Alkynes as Synthetic Equivalents of Ketones and Aldehydes: A Hidden Entry into Carbonyl Chemistry

 ,
, {kind=link}
{kind=link}
{kind=link}
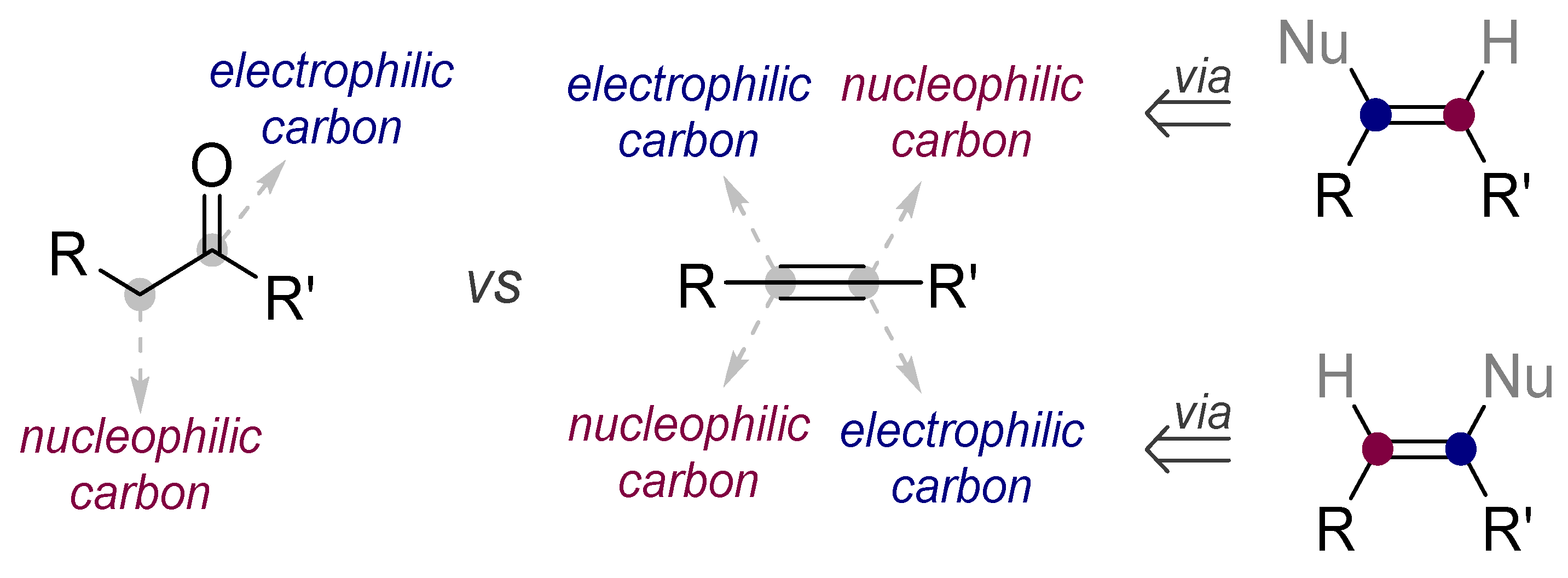{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
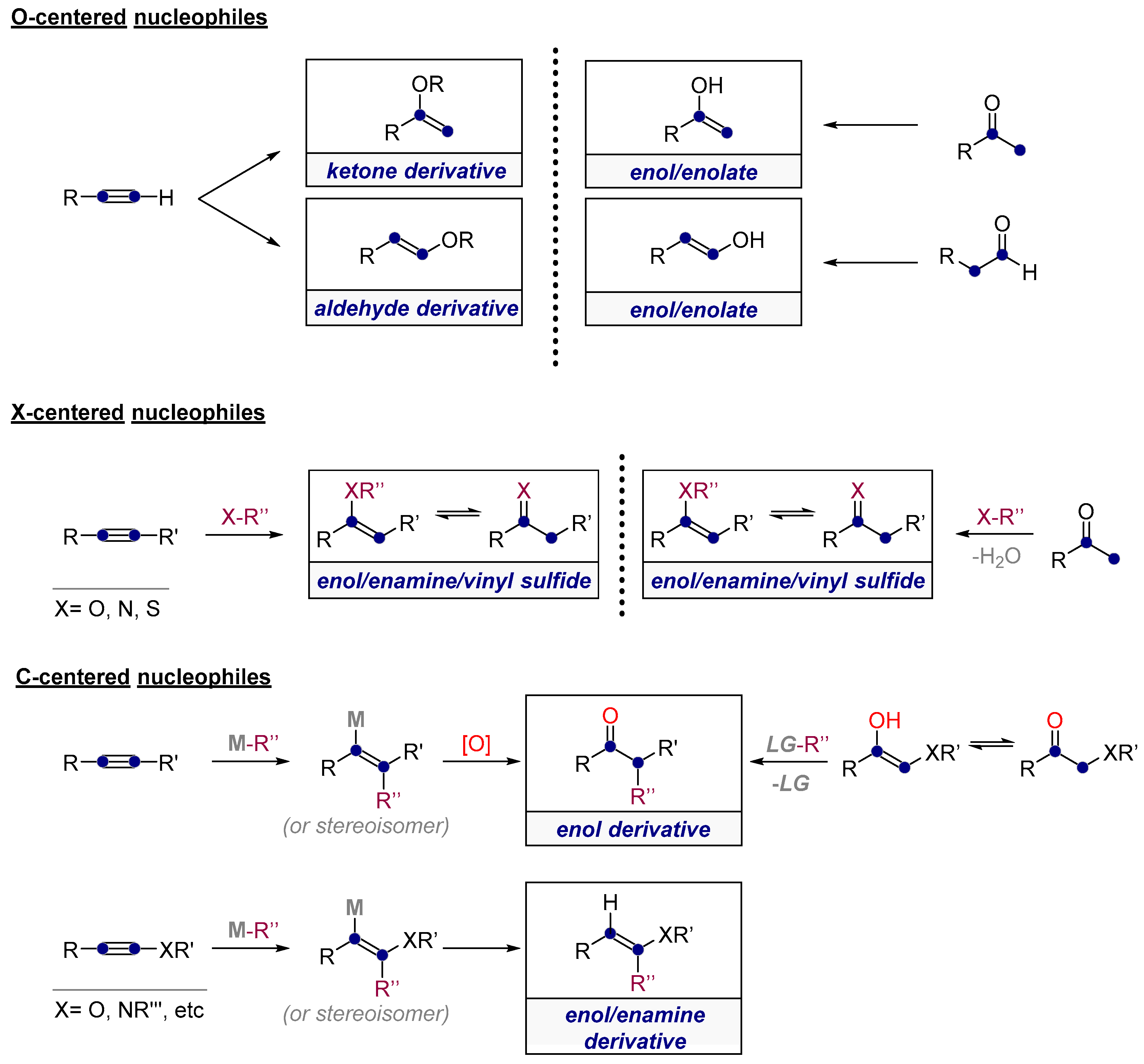
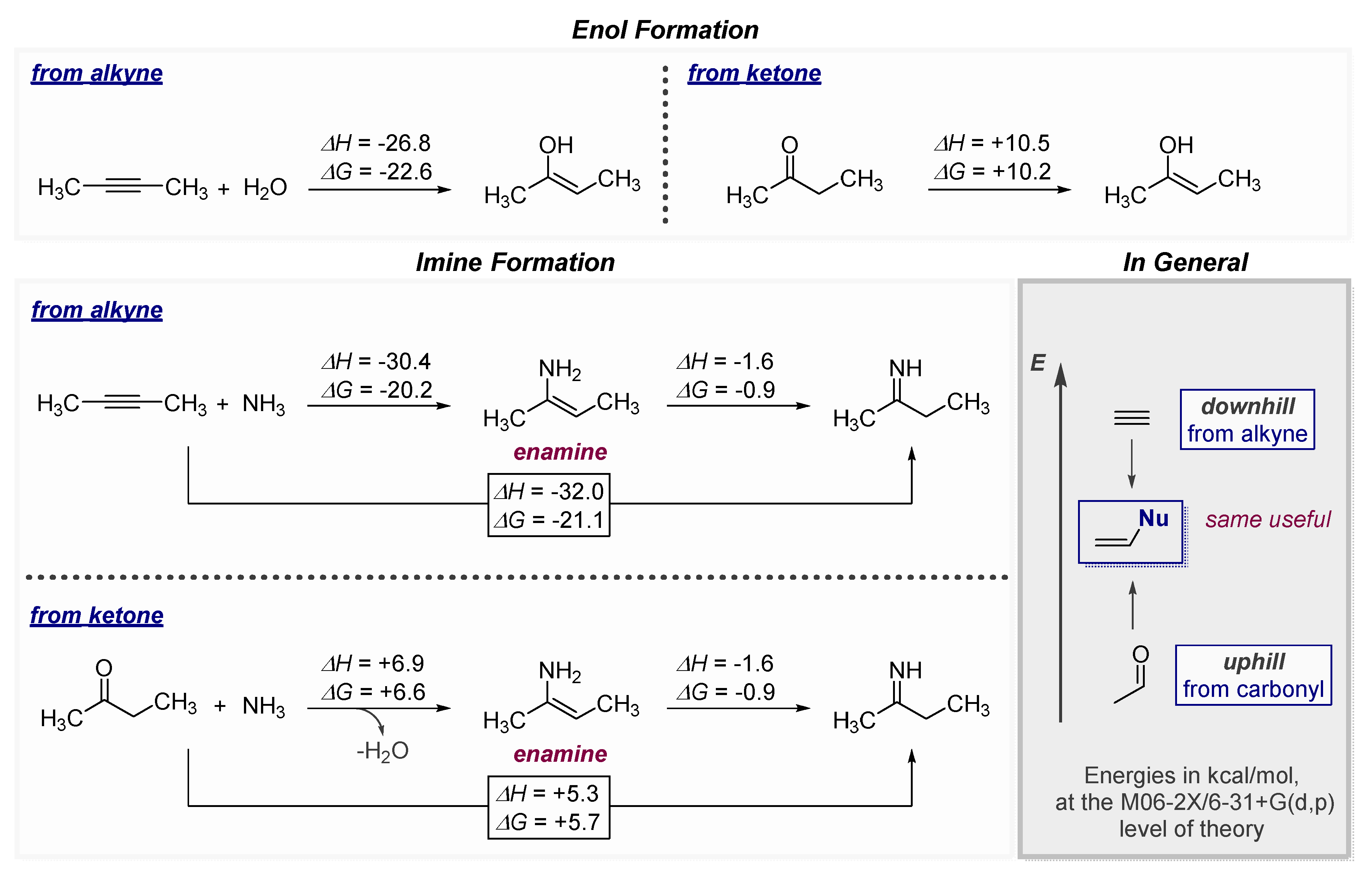
1.1. The High Energy of Alkynes Can be Used to Drive Difficult Transformations
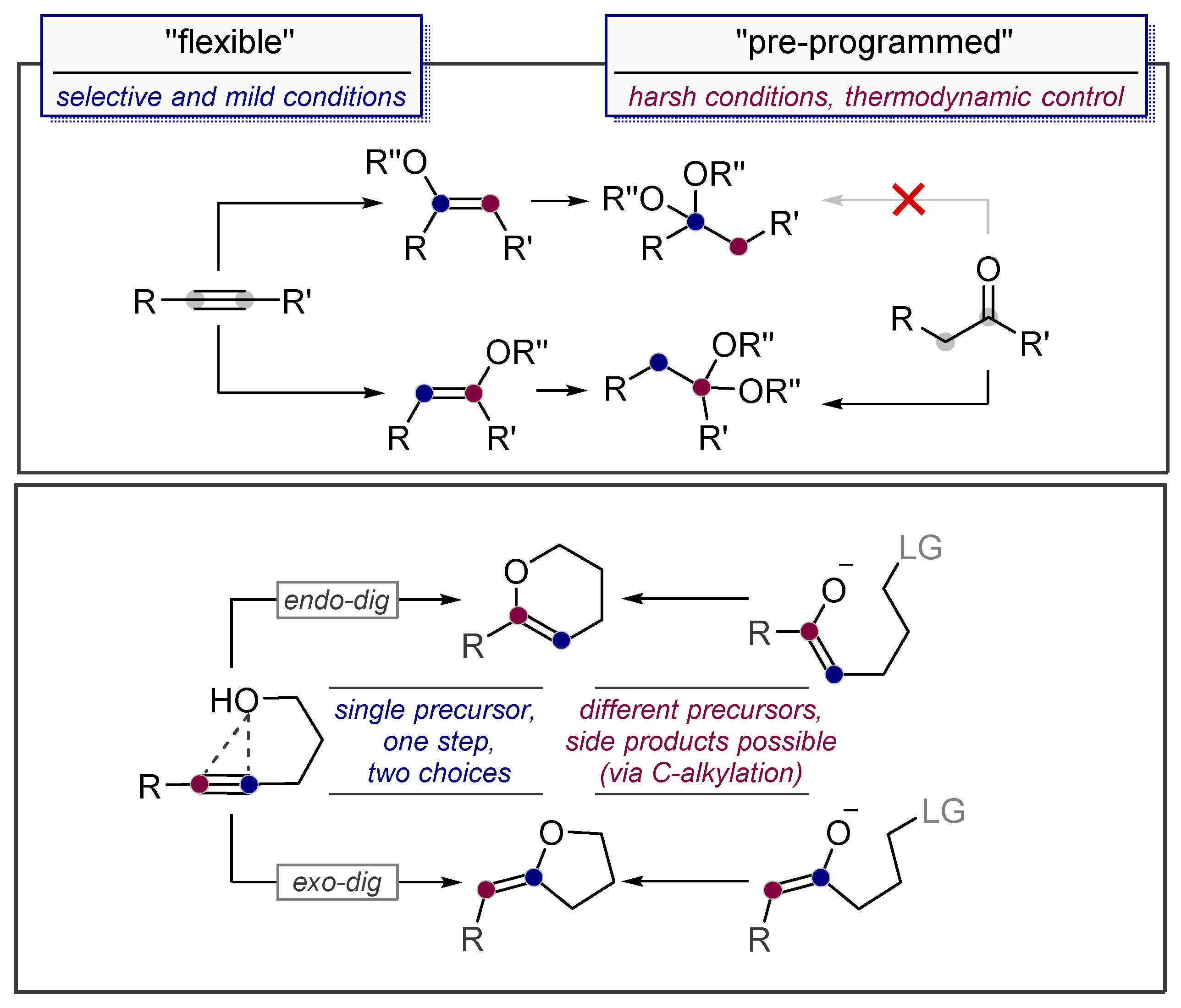
1.2. Low Polarization of Alkynes Endows Them with Flexible Selectivity
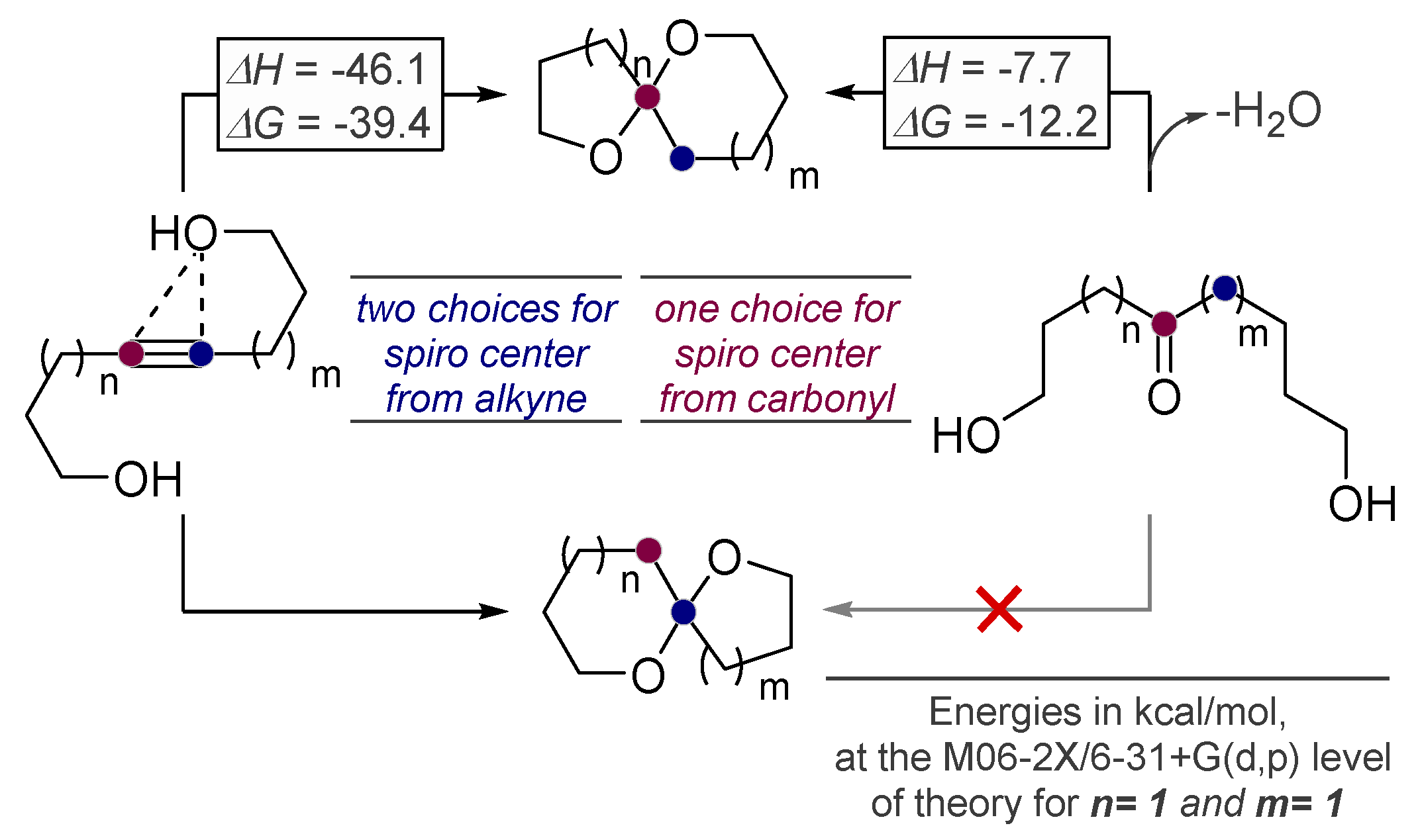
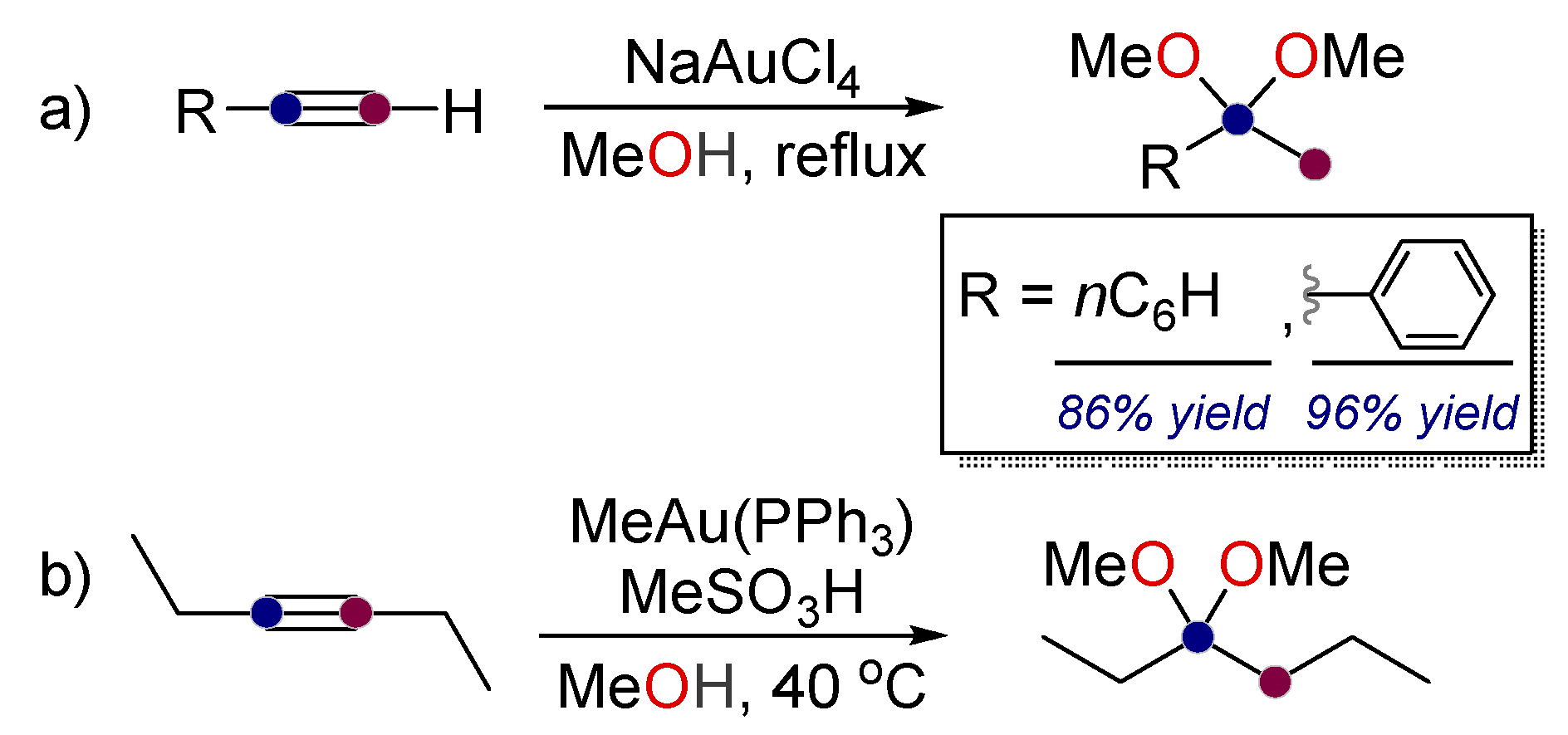
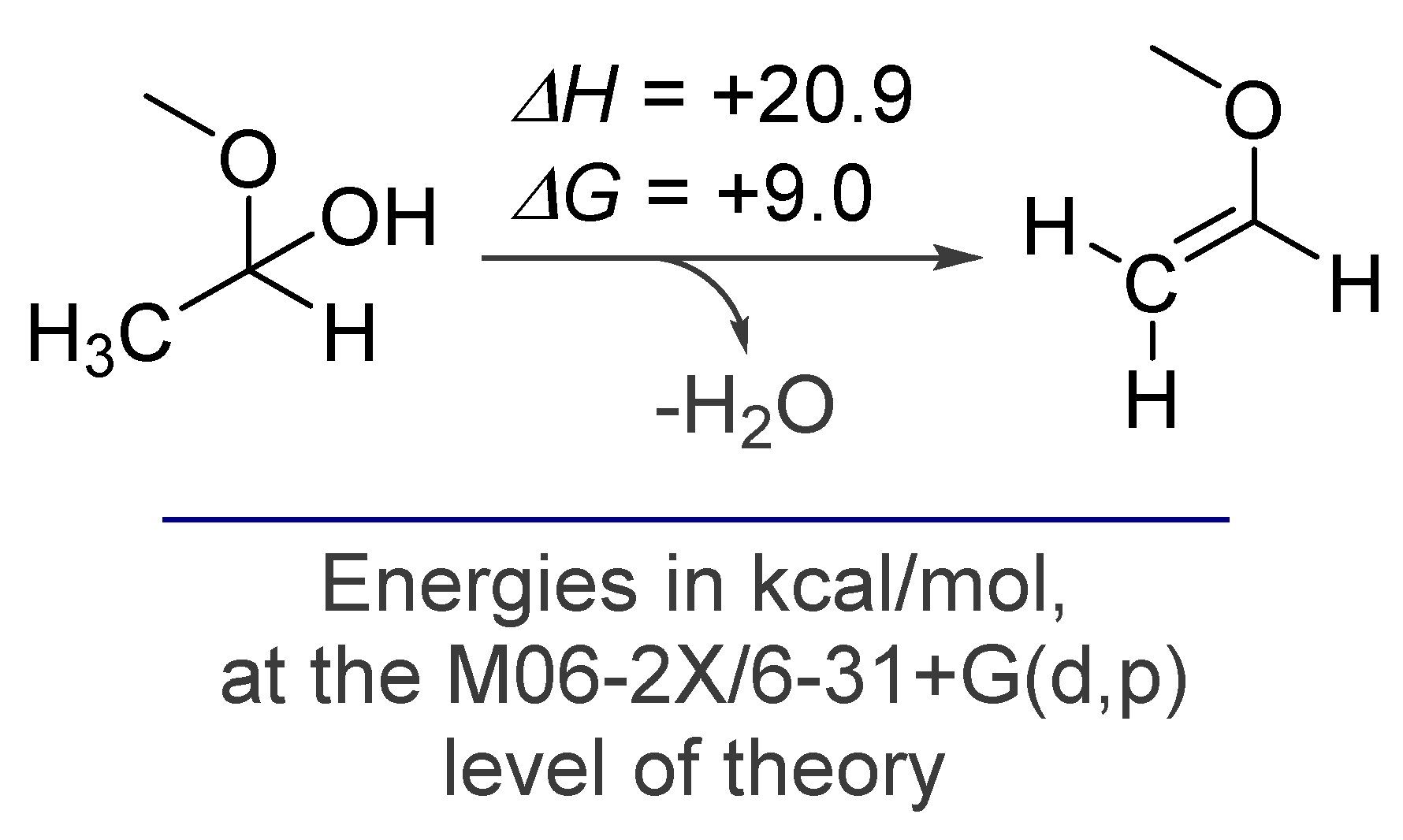
2. Alkynes in Ketal Formation
3. Vinyl Ether Generation from Alkynes
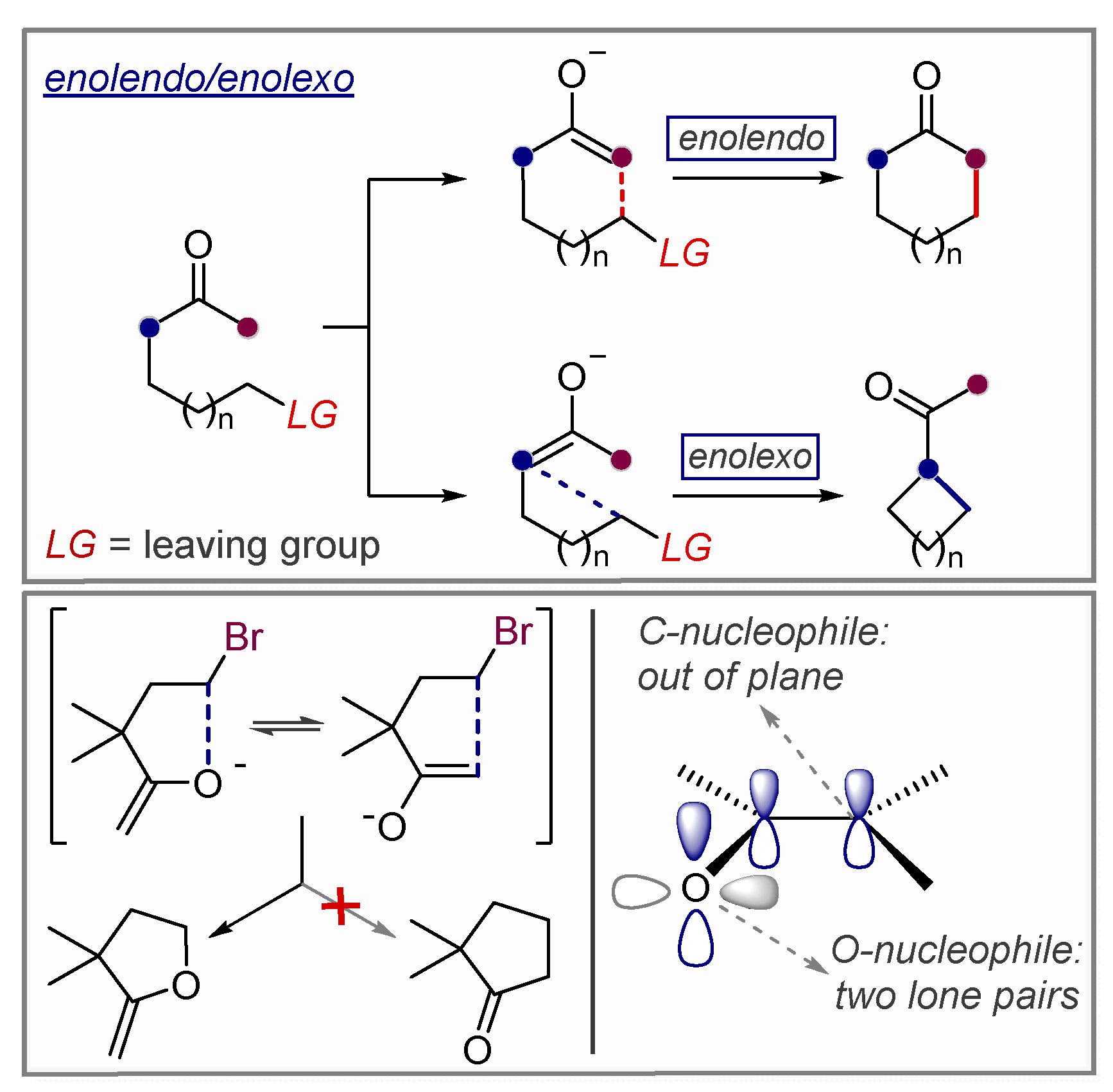
3.1. Anionic Cyclizations without Alkyne Preactivation
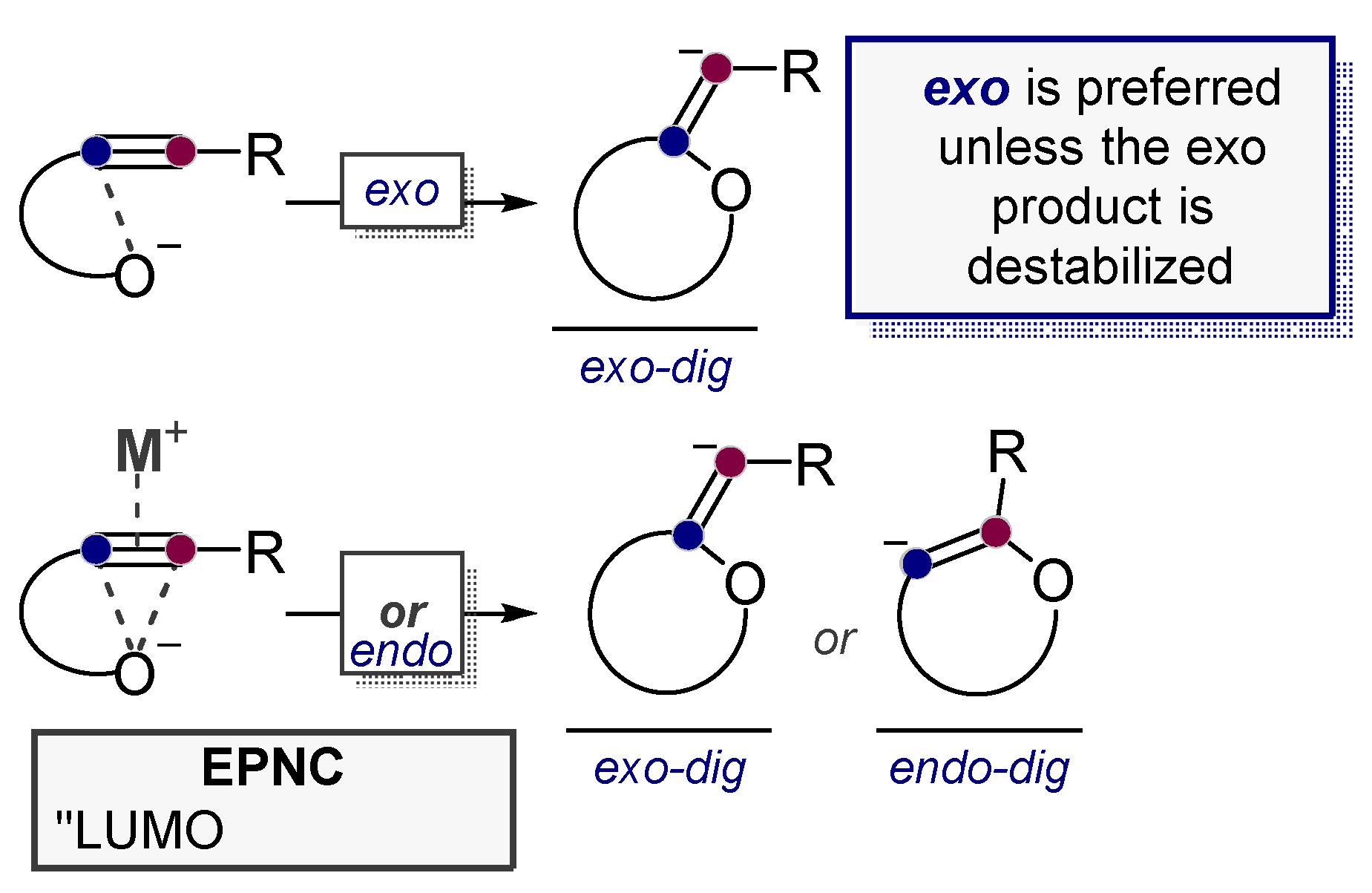
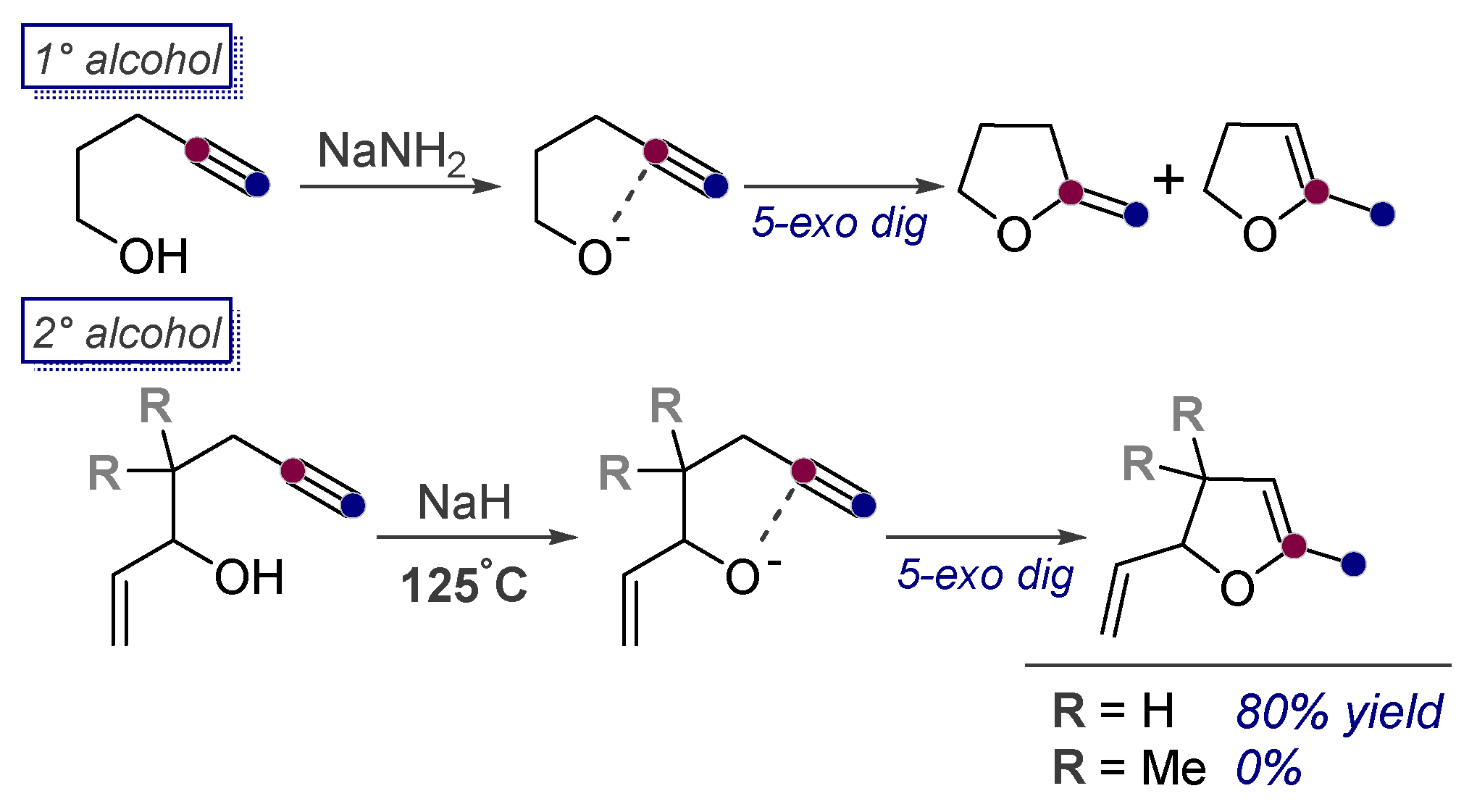
3.2. Use of Stereoelectronic Exo-Preference for Overriding Alkyne Polarizations
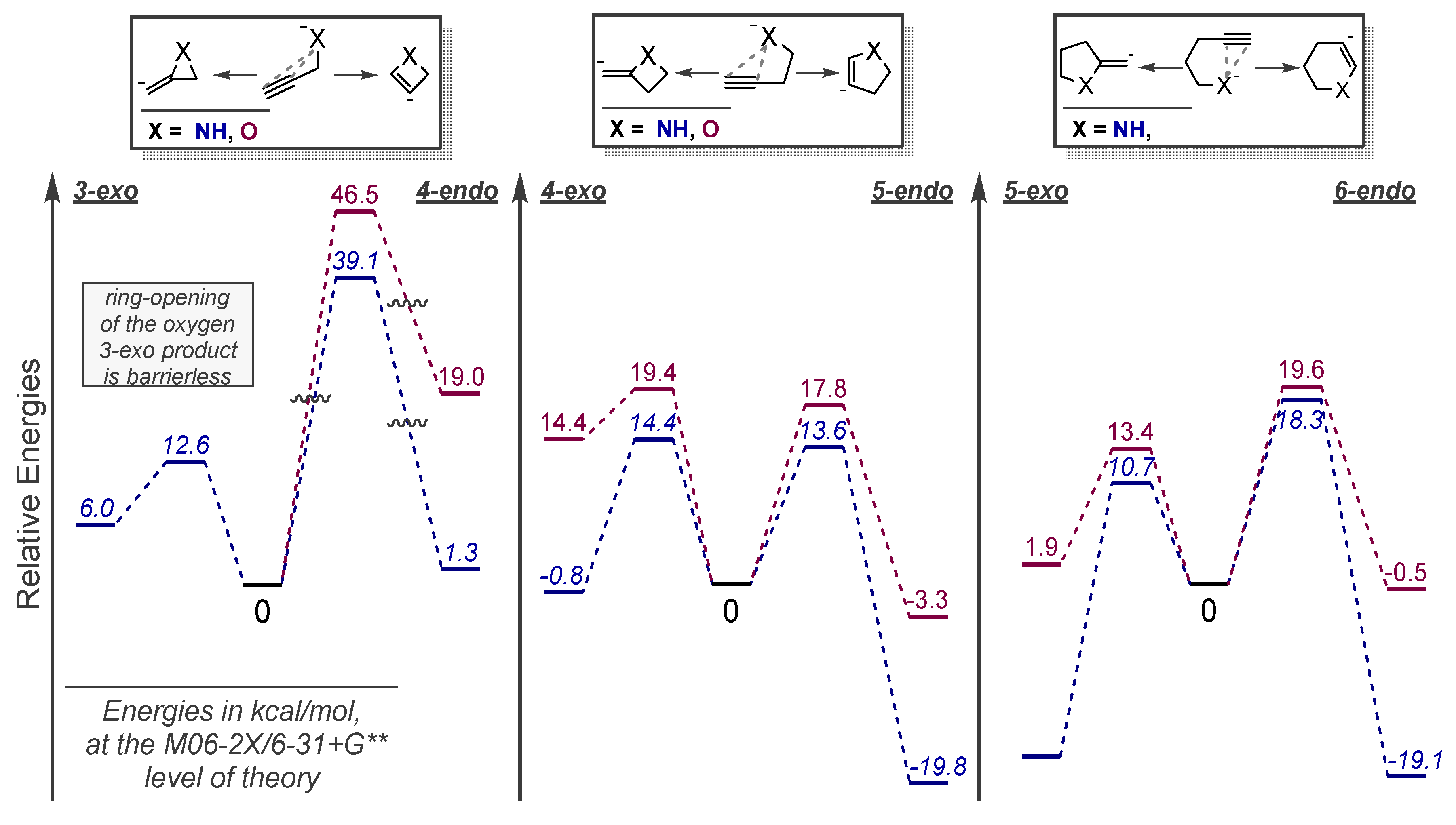
