Ginsenoside Re Inhibits ROS/ASK-1 Dependent Mitochondrial Apoptosis Pathway and Activation of Nrf2-Antioxidant Response in Beta-Amyloid-Challenged SH-SY5Y Cells

Abstract
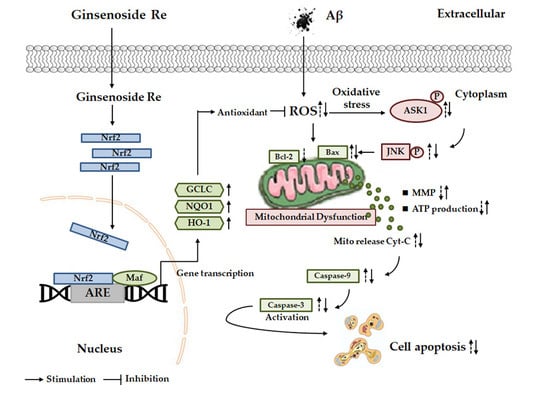
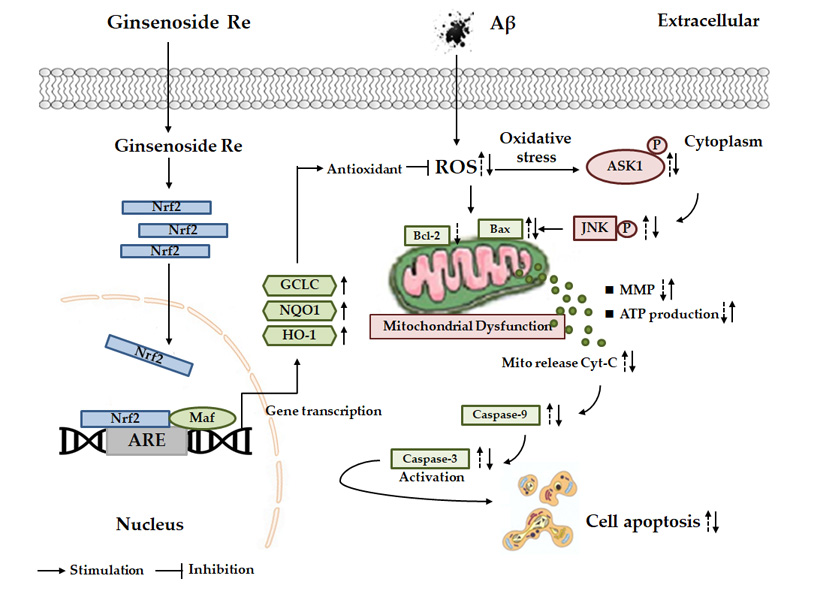
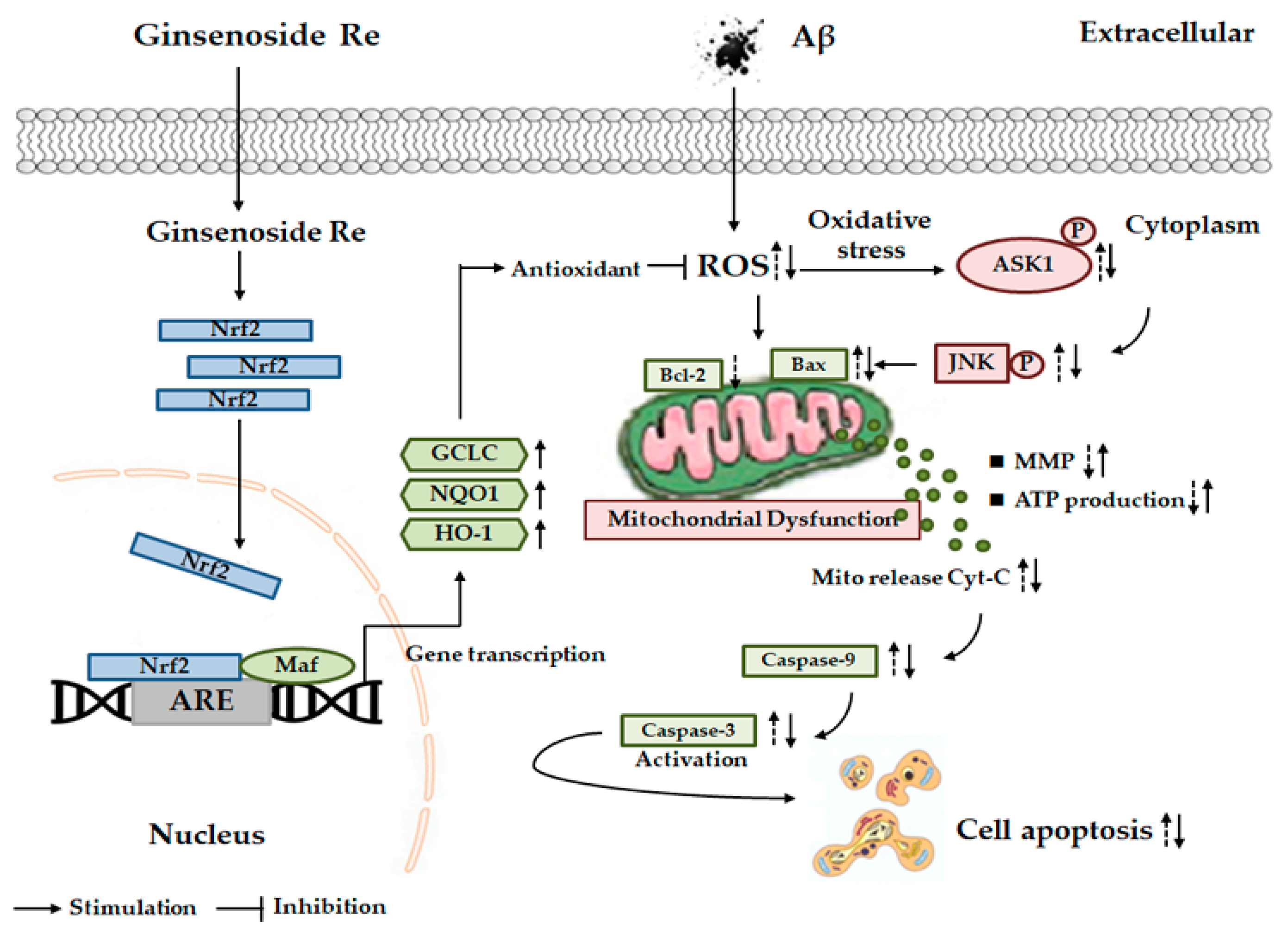
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
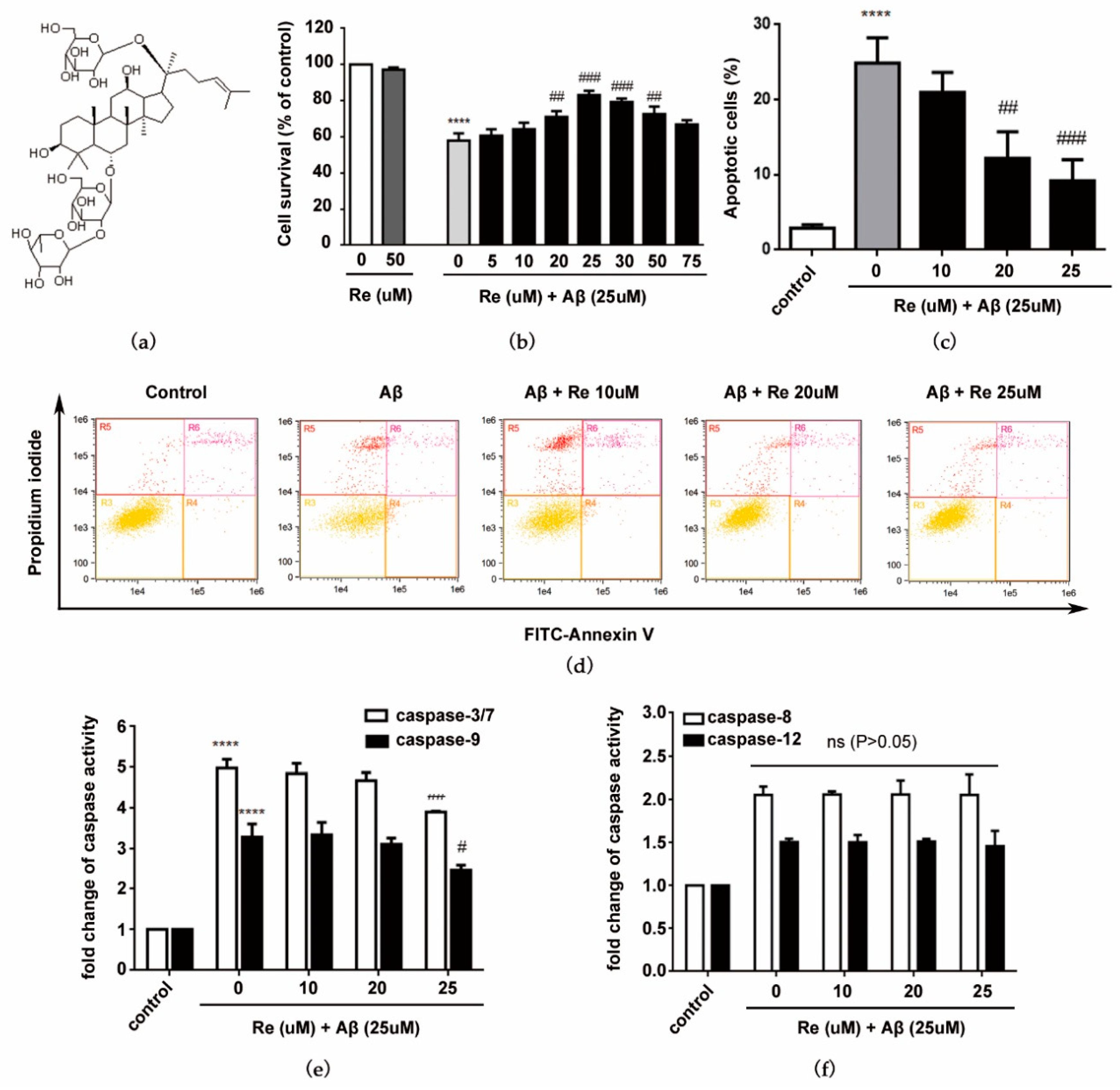
2.1. Ginsenoside Re Protected SH-SY5Y Cells against Aβ25–35-Induced Cytotoxicity
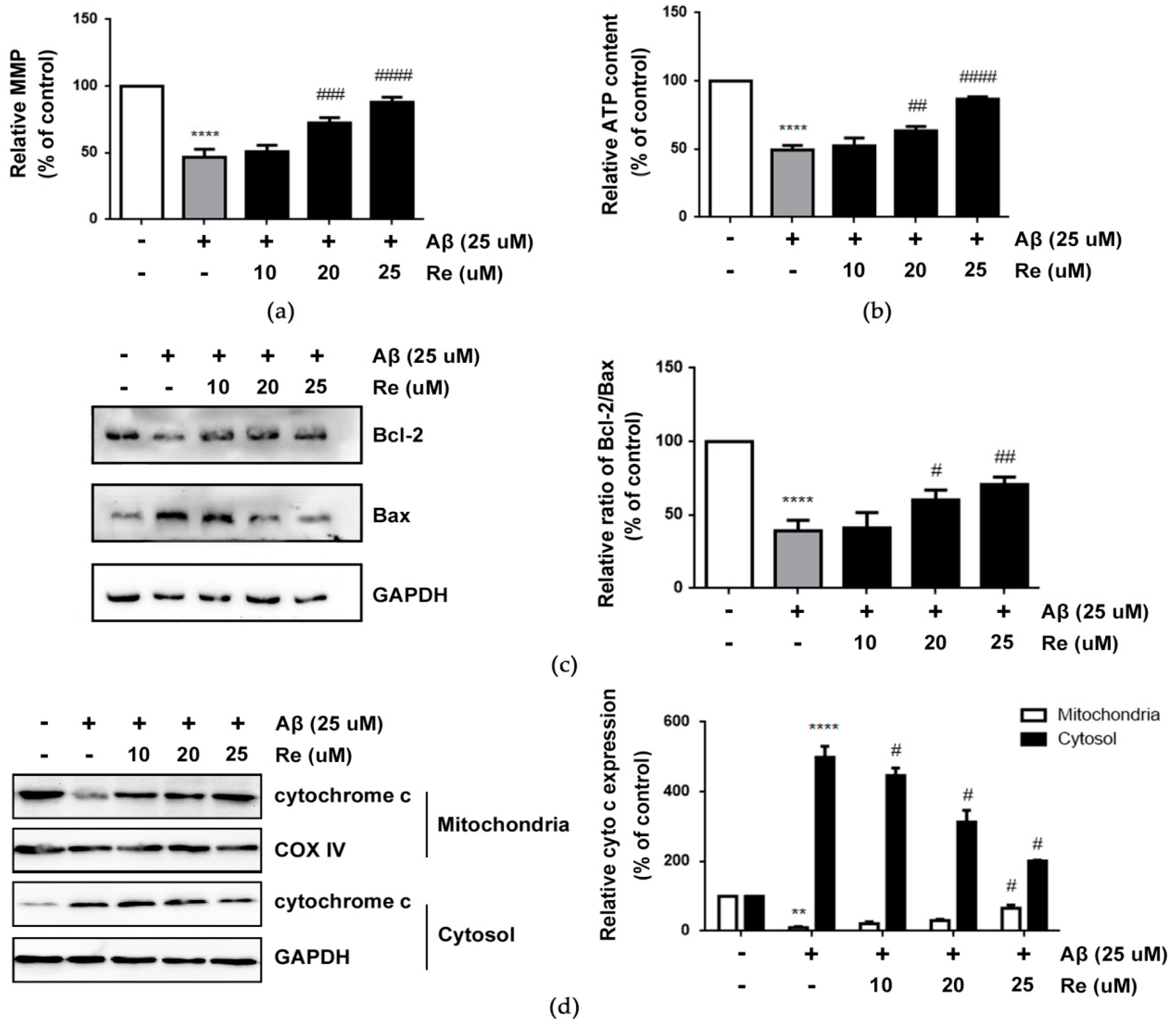
2.2. Ginsenoside Re Alleviated Mitochondrial Dysfunction and Prevented the Mitochondria-Mediated Apoptotic Pathway in SH-SY5Y Cells after Aβ25–35 Exposure
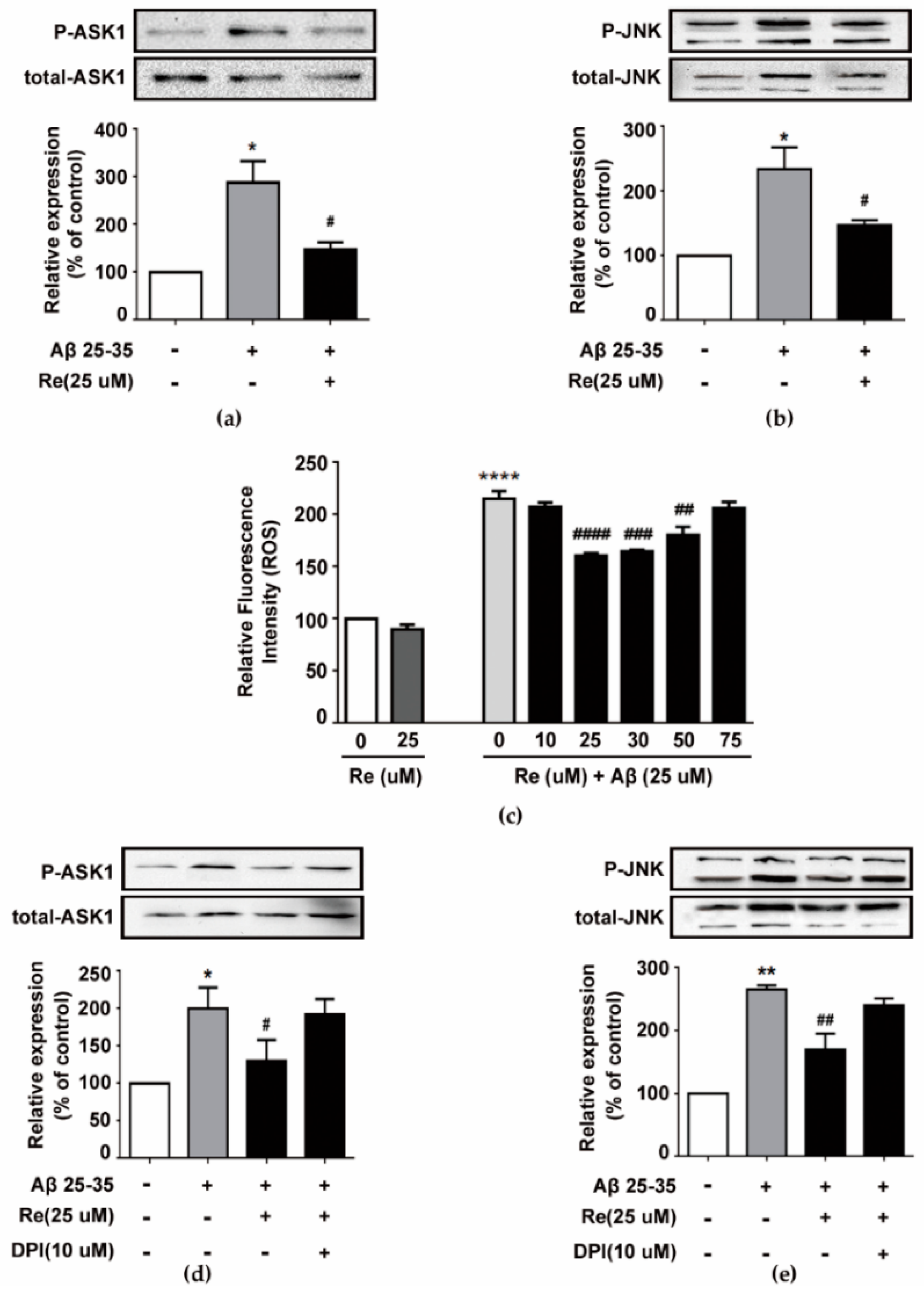
2.3. Ginsenoside Re Inhibited Aβ-induced ASK-1/JNK Activation in a ROS-Dependent Manner
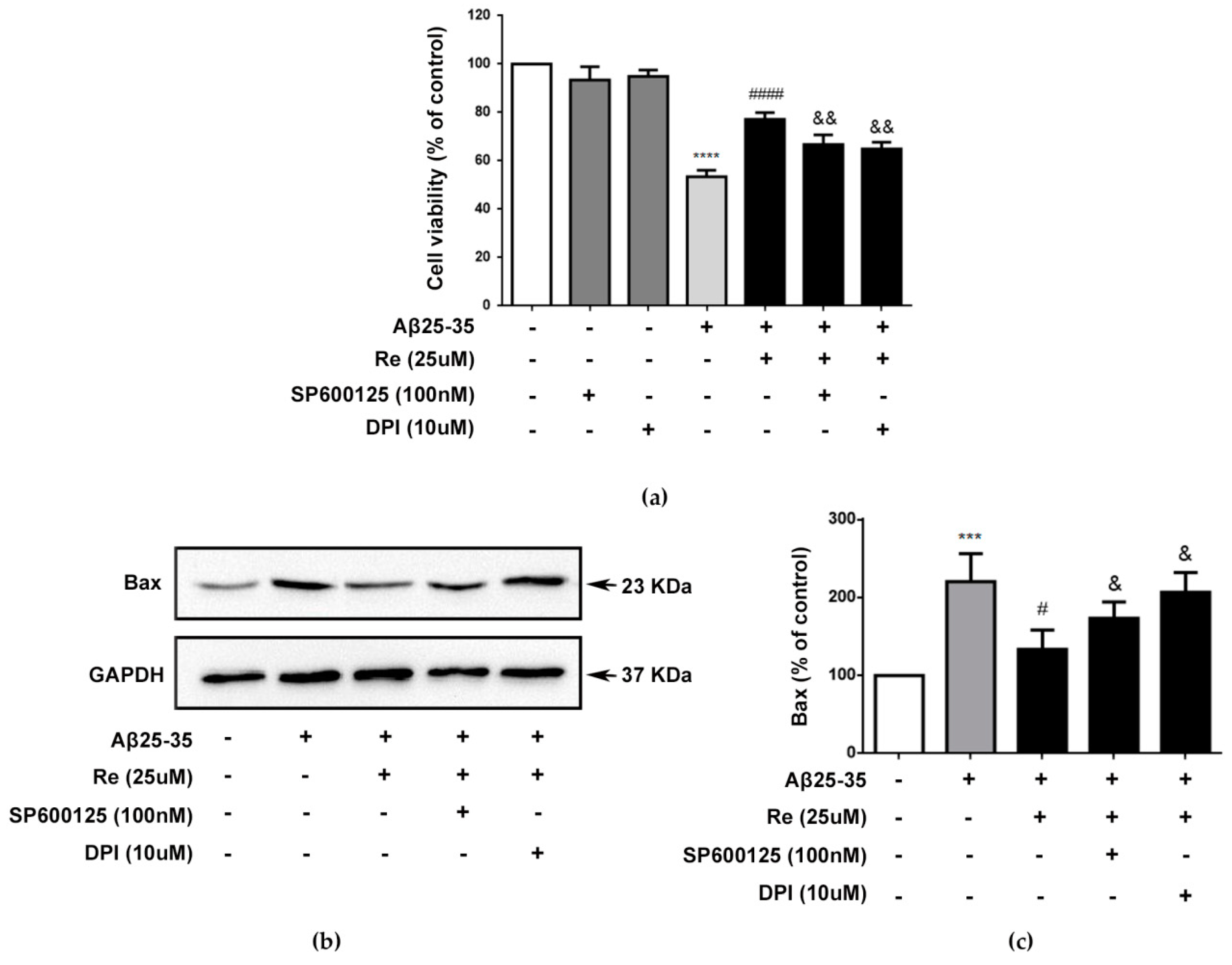
2.4. Inhibition of ROS-Dependent ASK-1/JNK Signaling Attenuated the Neuroprotective Effects of Ginsenoside Re
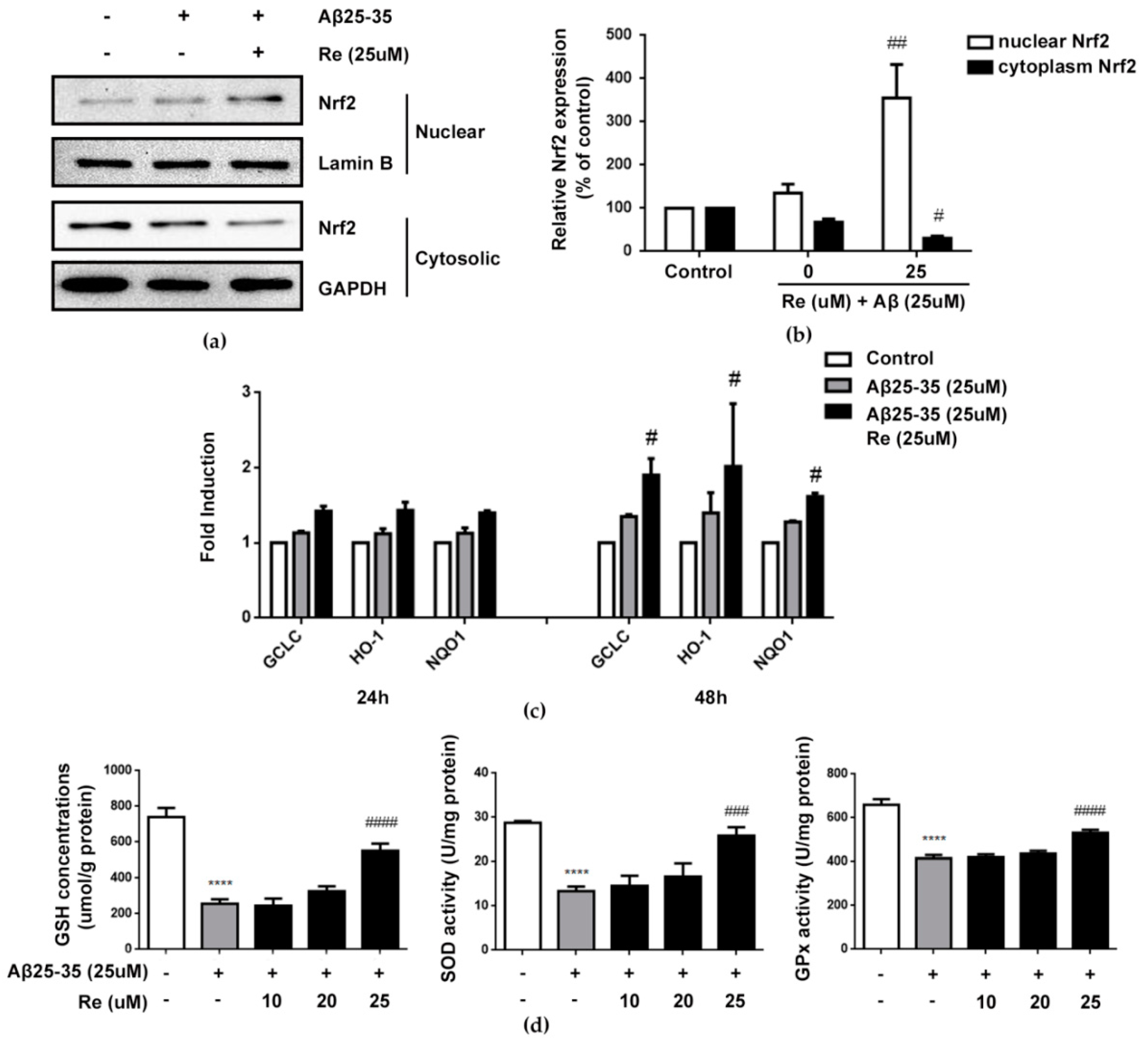
2.5. Ginsenoside Re Attenuated Aβ-Induced Cellular Oxidative Stress by Activating Nrf2-Antioxidant Signaling
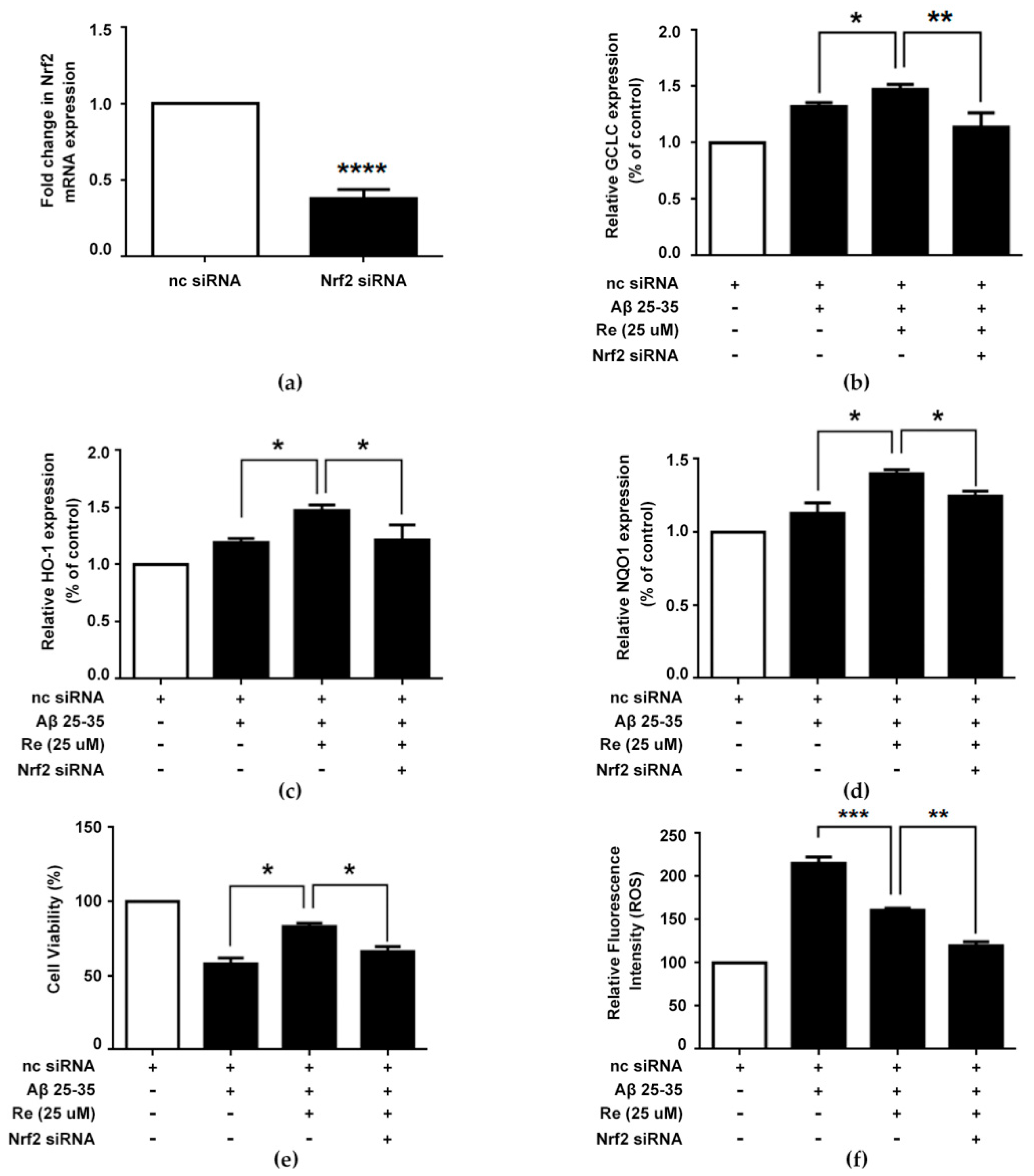
2.6. Ginsenoside Re Alleviated Aβ-Induced Cytotoxicity and ROS Generation in SH-SY5Y Cells in a Nrf2-Dependent Manner
3. Discussion
4. Materials and Methods
4.1. Drug Preparation
4.2. Cell Culture
4.3. Analysis of Cell Viability
4.4. Inhibitor Treatment and siRNA Transfection
4.5. Cell Apoptosis Analysis
4.6. Caspase Activity Assay
4.7. Mitochondrial Membrane Potential Analysis
4.8. Measurement of Cellular ATP Content
4.9. Intracellular ROS Generation Detection
4.10. Oxidative Stress Assays
4.11. Reverse-Transcription and Real-Time PCR
4.12. Protein Extraction and Western Blotting
4.13. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Scheltens, P.; Blennow, K.; Breteler, M.M.B.; de Strooper, B.; Frisoni, G.B.; Salloway, S.; Van der Flier, W.M. Alzheimer’s disease. Lancet 2016, 388, 505–517. [Google Scholar] [CrossRef]
