ng, S.; Wang, L.; Rosenberg, S.; Marlowe, C.K. Peptoids: A modular approach to drug discovery. Proc. Natl. Acad. Sci. USA 1992, 89, 9367–9371. [Google Scholar] [CrossRef] [PubMed]
- Combs, D.J.; Lokey, R.S. Extended peptoids: A new class of oligomers based on aromatic building blocks. Tetrahedron Lett. 2007, 48, 2679–2682. [Google Scholar] [CrossRef]
- Halpin, D.R.; Lee, J.A.; Wrenn, S.J.; Harbury, P.B. DNA display III. Solid-phase organic synthesis on unprotected DNA. PLoS Biol. 2004, 2, E175. [Google Scholar] [CrossRef]
- Wrenn, S.J.; Weisinger, R.M.; Halpin, D.R.; Harbury, P.B. Synthetic ligands discovered by in vitro selection. J. Am. Chem. Soc. 2007, 129, 13137–13143. [Google Scholar] [CrossRef]
- Schwochert, J.; Turner, R.; Thang, M.; Berkeley, R.F.; Ponkey, A.R.; Rodriguez, K.M.; Leung, S.S.F.; Khunte, B.; Goetz, G.; Limberakis, C.; et al. Peptide to Peptoid Substitutions Increase Cell Permeability in Cyclic Hexapeptides. Org. Lett. 2015, 17, 2928–2931. [Google Scholar] [CrossRef]
- Tan, N.C.; Yu, P.; Kwon, Y.-U.; Kodadek, T. High-throughput evaluation of relative cell permeability between peptoids and peptides. Bioorg. Med. Chem. 2008, 16, 5853–5861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akritopoulou-Zanze, I.; Hajduk, P.J. Kinase-targeted libraries: The design and synthesis of novel, potent, and selective kinase inhibitors. Drug Discov. Today 2009, 14, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Mincione, F.; Starnotti, M.; Menabuoni, L.; Scozzafava, A.; Casini, A.; Supuran, C.T. Carbonic anhydrase inhibitors: 4-sulfamoyl-benzenecarboxamides and 4-chloro-3-sulfamoyl-benzenecarboxamides with strong topical antiglaucoma properties. Bioorg. Med. Chem. Lett. 2001, 11, 1787–1791. [Google Scholar] [CrossRef]
- Tinti, M.; Nardozza, A.P.; Ferrari, E.; Sacco, F.; Corallino, S.; Castagnoli, L.; Cesareni, G. The 4G10, pY20 and p-TYR-100 antibody specificity: Profiling by peptide microarrays. N. Biotechnol. 2012, 29, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, R.; Baumann, G.; Gram, H. Catalytic specificity of phosphotyrosine kinases Blk, Lyn, c-Src and Syk as assessed by phage display. J. Mol. Biol. 1996, 260, 664–677. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.R.; Niu, J.; Lawrence, D.S. The Extraordinary Active Site Substrate Specificity of pp60c−src: A multiple specificity protein kinase. J. Biol. Chem. 1995, 270, 5375–5380. [Google Scholar] [CrossRef] [PubMed]
- Glenney, J.R.; Zokas, L. Novel tyrosine kinase substrates from Rous sarcoma virus-transformed cells are present in the membrane skeleton. J. Cell Biol. 1989, 108, 2401–2408. [Google Scholar] [CrossRef] [PubMed]
- Cabail, M.Z.; Chen, E.I.; Koller, A.; Miller, W.T. Auto-thiophosphorylation activity of Src tyrosine kinase. BMC Biochem. 2016, 17, 13. [Google Scholar] [CrossRef] [PubMed]
- Lorsch, J.R.; Herschlag, D. The DEAD Box Protein eIF4A. 1. A Minimal Kinetic and Thermodynamic Framework Reveals Coupled Binding of RNA and Nucleotide. Biochemistry 1998, 37, 2180–2193. [Google Scholar] [CrossRef] [PubMed]