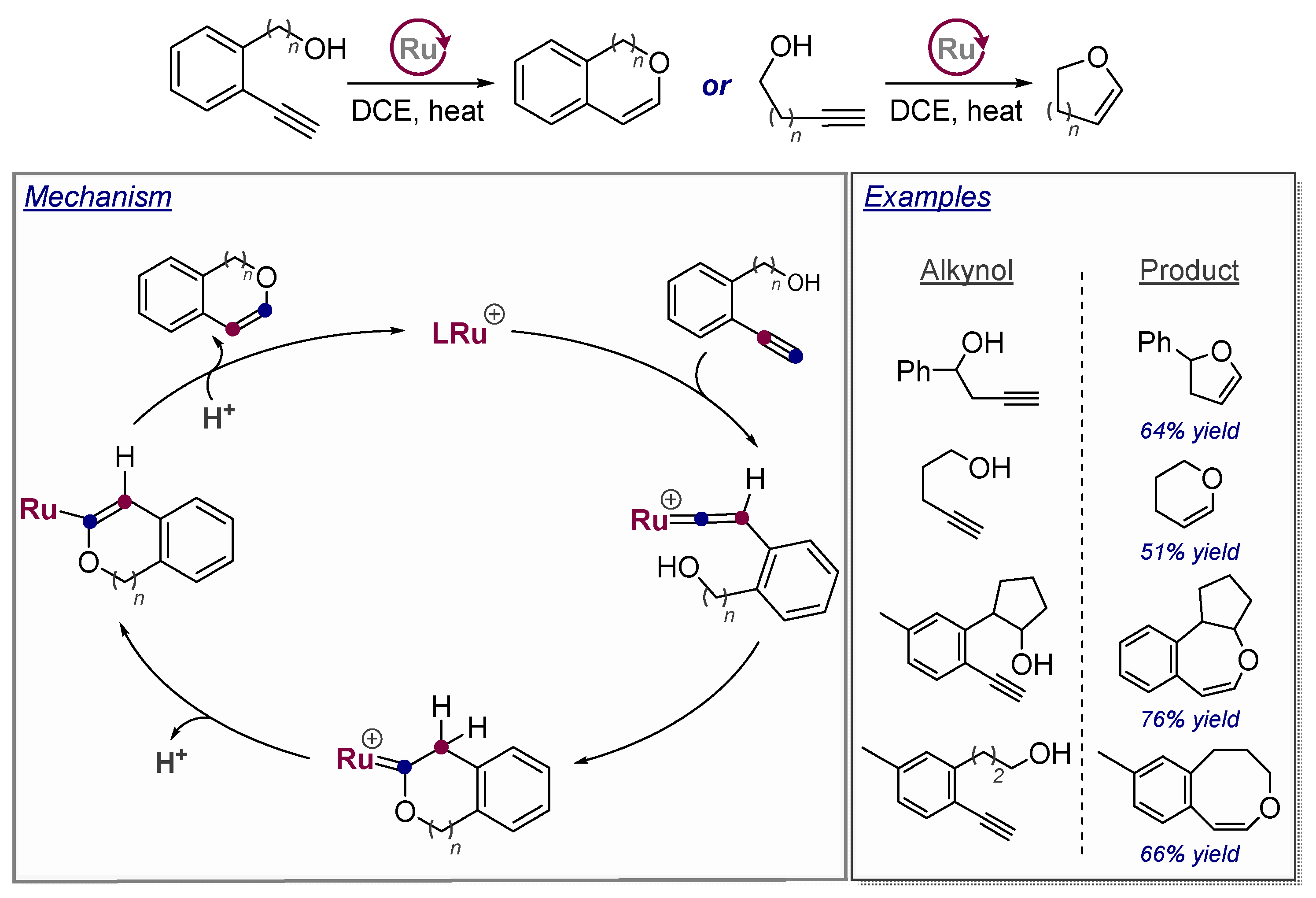
3.3. Use of Strain Effects to Favor 6-endo Selectivity
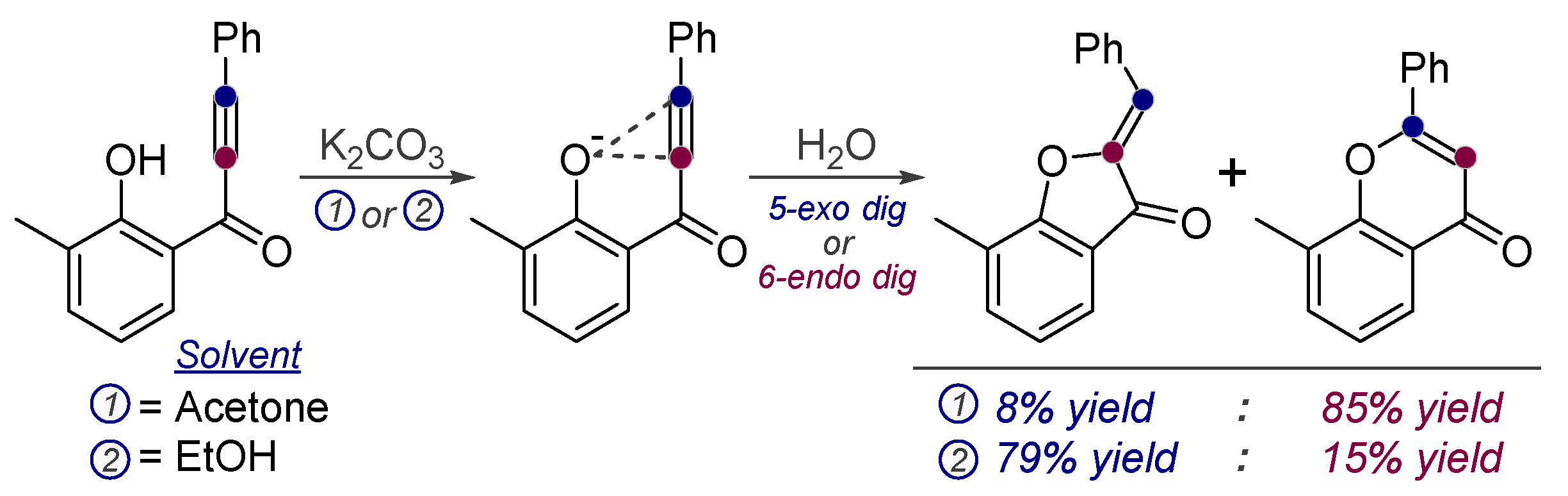
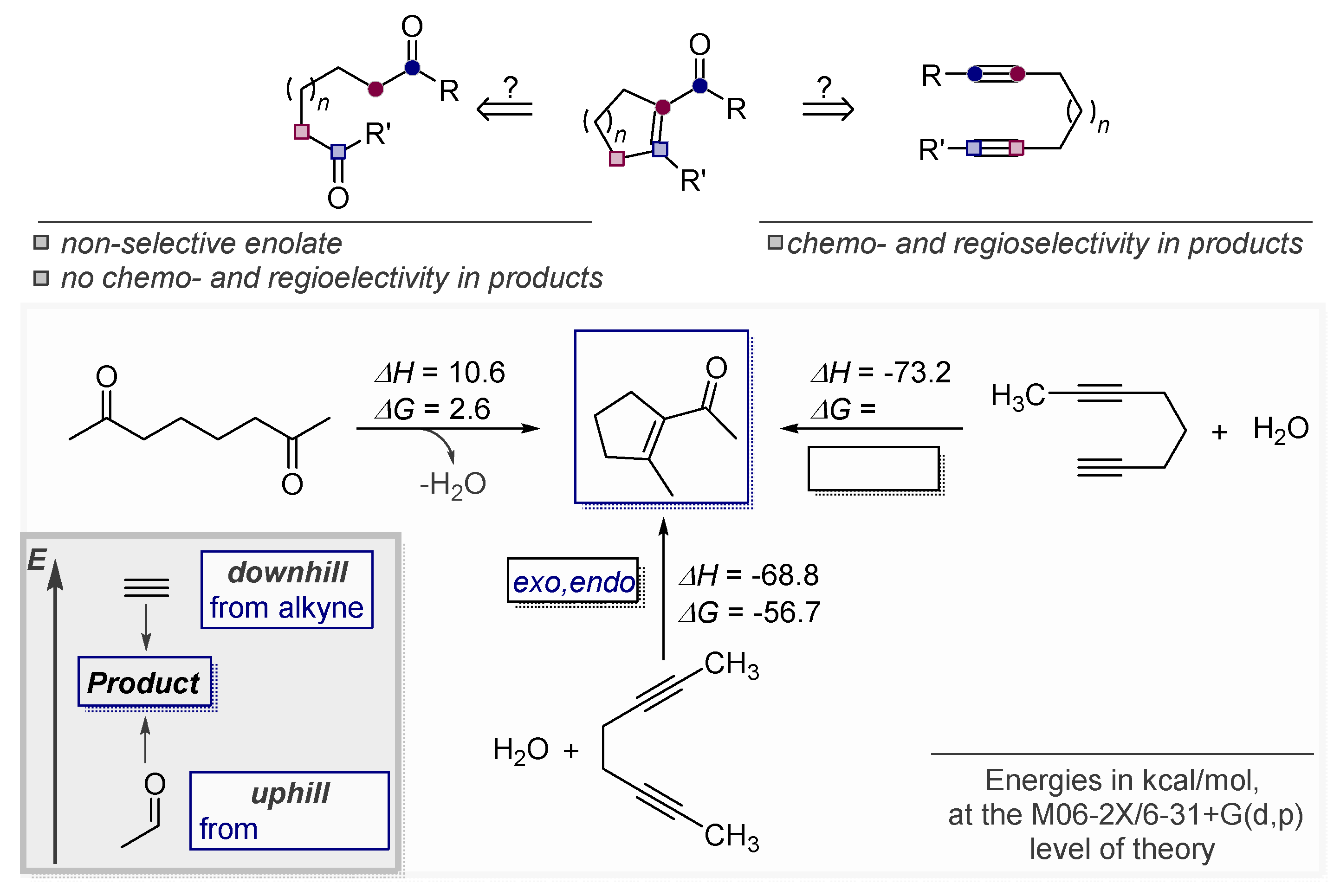
3.4. Controlling Regioselectivity Using Elements of Thermodynamic Control
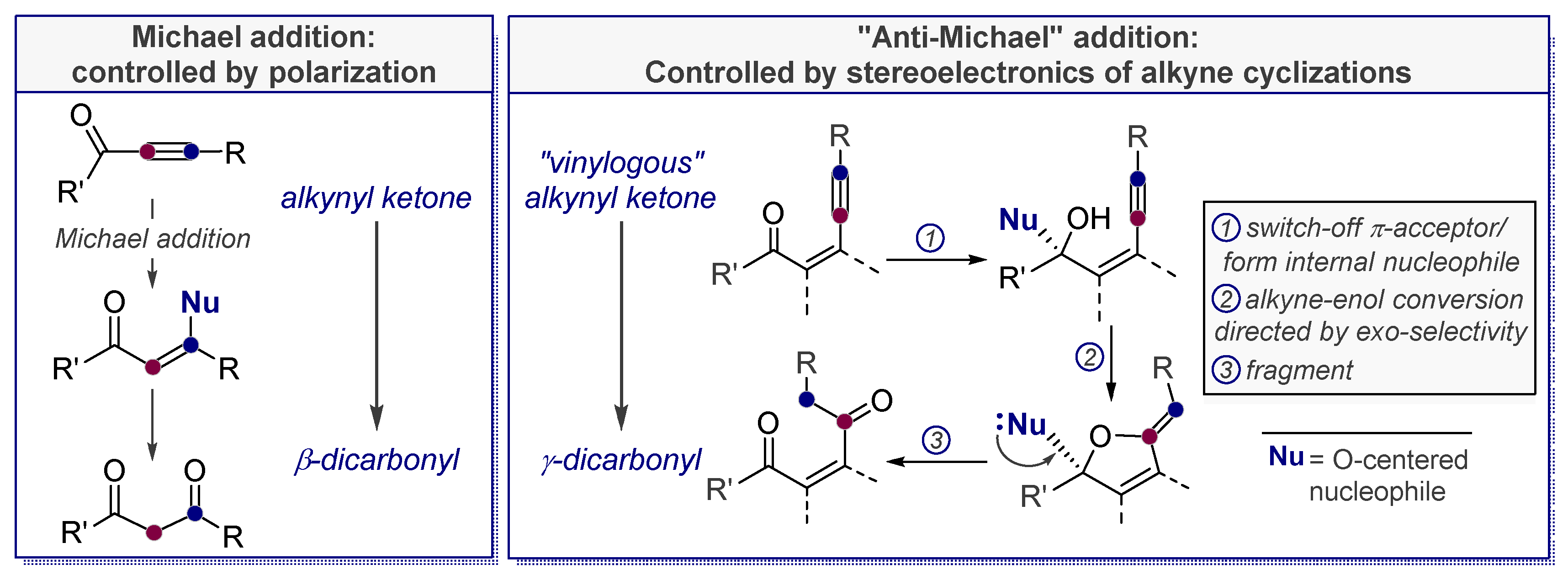
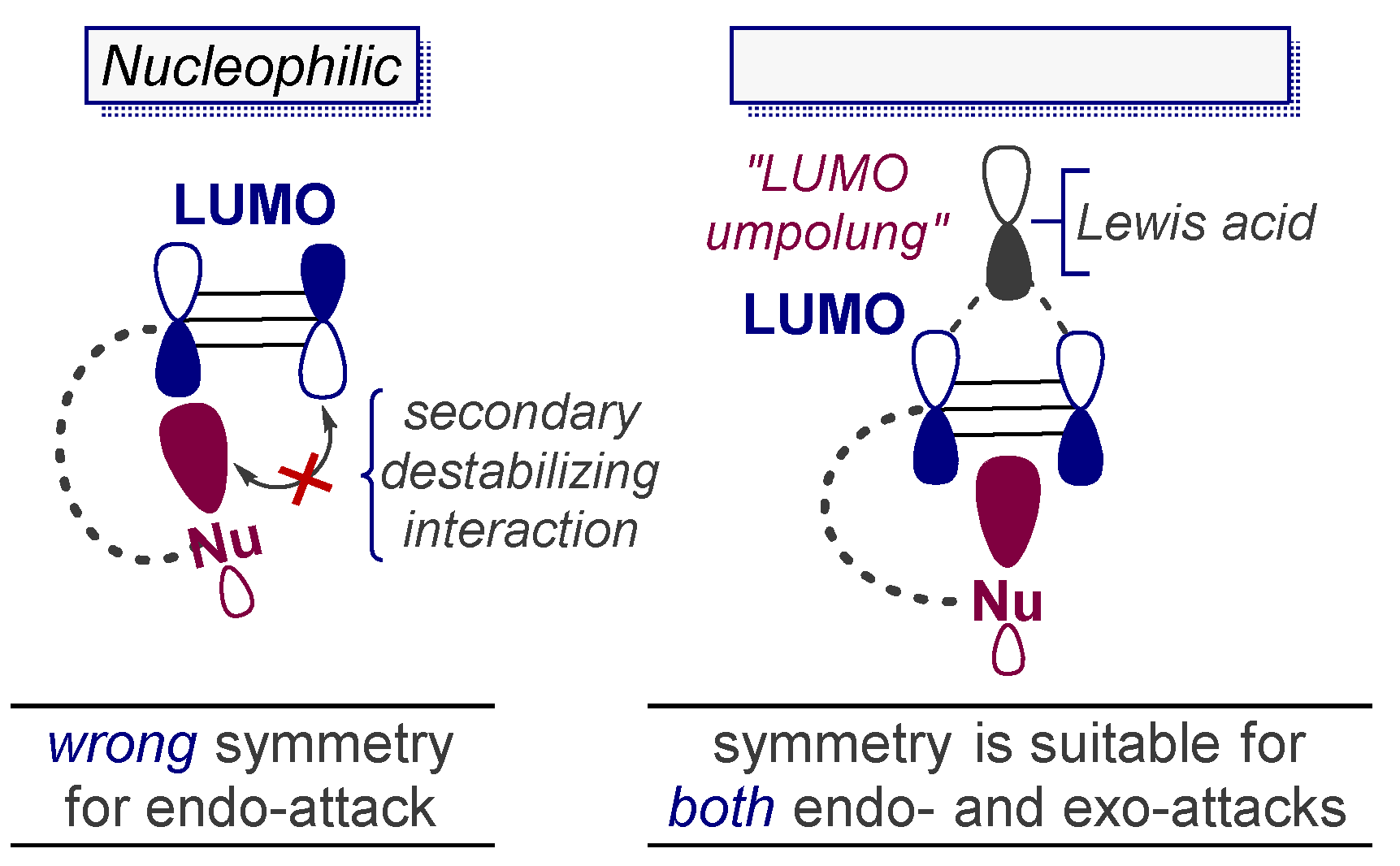
3.5. Electrophile-Promoted Nucleophilic Cyclizations (EPNC)—Rendering Endo-Cyclizations Possible through “LUMO Umpolung”
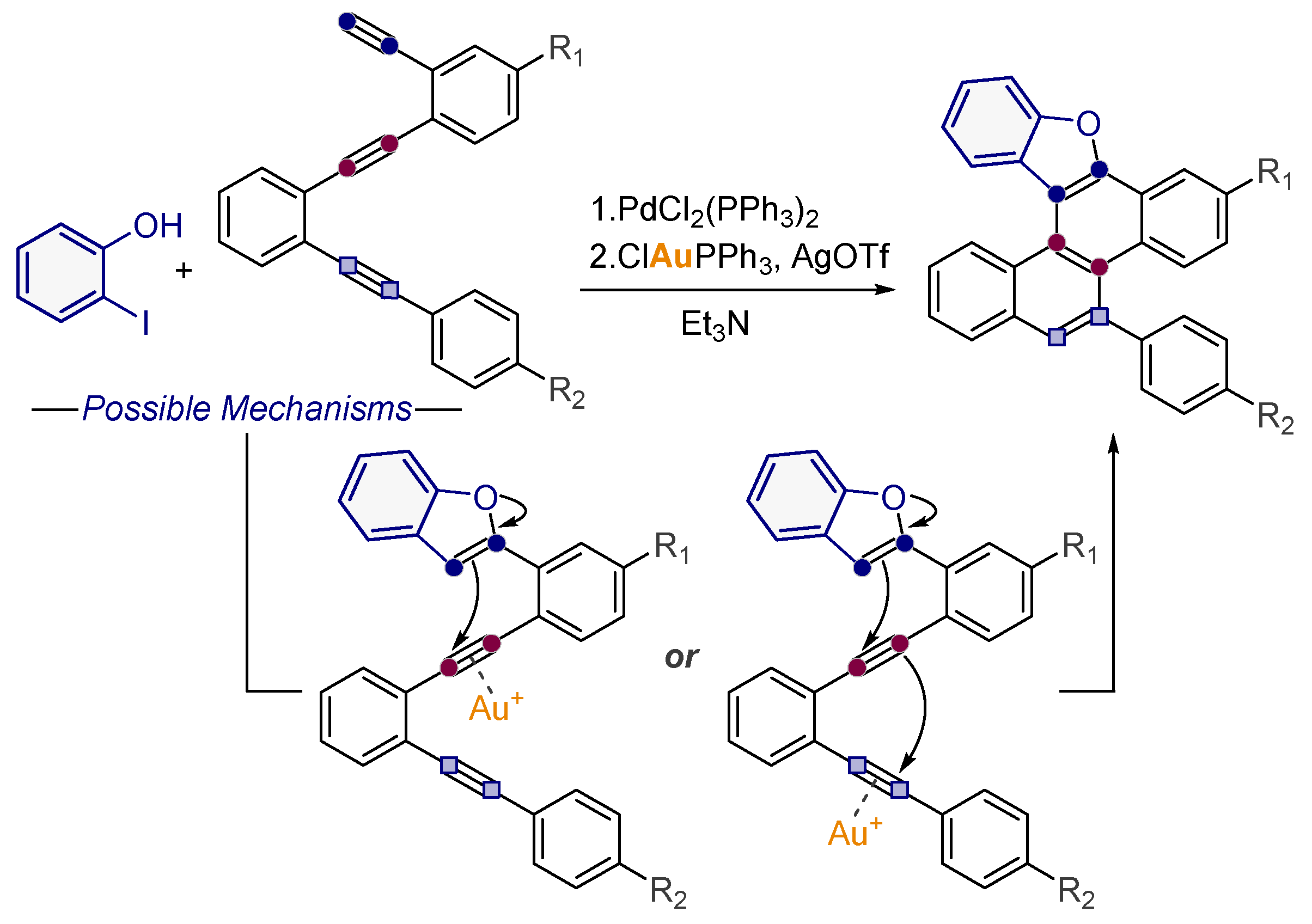
3.6. Endo-Cascade through Vinylidene Intermediates
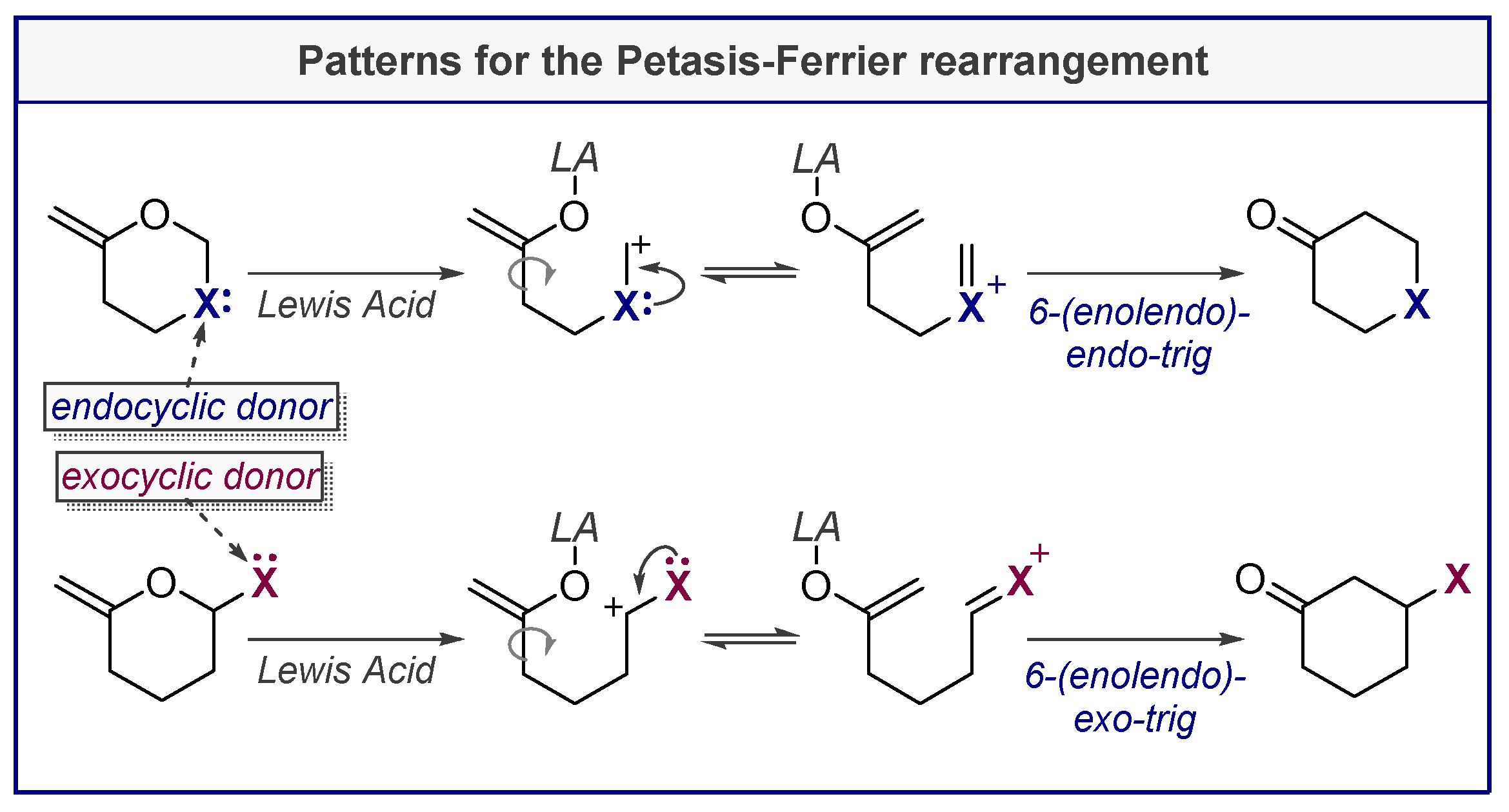
4. Petasis-Ferrier Rearrangement
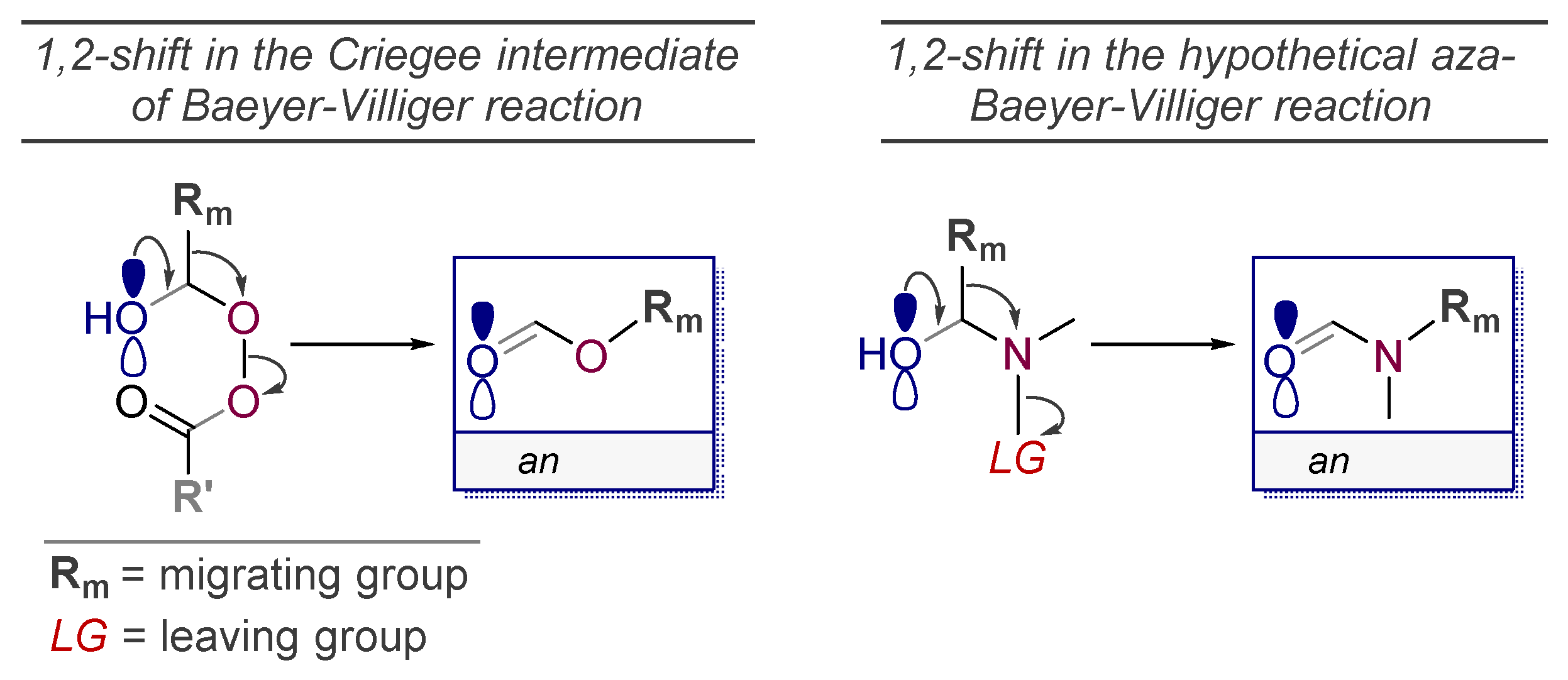
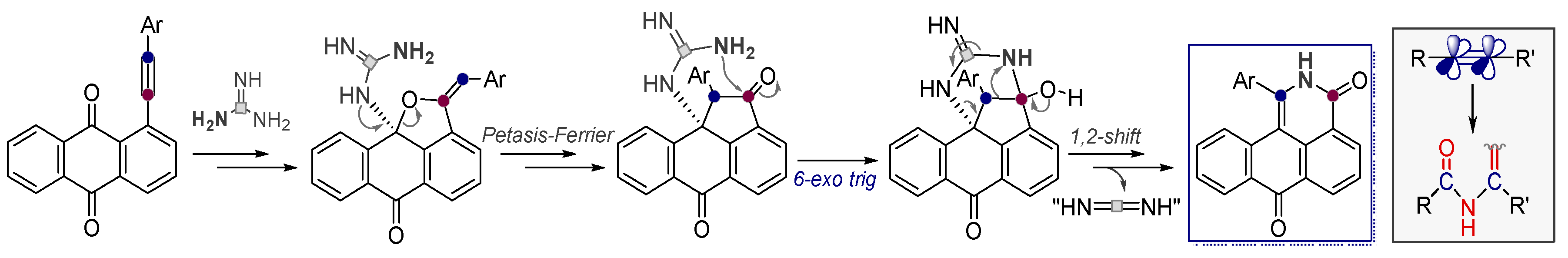
5. The “Oxidant-Free Nitrogen Baeyer-Villiger Rearrangement”
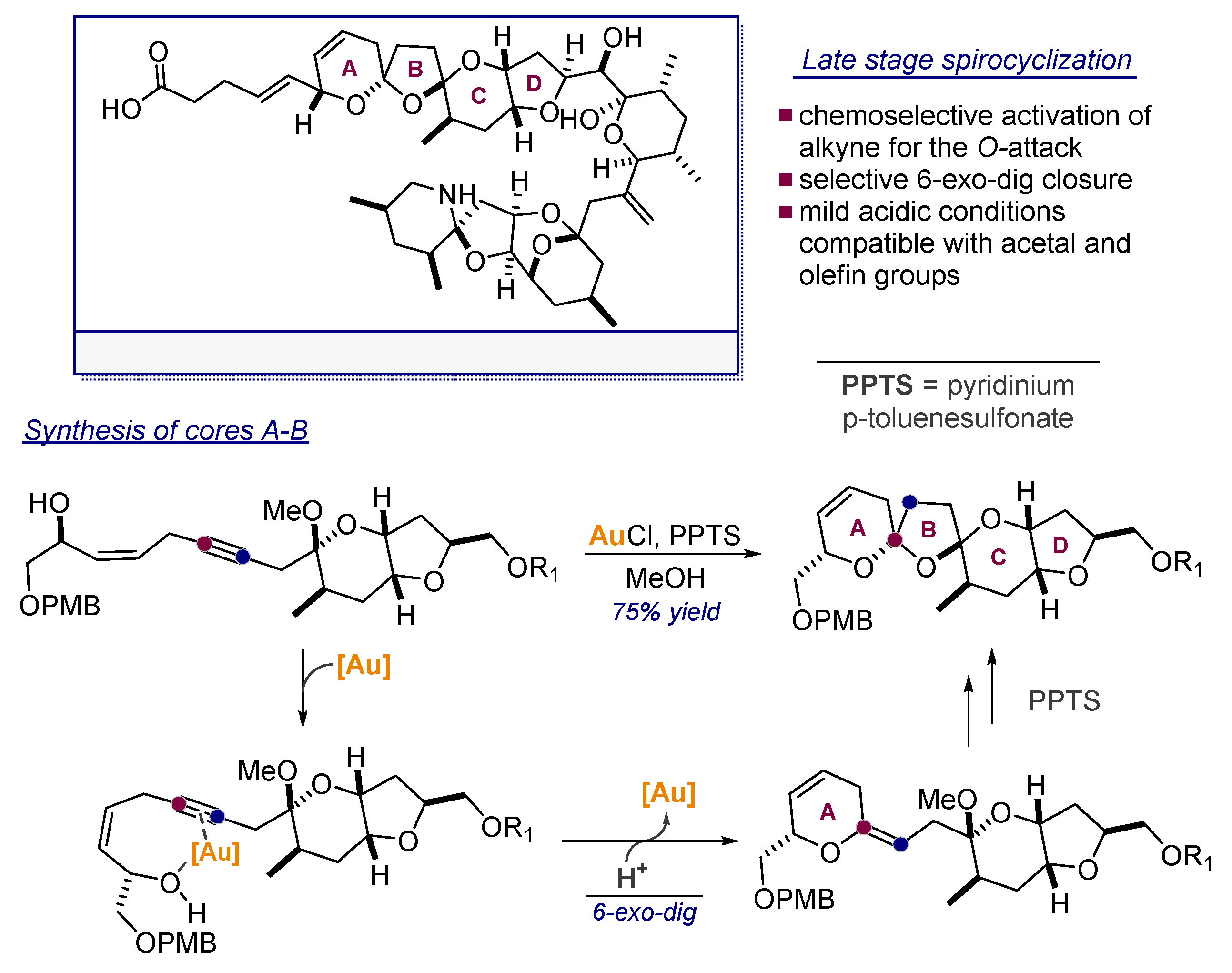
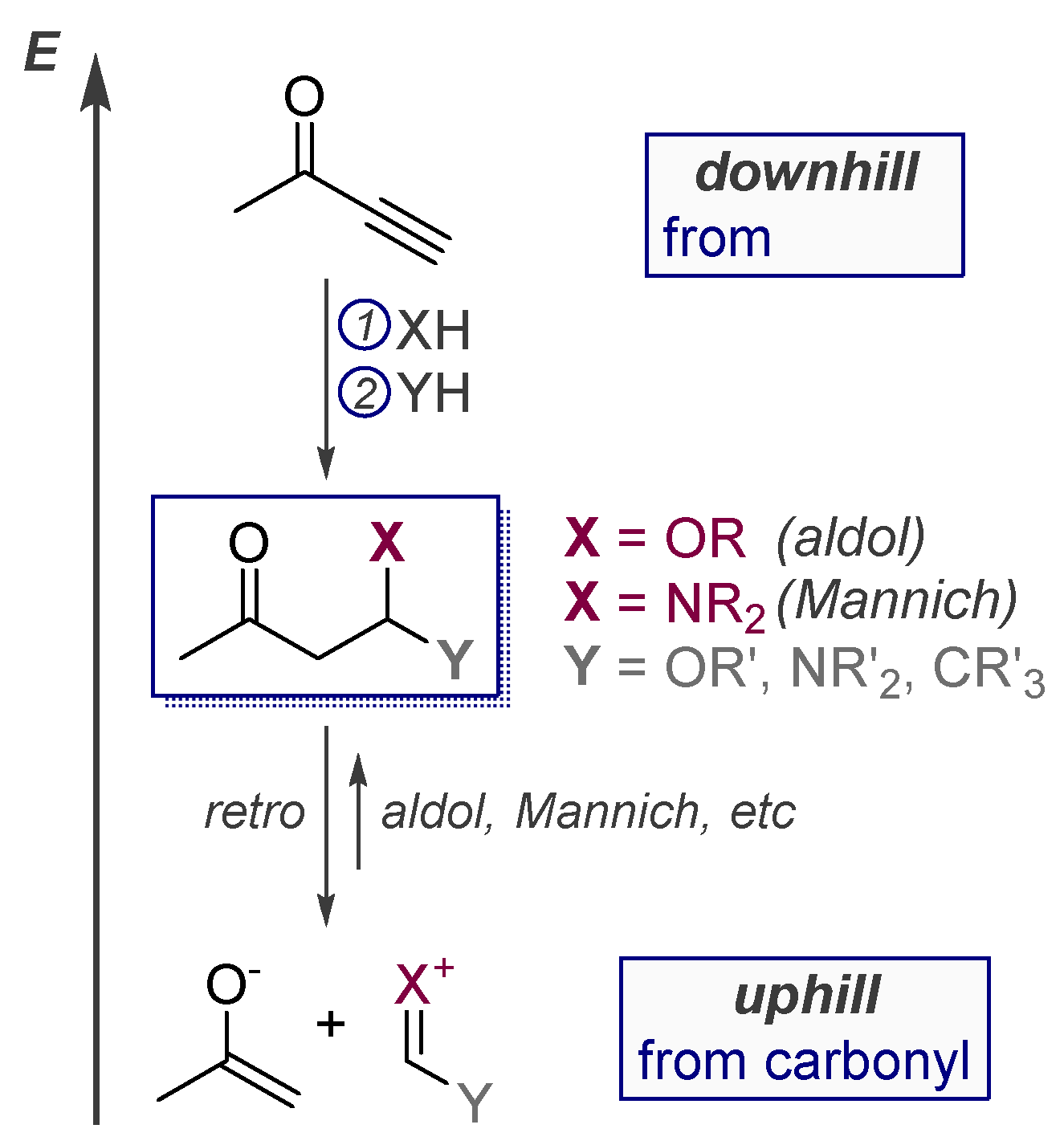
6. Alkynes as Carbonyl Surrogates in the Synthesis of Aldol Products
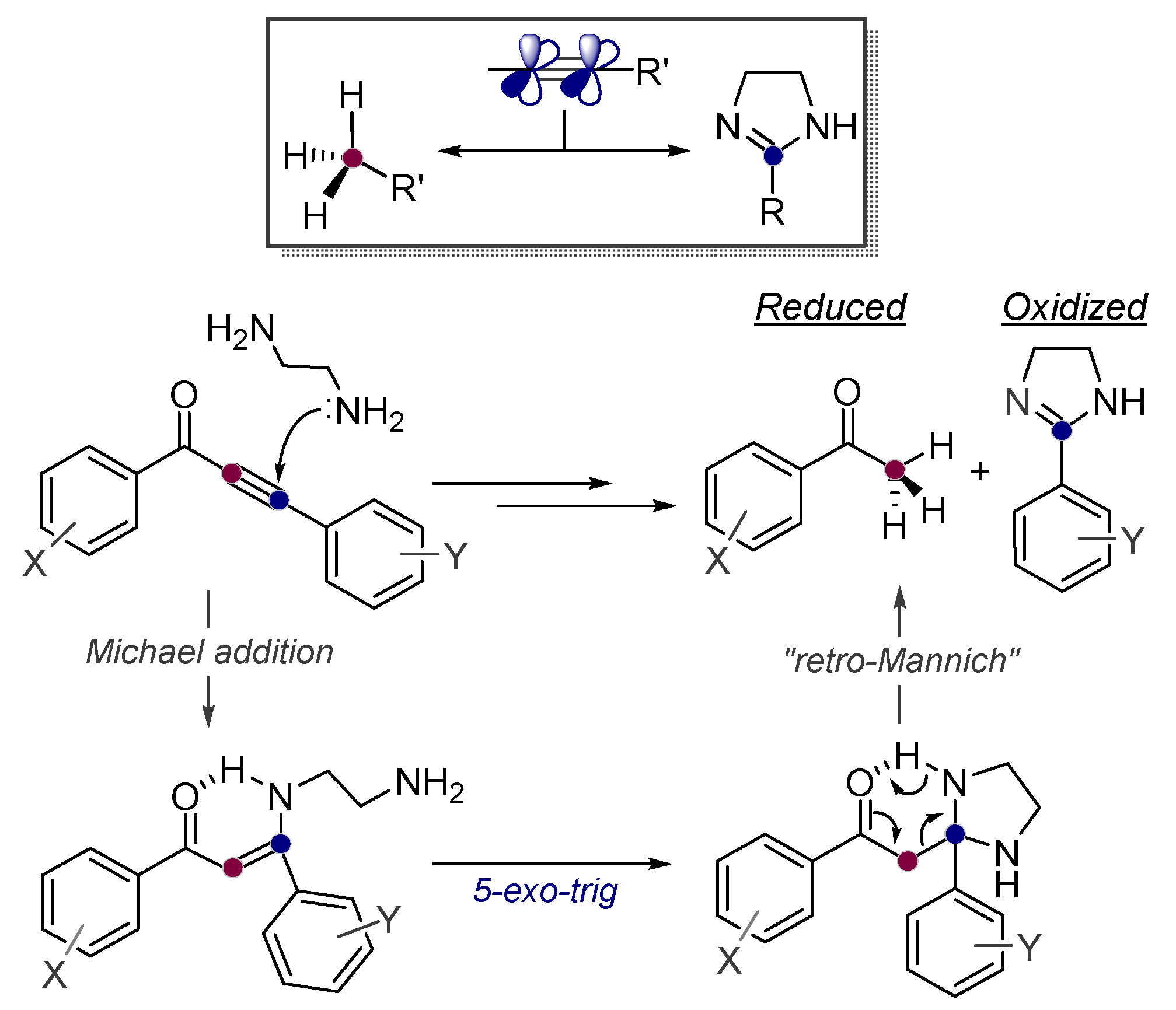
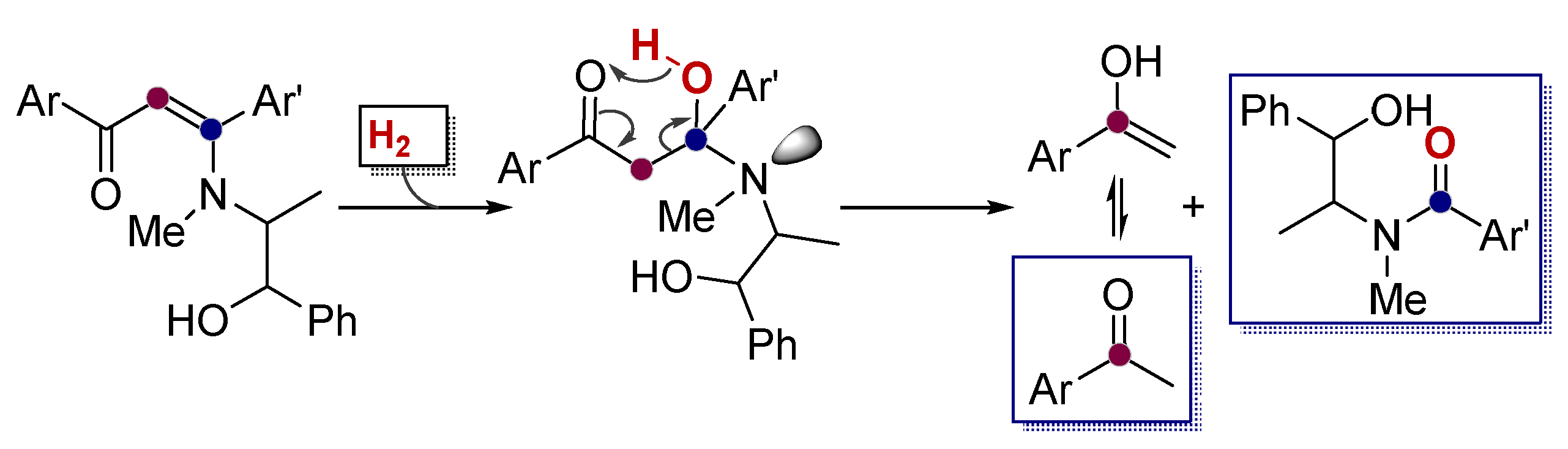
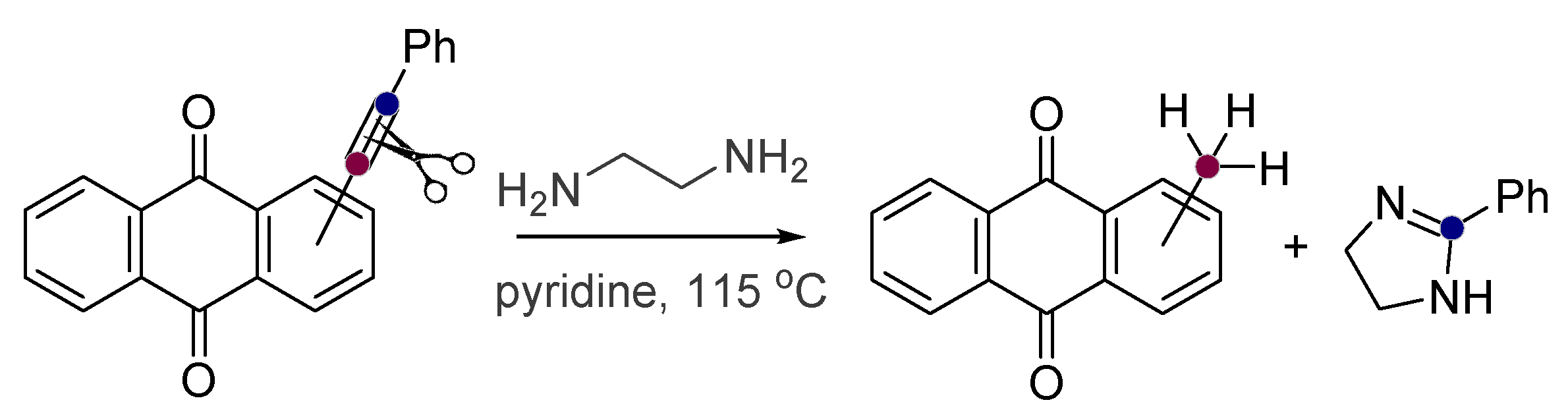