- Shah, H.; Albanese, E.; Duggan, C.; Rudan, I.; Langa, K.M.; Carrillo, M.C.; Chan, K.Y.; Joanette, Y.; Prince, M.; Rossor, M.; et al. Research priorities to reduce the global burden of dementia by 2025. Lancet Neurol. 2016, 15, 1285–1294. [Google Scholar] [CrossRef]
- Busciglio, J.; Lorenzo, A.; Yeh, J.; Yankner, B.A. Beta-amyloid fibrils induce tau phosphorylation and loss of microtubule binding. Neuron 1995, 14, 879–888. [Google Scholar] [CrossRef]
- Sciacca, M.F.M.; Tempra, C.; Scollo, F.; Milardi, D.; La Rosa, C. Amyloid growth and membrane damage: Current themes and emerging perspectives from theory and experiments on Abeta and hIAPP. Biochim. Biophys. Acta Biomembr. 2018, 1860, 1625–1638. [Google Scholar] [CrossRef] [PubMed]
- Grasso, G.; Santoro, A.M.; Lanza, V.; Sbardella, D.; Tundo, G.R.; Ciaccio, C.; Marini, S.; Coletta, M.; Milardi, D. The double faced role of copper in Aβ homeostasis: A survey on the interrelationship between metal dyshomeostasis, UPS functioning and autophagy in neurodegeneration. Coord. Chem. Rev. 2017, 347, 1–22. [Google Scholar] [CrossRef]
- Sevigny, J.; Chiao, P.; Bussiere, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Abeta plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Bloom, G.S. Amyloid-beta and tau: The trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 2014, 71, 505–508. [Google Scholar] [CrossRef]
- Barage, S.H.; Sonawane, K.D. Amyloid cascade hypothesis: Pathogenesis and therapeutic strategies in Alzheimer’s disease. Neuropeptides 2015, 52, 1–18. [Google Scholar] [CrossRef]
- Xu, T.; Niu, C.; Zhang, X.; Dong, M. beta-Ecdysterone protects SH-SY5Y cells against beta-amyloid-induced apoptosis via c-Jun N-terminal kinase- and Akt-associated complementary pathways. Lab. Investig. J. Tech. Methods Pathol. 2018, 98, 489–499. [Google Scholar] [CrossRef]
- Radi, E.; Formichi, P.; Battisti, C.; Federico, A. Apoptosis and oxidative stress in neurodegenerative diseases. J. Alzheimers Dis. JAD 2014, 42 (Suppl. S3), S125–S152. [Google Scholar] [CrossRef]