- Kuai, L.; O’Keeffe, T.; Arico-Muendel, C. Randomness in DNA Encoded Library Selection Data Can Be Modeled for More Reliable Enrichment Calculation. SLAS Discov. Adv. Life Sci. 2018, 23, 405–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmermann, G.; Li, Y.; Rieder, U.; Mattarella, M.; Neri, D.; Scheuermann, J. Hit-Validation Methodologies for Ligands Isolated from DNA-Encoded Chemical Libraries. Chembiochem 2017, 18, 853–857. [Google Scholar] [CrossRef] [PubMed]
- Barker, S.; Kassel, D.B.; Weigl, D.; Huang, X.; Luther, M.; Knight, W.B. Characterization of pp60c-src Tyrosine Kinase Activities Using a Continuous Assay: Autoactivation of the Enzyme Is an Intermolecular Autophosphorylation Process. Biochemistry 1995, 34, 14843–14851. [Google Scholar] [CrossRef] [PubMed]
- Masterson, L.R.; Mascioni, A.; Traaseth, N.J.; Taylor, S.S.; Veglia, G. Allosteric cooperativity in protein kinase A. Proc. Natl. Acad. Sci. USA 2008, 105, 506–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogtherr, M.; Saxena, K.; Hoelder, S.; Grimme, S.; Betz, M.; Schieborr, U.; Pescatore, B.; Robin, M.; Delarbre, L.; Langer, T.; et al. NMR Characterization of Kinase p38 Dynamics in Free and Ligand-Bound Forms. Angew. Chemie Int. Ed. 2006, 45, 993–997. [Google Scholar] [CrossRef] [PubMed]
- Klein, T.; Vajpai, N.; Phillips, J.J.; Davies, G.; Holdgate, G.A.; Phillips, C.; Tucker, J.A.; Norman, R.A.; Scott, A.D.; Higazi, D.R.; et al. Structural and dynamic insights into the energetics of activation loop rearrangement in FGFR1 kinase. Nat. Commun. 2015, 6, 7877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pucheta-Martínez, E.; Saladino, G.; Morando, M.A.; Martinez-Torrecuadrada, J.; Lelli, M.; Sutto, L.; D’Amelio, N.; Gervasio, F.L. An Allosteric Cross-Talk Between the Activation Loop and the ATP Binding Site Regulates the Activation of Src Kinase. Sci. Rep. 2016, 6, 24235. [Google Scholar] [CrossRef] [PubMed]
- Tong, M.; Pelton, J.G.; Gill, M.L.; Zhang, W.; Picart, F.; Seeliger, M.A. Survey of solution dynamics in Src kinase reveals allosteric cross talk between the ligand binding and regulatory sites. Nat. Commun. 2017, 8, 2160. [Google Scholar] [CrossRef] [PubMed]
- Cowan-Jacob, S.W.; Fendrich, G.; Manley, P.W.; Jahnke, W.; Fabbro, D.; Liebetanz, J.; Meyer, T. The Crystal Structure of a c-Src Complex in an Active Conformation Suggests Possible Steps in c-Src Activation. Structure 2005, 13, 861–871. [Google Scholar] [CrossRef] [Green Version]
- Wendt, M.K.; Smith, J.A.; Schiemann, W.P. Transforming growth factor-beta-induced epithelial-mesenchymal transition facilitates epidermal growth factor-dependent breast cancer progression. Oncogene 2010, 29, 6485–6498. [Google Scholar] [CrossRef]
- Balanis, N.; Wendt, M.K.; Schiemann, B.J.; Wang, Z.; Schiemann, W.P.; Carlin, C.R. Epithelial to mesenchymal transition promotes breast cancer progression via a fibronectin-dependent STAT3 signaling pathway. J. Biol. Chem. 2013, 288, 17954–17967. [Google Scholar] [CrossRef]
- Armstrong, R.N.; Kondo, H.; Kaiser, E.T. Cyclic AMP-dependent ATPase activity of bovine heart protein kinase. Proc. Natl. Acad. Sci. USA 1979, 76, 722–725. [Google Scholar] [CrossRef] [PubMed]