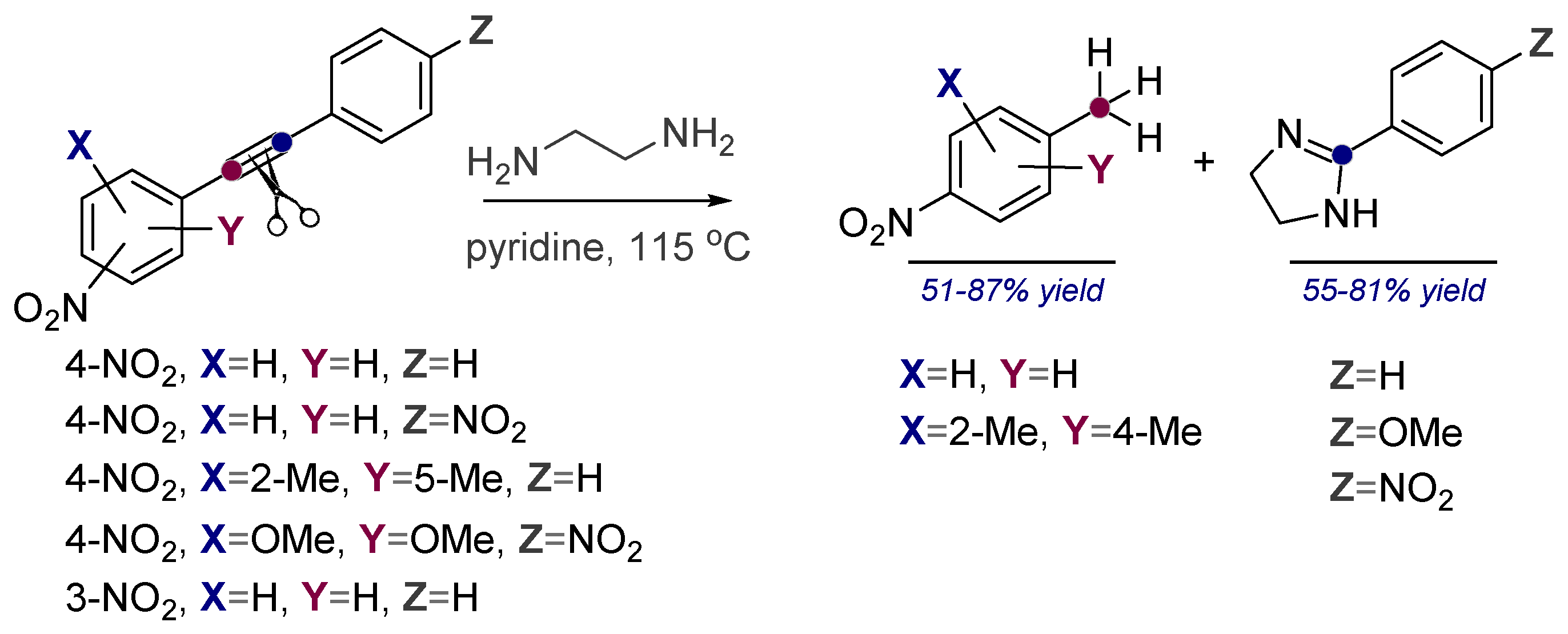
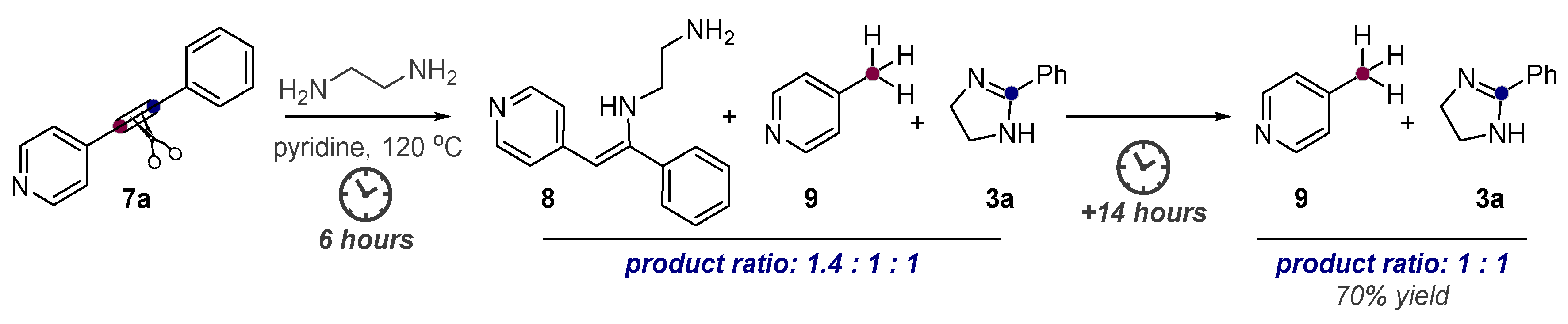
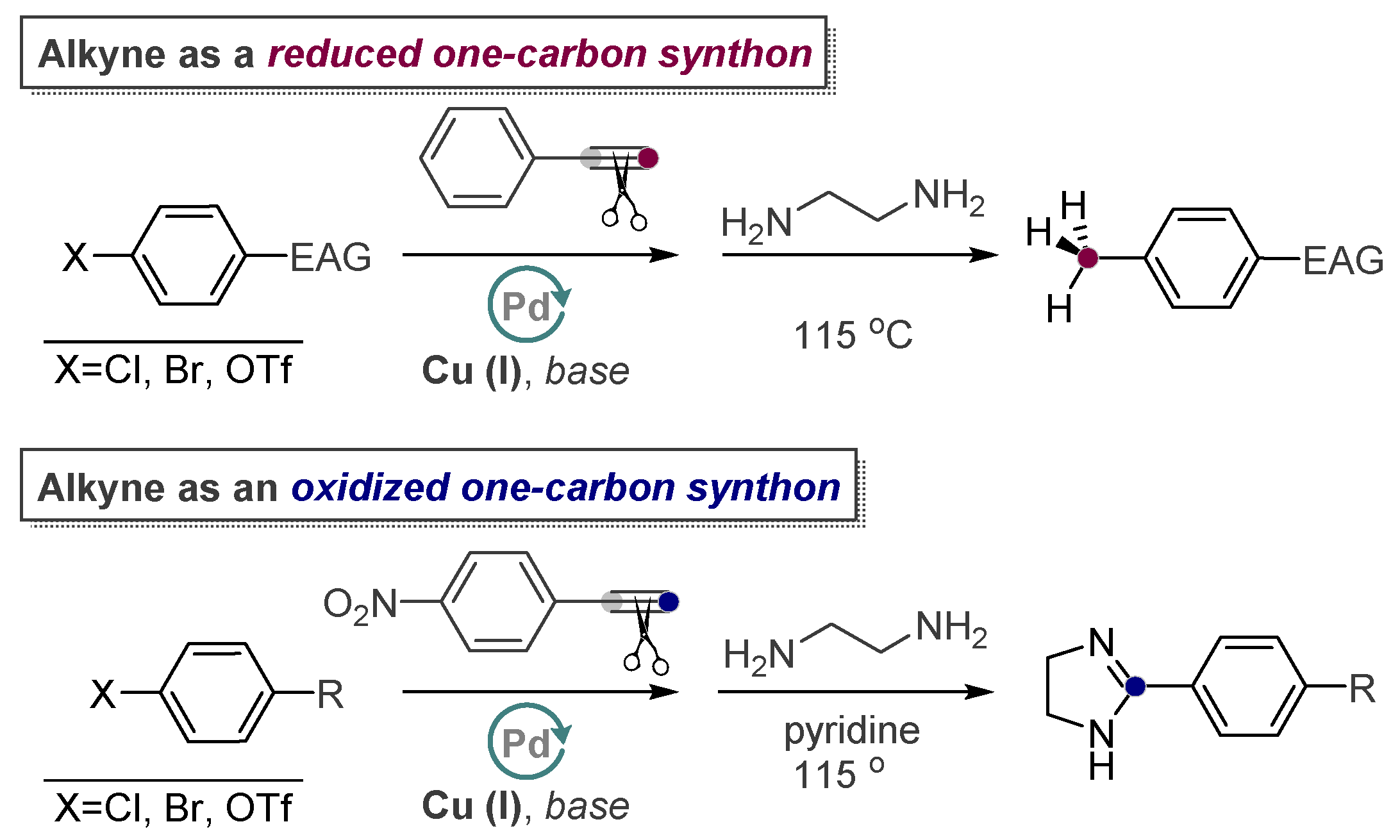
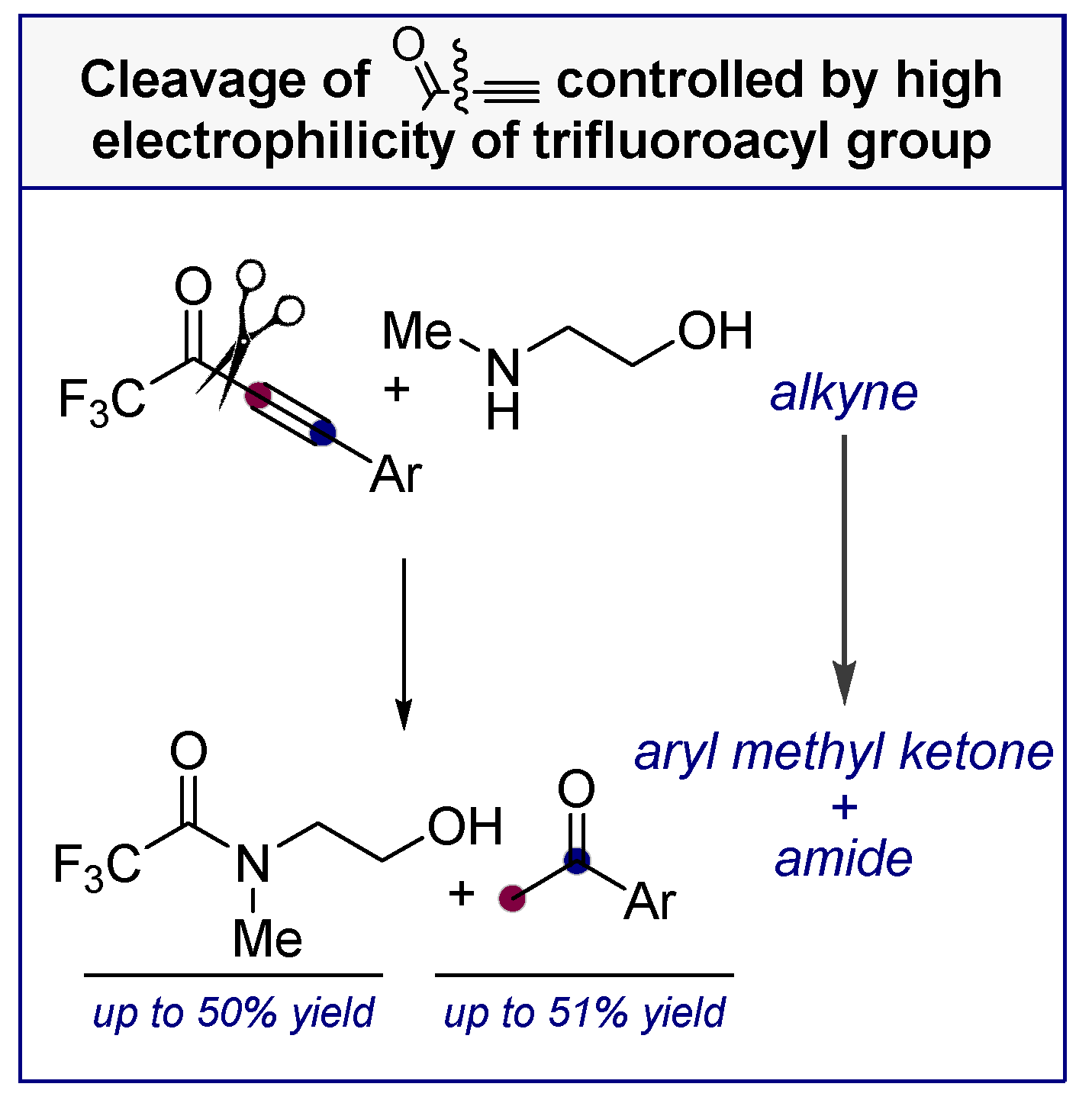
Alkynes in Retro-Aldol and Retro-Mannich Fragmentations
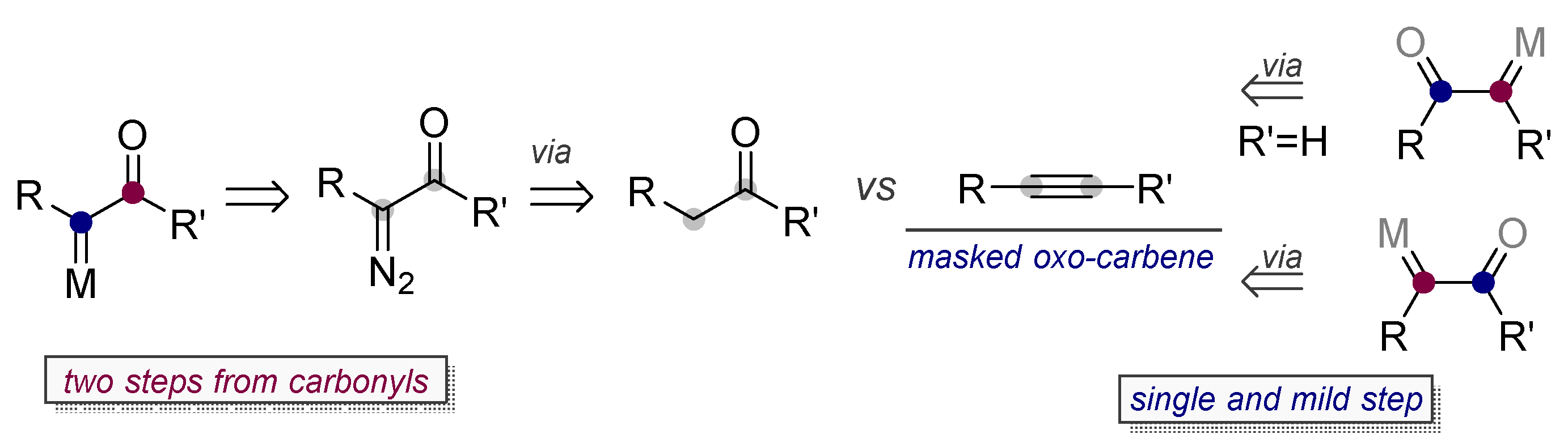
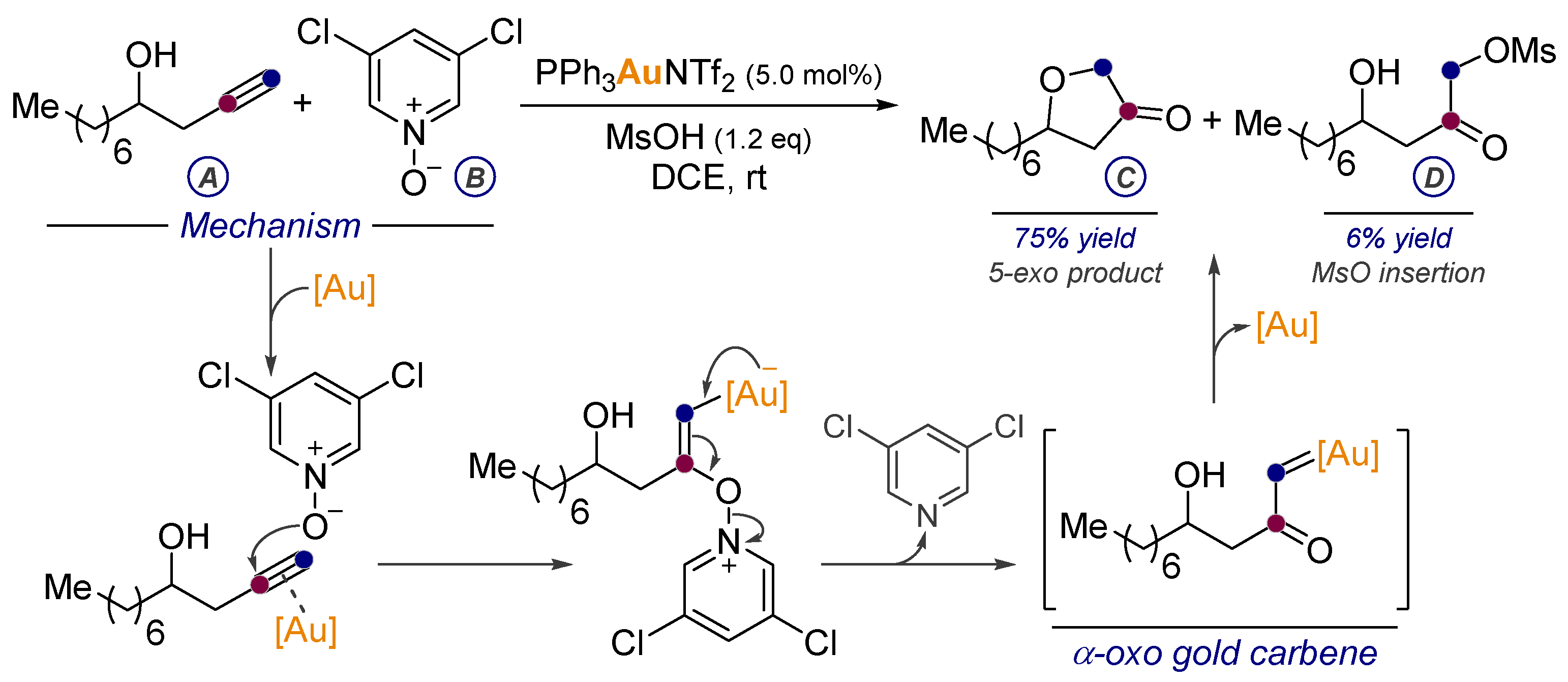
7. Alkynes in the Synthesis of α-oxo Gold Carbenes
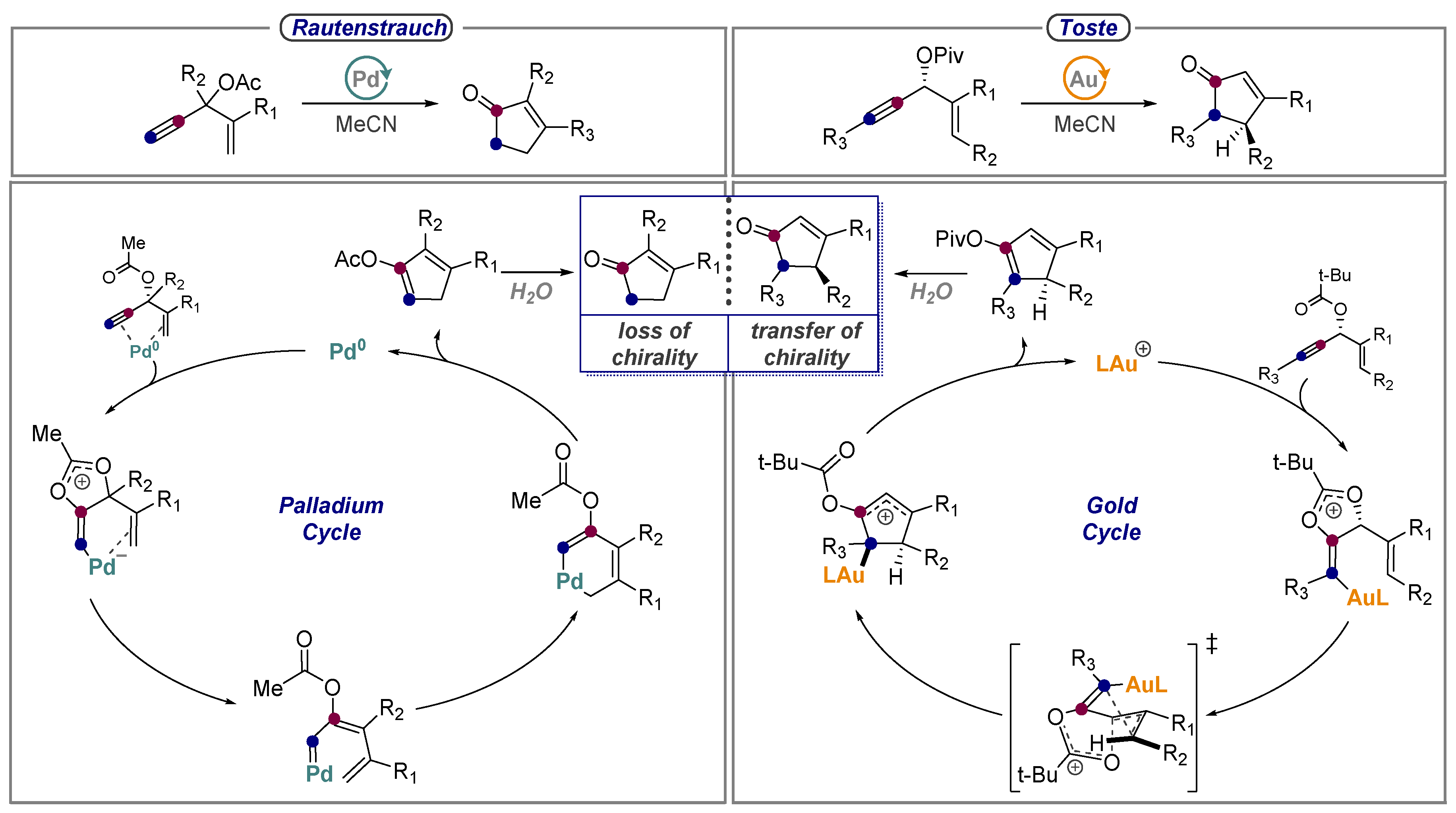
8. Alkynes as Carbonyls in the Rautenstrauch Rearrangement
9. Converting Alkynes to Carbonyls Via Pericyclic Reactions
10. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References and Notes
- Chernick, E.T.; Tykwinski, R.R. Carbon-rich nanostructures: The conversion of acetylenes into materials. J. Phys. Org. Chem. 2013, 26, 742–749. [Google Scholar] [CrossRef]
- Diederich, F.; Stang, P.J.; Tykwinski, R.R. Acetylene Chemistry: Chemistry, Biology and Material Science; Wiley-VCH: Weinheim, Germany, 2005. [Google Scholar]