- Mufson, E.J.; He, B.; Nadeem, M.; Perez, S.E.; Counts, S.E.; Leurgans, S.; Fritz, J.; Lah, J.; Ginsberg, S.D.; Wuu, J.; et al. Hippocampal proNGF signaling pathways and beta-amyloid levels in mild cognitive impairment and Alzheimer disease. J. Neuropathol. Exp. Neurol. 2012, 71, 1018–1029. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, C.; Xing, G.; Zhou, L.; Dong, M.; Geng, Y.; Li, X.; Li, J.; Wang, G.; Zou, D.; et al. Beta-asarone attenuates neuronal apoptosis induced by Beta amyloid in rat hippocampus. Yakugaku Zasshi J. Pharm. Soc. Jpn. 2010, 130, 737–746. [Google Scholar] [CrossRef]
- Zhou, Y.; Xie, N.; Li, L.; Zou, Y.; Zhang, X.; Dong, M. Puerarin alleviates cognitive impairment and oxidative stress in APP/PS1 transgenic mice. Int. J. Neuropsychopharmacol. 2014, 17, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, M.; Coppede, F.; Murri, L.; Siciliano, G. Mitochondrial cascade hypothesis of Alzheimer’s disease: Myth or reality? Antioxid. Redox Signal. 2007, 9, 1631–1646. [Google Scholar] [CrossRef] [PubMed]
- Bosetti, F.; Brizzi, F.; Barogi, S.; Mancuso, M.; Siciliano, G.; Tendi, E.A.; Murri, L.; Rapoport, S.I.; Solaini, G. Cytochrome c oxidase and mitochondrial F1F0-ATPase (ATP synthase) activities in platelets and brain from patients with Alzheimer’s disease. Neurobiol. Aging 2002, 23, 371–376. [Google Scholar] [CrossRef]
- Devi, L.; Prabhu, B.M.; Galati, D.F.; Avadhani, N.G.; Anandatheerthavarada, H.K. Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer’s disease brain is associated with mitochondrial dysfunction. J. Neurosci. Off. J. Soc. Neurosci. 2006, 26, 9057–9068. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Oh, Y.; Kim, K.; Jeong, S.; Chon, S.; Kim, D.; Jung, M.H.; Pak, Y.K.; Ha, J.; Kang, I.; et al. Cyclophilin A regulates JNK/p38-MAPK signaling through its physical interaction with ASK1. Biochem. Biophys. Res. Commun. 2015, 464, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Qiu, C.; Wang, Y.P.; Pan, X.D.; Liu, X.Y.; Chen, Z.; Liu, L.B. Exendin-4 protects Abeta(1-42) oligomer-induced PC12 cell apoptosis. Am. J. Transl. Res. 2016, 8, 3540–3548. [Google Scholar]