- Paudel, H.K.; Carlson, G.M. The ATPase activity of phosphorylase kinase is regulated in parallel with its protein kinase activity. J. Biol. Chem. 1991, 266, 16524–16529. [Google Scholar] [PubMed]
- Denton, K.E.; Krusemark, C.J. Crosslinking of DNA-linked ligands to target proteins for enrichment from DNA-encoded libraries. Med. Chem. Commun. 2016, 7, 2020–2027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeAngelis, M.M.; Wang, D.G.; Hawkins, T.L. Solid-phase reversible immobilization for the isolation of PCR products. Nucleic Acids Res. 1995, 23, 4742–4743. [Google Scholar] [CrossRef] [PubMed]
- Satz, A.L.; Cai, J.; Chen, Y.; Goodnow, R.; Gruber, F.; Kowalczyk, A.; Petersen, A.; Naderi-Oboodi, G.; Orzechowski, L.; Strebel, Q. DNA Compatible Multistep Synthesis and Applications to DNA Encoded Libraries. Bioconjug. Chem. 2015, 26, 1623–1632. [Google Scholar] [CrossRef] [PubMed]
- Denton, K.E.; Wang, S.; Gignac, M.C.; Milosevich, N.; Hof, F.; Dykhuizen, E.C.; Krusemark, C.J. Robustness of In Vitro Selection Assays of DNA-Encoded Peptidomimetic Ligands to CBX7 and CBX8. SLAS Discov. 2018, 23. [Google Scholar] [CrossRef]
- Kim, D.; Jetson, R.R.; Krusemark, C.J. A DNA-assisted immunoassay for enzyme activity via a DNA-linked, activity-based probe. Chem. Commun. 2017, 53, 9474–9477. [Google Scholar] [CrossRef] [PubMed]
- Seeliger, M.A.; Young, M.; Henderson, M.N.; Pellicena, P.; King, D.S.; Falick, A.M.; Kuriyan, J. High yield bacterial expression of active c-Abl and c-Src tyrosine kinases. Protein Sci. 2005, 14, 3135–3139. [Google Scholar] [CrossRef] [Green Version]
Sample Availability: Samples of all off-DNA synthesized compounds are available from the authors. |











{kind=link}
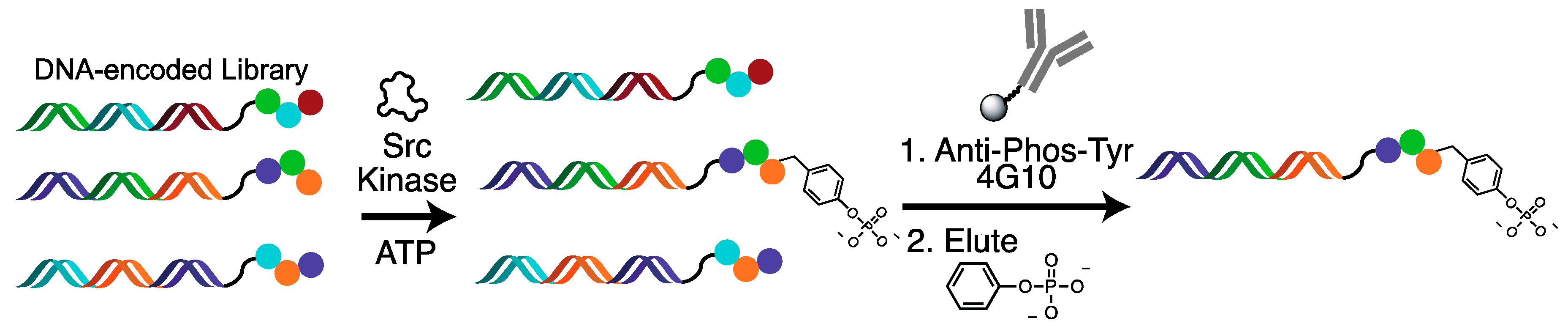{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Km | kcat | kcat/Km |  | |
| Compound | (µM) | (min−1) | (min−1 µM−1) | |
| SrcDEL5 | 33 | 350 | 11 | |
| SrcDEL8 | 19 | 150 | 7.8 | |
| SrcDEL10 | 18 | 82 | 4.6 | |
| SrcDEL14 | 13 | 45 | 3.3 | |
| SrcDEL16 | 18 | 32 | 1.8 | |
| SrcDEL21 | 28 | 86 | 3.0 | |
| LYNtide (DEDIYGVLP) | 5.2 | 390 | 74 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, D.; Sun, Y.; Xie, D.; Denton, K.E.; Chen, H.; Lin, H.; Wendt, M.K.; Post, C.B.; Krusemark, C.J. Application of a Substrate-Mediated Selection with c-Src Tyrosine Kinase to a DNA-Encoded Chemical Library. Molecules 2019, 24, 2764. https://doi.org/10.3390/molecules24152764
Kim D, Sun Y, Xie D, Denton KE, Chen H, Lin H, Wendt MK, Post CB, Krusemark CJ. Application of a Substrate-Mediated Selection with c-Src Tyrosine Kinase to a DNA-Encoded Chemical Library. Molecules. 2019; 24(15):2764. https://doi.org/10.3390/molecules24152764
Chicago/Turabian StyleKim, Dongwook, Yixing Sun, Dan Xie, Kyle E. Denton, Hao Chen, Hang Lin, Michael K. Wendt, Carol Beth Post, and Casey J. Krusemark. 2019. "Application of a Substrate-Mediated Selection with c-Src Tyrosine Kinase to a DNA-Encoded Chemical Library" Molecules 24, no. 15: 2764. https://doi.org/10.3390/molecules24152764