- Alabugin, I.V.; Gonzalez-Rodriguez, E. Alkyne Origami: Folding Oligoalkynes into Polyaromatics. Acc. Chem. Res. 2018, 51, 1206–1219. [Google Scholar] [CrossRef] [PubMed]
- Alabugin, I.V.; Gold, B. “Two Functional Groups in One Package”: Using Both Alkyne π-Bonds in Cascade Transformations. J. Org. Chem. 2013, 78, 7777–7784. [Google Scholar] [CrossRef] [PubMed]
- Zeidan, T.; Kovalenko, S.V.; Manoharan, M.; Clark, R.J.; Ghiviriga, I.; Alabugin, I.V. Triplet acetylenes as Synthetic Equivalents of 1,2-Dicarbenes. Phantom n,π* State Controls Reactivity in Triplet Photocycloaddition. J. Am. Chem. Soc. 2005, 127, 4270–4285. [Google Scholar] [CrossRef] [PubMed]
- Senese, A.D.; Chalifoux, W.A. Nanographene and Graphene Nanoribbon Synthesis via Alkyne Benzannulations. Molecules 2019, 24, 118. [Google Scholar] [CrossRef] [PubMed]
- Hein, S.J.; Lehnherr, D.; Arslan, H.; Uribe-Romo, F.J.; Dichtel, W.R. Alkyne Benzannulation Reactions for the Synthesis of Novel Aromatic Architectures. Acc. Chem. Res. 2017, 50, 2776–2788. [Google Scholar] [CrossRef] [PubMed]