- Pilkington, A.W.T.; Donohoe, G.C.; Akhmedov, N.G.; Ferrebee, T.; Valentine, S.J.; Legleiter, J. Hydrogen Peroxide Modifies Abeta-Membrane Interactions with Implications for Abeta40 Aggregation. Biochemistry 2019, 58, 2893–2905. [Google Scholar] [CrossRef]
- La Rosa, C.; Scalisi, S.; Lolicato, F.; Pannuzzo, M.; Raudino, A. Lipid-assisted protein transport: A diffusion-reaction model supported by kinetic experiments and molecular dynamics simulations. J. Chem. Phys. 2016, 144, 184901. [Google Scholar] [CrossRef]
- Scollo, F.; Tempra, C.; Lolicato, F.; Sciacca, M.F.M.; Raudino, A.; Milardi, D.; La Rosa, C. Phospholipids Critical Micellar Concentrations Trigger Different Mechanisms of Intrinsically Disordered Proteins Interaction with Model Membranes. J. Phys. Chem. Lett. 2018, 9, 5125–5129. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Sun, Q.; Chen, S. Oxidative stress: A major pathogenesis and potential therapeutic target of antioxidative agents in Parkinson’s disease and Alzheimer’s disease. Prog. Neurobiol. 2016, 147, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Mokhtari, Z.; Baluchnejadmojarad, T.; Nikbakht, F.; Mansouri, M.; Roghani, M. Riluzole ameliorates learning and memory deficits in Abeta25-35-induced rat model of Alzheimer’s disease and is independent of cholinoceptor activation. Biomed. Pharmacother. Biomed. Pharmacother. 2017, 87, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Sciacca, M.F.M.; Romanucci, V.; Zarrelli, A.; Monaco, I.; Lolicato, F.; Spinella, N.; Galati, C.; Grasso, G.; D’Urso, L.; Romeo, M.; et al. Inhibition of Abeta Amyloid Growth and Toxicity by Silybins: The Crucial Role of Stereochemistry. ACS Chem. Neurosci. 2017, 8, 1767–1778. [Google Scholar] [CrossRef] [PubMed]
- Malishev, R.; Shaham-Niv, S.; Nandi, S.; Kolusheva, S.; Gazit, E.; Jelinek, R. Bacoside-A, an Indian Traditional-Medicine Substance, Inhibits beta-Amyloid Cytotoxicity, Fibrillation, and Membrane Interactions. ACS Chem. Neurosci. 2017, 8, 884–891. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Cao, H.; Cui, X.; Zheng, W.; Wang, S.; Yu, J.; Chen, Z. Silymarin’s Inhibition and Treatment Effects for Alzheimer’s Disease. Molecules 2019, 24, 1748. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Pradhan, N.; Bera, S.; Datta, A.; Krishnamoorthy, J.; Jana, N.R.; Bhunia, A. Inhibition and Degradation of Amyloid Beta (Abeta40) Fibrillation by Designed Small Peptide: A Combined Spectroscopy, Microscopy, and Cell Toxicity Study. ACS Chem. Neurosci. 2017, 8, 718–722. [Google Scholar] [CrossRef]
- Cho, I.H. Effects of Panax ginseng in Neurodegenerative Diseases. J. Ginseng Res. 2012, 36, 342–353. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Kim, P.; Shin, C.Y. A comprehensive review of the therapeutic and pharmacological effects of ginseng and ginsenosides in central nervous system. J. Ginseng Res. 2013, 37, 8–29. [Google Scholar] [CrossRef] [Green Version]
- Tachikawa, E.; Kudo, K.; Harada, K.; Kashimoto, T.; Miyate, Y.; Kakizaki, A.; Takahashi, E. Effects of ginseng saponins on responses induced by various receptor stimuli. Eur. J. Pharmacol. 1999, 369, 23–32. [Google Scholar] [CrossRef]
- Chen, L.M.; Zhou, X.M.; Cao, Y.L.; Hu, W.X. Neuroprotection of ginsenoside Re in cerebral ischemia-reperfusion injury in rats. J. Asian Nat. Prod. Res. 2008, 10, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Cho, W.C.; Chung, W.S.; Lee, S.K.; Leung, A.W.; Cheng, C.H.; Yue, K.K. Ginsenoside Re of Panax ginseng possesses significant antioxidant and antihyperlipidemic efficacies in streptozotocin-induced diabetic rats. Eur. J. Pharmacol. 2006, 550, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.T.; Shao, Z.H.; Vanden Hoek, T.L.; Chang, W.T.; Li, J.; Mehendale, S.; Wang, C.Z.; Hsu, C.W.; Becker, L.B.; Yin, J.J.; et al. Antioxidant effects of ginsenoside Re in cardiomyocytes. Eur. J. Pharmacol. 2006, 532, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Eckman, E.A.; Eckman, C.B. Reductions in levels of the Alzheimer’s amyloid beta peptide after oral administration of ginsenosides. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2006, 20, 1269–1271. [Google Scholar]