- Trost, B.M.; Li, C.J. (Eds.) Modern Alkyne Chemistry: Catalytic and Atom-Economic Transformations; Wiley-VCH: Weinheim, Germany, 2014. [Google Scholar]
- Scott, L.T.; Hashemi, M.M.; Meyer, D.T.; Warren, H.B. Corannulene. A convenient new synthesis. J. Am. Chem. Soc. 1991, 113, 7082–7084. [Google Scholar] [CrossRef]
- Jordan, R.S.; Wang, Y.; McCurdy, R.D.; Yeung, M.T.; Marsh, K.L.; Khan, S.I.; Kaner, R.B.; Rubin, Y. Synthesis of Graphene Nanoribbons via the Topochemical Polymerization and Subsequent Aromatization of a Diacetylene Precursor. Chem 2016, 1, 78–90. [Google Scholar] [CrossRef]
- Jordan, R.S.; Li, Y.L.; Lin, C.-W.; McCurdy, R.D.; Lin, J.B.; Brosmer, J.L.; Marsh, K.L.; Khan, S.I.; Houk, K.N.; Kaner, R.B. Synthesis of N = 8 Armchair Graphene Nanoribbons from Four Distinct Polydiacetylenes. J. Am. Chem. Soc. 2017, 139, 15878–15890. [Google Scholar] [CrossRef]
- Goldfinger, M.B.; Swager, T.M. Fused Polycyclic Aromatics via Electrophile-Induced Cyclization Reactions: Application to the Synthesis of Graphite Ribbons. J. Am. Chem. Soc. 1994, 116, 7895–7896. [Google Scholar] [CrossRef]