- Kim, R.; Emi, M.; Tanabe, K. Role of mitochondria as the gardens of cell death. Cancer Chemother. Pharmacol. 2006, 57, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Martinou, J.C.; Youle, R.J. Mitochondria in apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev. Cell 2011, 21, 92–101. [Google Scholar] [CrossRef]
- Wang, J.Z.; Wang, Z.H. Senescence may mediate conversion of tau phosphorylation-induced apoptotic escape to neurodegeneration. Exp. Gerontol. 2015, 68, 82–86. [Google Scholar] [CrossRef]
- Arunachalam, S.; Kim, S.Y.; Lee, S.H.; Lee, Y.H.; Kim, M.S.; Yun, B.S.; Yi, H.K.; Hwang, P.H. Davallialactone protects against adriamycin-induced cardiotoxicity in vitro and in vivo. J. Nat. Med. 2012, 66, 149–157. [Google Scholar] [CrossRef]
- Chang, W.T.; Li, J.; Haung, H.H.; Liu, H.; Han, M.; Ramachandran, S.; Li, C.Q.; Sharp, W.W.; Hamann, K.J.; Yuan, C.S.; et al. Baicalein protects against doxorubicin-induced cardiotoxicity by attenuation of mitochondrial oxidant injury and JNK activation. J. Cell. Biochem. 2011, 112, 2873–2881. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Chang, F.; Li, F.; Fu, H.; Wang, J.; Zhang, S.; Zhao, J.; Yin, D. Palmitate promotes autophagy and apoptosis through ROS-dependent JNK and p38 MAPK. Biochem. Biophys. Res. Commun. 2015, 463, 262–267. [Google Scholar] [CrossRef]
- Wang, Z.Y.; Liu, J.G.; Li, H.; Yang, H.M. Pharmacological Effects of Active Components of Chinese Herbal Medicine in the Treatment of Alzheimer’s Disease: A Review. Am. J. Chin. Med. 2016, 44, 1525–1541. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.T.; Zheng, X.W.; Chen, S.; Shan, C.S.; Xu, Q.Q.; Zhu, J.Z.; Bao, X.Y.; Lin, Y.; Zheng, G.Q.; Wang, Y. Chinese herbal medicine for Alzheimer’s disease: Clinical evidence and possible mechanism of neurogenesis. Biochem. Pharmacol. 2017, 141, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.S.; Kang, M.S.; Lee, K.Y.; Lee, M.J.; Wang, L.; Kim, H.J. Therapeutic effects of mesenchymal stem cells and hyaluronic Acid injection on osteochondral defects in rabbits’ knees. Knee Surg. Relat. Res. 2012, 24, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Ji, Z.N.; Dong, T.T.; Ye, W.C.; Choi, R.C.; Lo, C.K.; Tsim, K.W. Ginsenoside Re attenuate beta-amyloid and serum-free induced neurotoxicity in PC12 cells. J. Ethnopharmacol. 2006, 107, 48–52. [Google Scholar] [CrossRef]
- Liu, Q.; Zhao, B. Nicotine attenuates beta-amyloid peptide-induced neurotoxicity, free radical and calcium accumulation in hippocampal neuronal cultures. Br. J. Pharmacol. 2004, 141, 746–754. [Google Scholar] [CrossRef]
- Manczak, M.; Anekonda, T.S.; Henson, E.; Park, B.S.; Quinn, J.; Reddy, P.H. Mitochondria are a direct site of A beta accumulation in Alzheimer’s disease neurons: Implications for free radical generation and oxidative damage in disease progression. Hum. Mol. Genet. 2006, 15, 1437–1449. [Google Scholar] [CrossRef]
- Soga, M.; Matsuzawa, A.; Ichijo, H. Oxidative Stress-Induced Diseases via the ASK1 Signaling Pathway. Int. J. Cell Biol. 2012, 2012, 439587. [Google Scholar] [CrossRef]
- Joshi, G.; Johnson, J.A. The Nrf2-ARE pathway: A valuable therapeutic target for the treatment of neurodegenerative diseases. Recent Pat. CNS Drug Discov. 2012, 7, 218–229. [Google Scholar] [CrossRef]
- Cui, G.; Luk, S.C.; Li, R.A.; Chan, K.K.; Lei, S.W.; Wang, L.; Shen, H.; Leung, G.P.; Lee, S.M. Cytoprotection of baicalein against oxidative stress-induced cardiomyocytes injury through the Nrf2/Keap1 pathway. J. Cardiovasc. Pharmacol. 2015, 65, 39–46. [Google Scholar] [CrossRef]
- Ramsey, C.P.; Glass, C.A.; Montgomery, M.B.; Lindl, K.A.; Ritson, G.P.; Chia, L.A.; Hamilton, R.L.; Chu, C.T.; Jordan-Sciutto, K.L. Expression of Nrf2 in neurodegenerative diseases. J. Neuropathol. Exp. Neurol. 2007, 66, 75–85. [Google Scholar] [CrossRef]
- Sarkar, B.; Dhiman, M.; Mittal, S.; Mantha, A.K. Curcumin revitalizes Amyloid beta (25-35)-induced and organophosphate pesticides pestered neurotoxicity in SH-SY5Y and IMR-32 cells via activation of APE1 and Nrf2. Metab. Brain Dis. 2017, 32, 2045–2061. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Zhang, J.; Xiang, J.; Li, Y.; Wu, D.; Xu, J. Calcitriol inhibits ROS-NLRP3-IL-1beta signaling axis via activation of Nrf2-antioxidant signaling in hyperosmotic stress stimulated human corneal epithelial cells. Redox Biol. 2018, 21, 101093. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Li, C.; Chen, S.; Li, Z.; Jia, X.; Wang, K.; Bao, J.; Liang, Y.; Wang, X.; Chen, M.; et al. Berberine protects against 6-OHDA-induced neurotoxicity in PC12 cells and zebrafish through hormetic mechanisms involving PI3K/AKT/Bcl-2 and Nrf2/HO-1 pathways. Redox Biol. 2017, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, M.; Bai, X.; Yu, S.; Zhao, W.; Qiao, J.; Liu, Y.; Zhao, D.; Wang, J.; Wang, S. Ginsenoside Re Inhibits ROS/ASK-1 Dependent Mitochondrial Apoptosis Pathway and Activation of Nrf2-Antioxidant Response in Beta-Amyloid-Challenged SH-SY5Y Cells. Molecules 2019, 24, 2687. https://doi.org/10.3390/molecules24152687
Liu M, Bai X, Yu S, Zhao W, Qiao J, Liu Y, Zhao D, Wang J, Wang S. Ginsenoside Re Inhibits ROS/ASK-1 Dependent Mitochondrial Apoptosis Pathway and Activation of Nrf2-Antioxidant Response in Beta-Amyloid-Challenged SH-SY5Y Cells. Molecules. 2019; 24(15):2687. https://doi.org/10.3390/molecules24152687
Chicago/Turabian StyleLiu, Meichen, Xueyuan Bai, Shiting Yu, Wenxue Zhao, Juhui Qiao, Ying Liu, Daqing Zhao, Jiawen Wang, and Siming Wang. 2019. "Ginsenoside Re Inhibits ROS/ASK-1 Dependent Mitochondrial Apoptosis Pathway and Activation of Nrf2-Antioxidant Response in Beta-Amyloid-Challenged SH-SY5Y Cells" Molecules 24, no. 15: 2687. https://doi.org/10.3390/molecules24152687