- Goldfinger, M.B.; Crawford, K.B.; Swager, T.M. Synthesis of Ethynyl-Substituted Quinquephenyls and Conversion to Extended Fused-Ring Structures. J. Org. Chem. 1998, 63, 1676–1686. [Google Scholar] [CrossRef]
- Feng, X.; Pisula, W.; Müllen, K. From Helical to Staggered Stacking of Zigzag Nanographenes. J. Am. Chem. Soc. 2007, 129, 14116–14117. [Google Scholar] [CrossRef]
- Mukherjee, A.; Pati, K.; Liu, R.-S. A Convenient Synthesis of Tetrabenzo-[de,hi,mn,qr]naphthacene from Readily Available 1,2-Di(phenanthren4-yl)ethyne. J. Org. Chem. 2009, 74, 6311–6314. [Google Scholar] [CrossRef]
- Mohamed, R.K.; Mondal, S.; Guerrera, J.V.; Eaton, T.M.; Albrecht-Schmitt, T.E.; Shatruk, M.; Alabugin, I.V. Alkynes as Linchpins for the Additive Annulation of Biphenyls: Convergent Construction of Functionalized Fused Helicenes. Angew. Chem. Int. Ed. 2016, 55, 12054–12058. [Google Scholar] [CrossRef]
- Tsvetkov, N.P.; Gonzalez-Rodriguez, E.; Hughes, A.; dos Passos Gomes, G.; White, F.D.; Kuriakose, F.; Alabugin, I.V. Radical Alkyne Peri-annulations for Synthesis of Functionalized Phenalenes, Benzanthrenes, and Olympicene. Angew. Chem. Int. Ed. 2018, 57, 3651–3655. [Google Scholar] [CrossRef]
- Mohamed, R.; Mondal, S.; Gold, B.; Evoniuk, C.J.; Banerjee, T.; Hanson, K.; Alabugin, I.V. Alkenes as Alkyne Equivalents in Radical Cascades Terminated by Fragmentations: Overcoming Stereoelectronic Restrictions on Ring Expansions for the Preparation of Expanded Polyaromatics. J. Am. Chem. Soc. 2015, 137, 6335–6349. [Google Scholar] [CrossRef]
- Yang, W.; Monteiro, J.H.S.K.; de Bettencourt-Dias, A.; Catalano, V.J.; Chalifoux, W.A. Pyrenes, Peropyrenes, and Teropyrenes: Synthesis, Structures, and Photophysical Properties. Angew. Chem. Int. Ed. 2016, 55, 10427–10430. [Google Scholar] [CrossRef]
- Yang, W.; Longhi, G.; Abbate, S.; Lucotti, A.; Tommasini, M.; Villani, C.; Catalano, V.J.; Lykhin, A.O.; Varganov, S.A.; Chalifoux, W.A. Chiral Peropyrene: Synthesis, Structure, and Properties. J. Am. Chem. Soc. 2017, 139, 13102–13109. [Google Scholar] [CrossRef]
- Yang, W.; Bam, R.; Catalano, V.J.; Chalifoux, W.A. Highly Regioselective Domino Benzannulation Reaction of Buta-1,3-diynes to Construct Irregular Nanographenes. Angew. Chem. Int. Ed. 2018, 57, 14773–14777. [Google Scholar] [CrossRef]
- Ozaki, K.; Murai, K.; Matsuoka, W.; Kawasumi, K.; Ito, H.; Itami, K. One-Step Annulative π-Extension of Alkynes with Dibenzosiloles or Dibenzogermoles by Palladium/o-chloranil Catalysis. Angew. Chem. Int. Ed. 2017, 56, 1361–1364. [Google Scholar] [CrossRef]
- Ito, H.; Ozaki, K.; Itami, K. Annulative π-Extension (APEX): Rapid Access to Fused Arenes, Heteroarenes, and Nanographenes. Angew. Chem. Int. Ed. 2017, 56, 11144–11164. [Google Scholar] [CrossRef] [Green Version]
- Marek, I.; Minko, Y.; Pasco, M.; Mejuch, T.; Gilboa, N.; Chechik, H.; Das, P.J. All-Carbon Quaternary Stereogenic Centers in Acyclic Systems through the Creation of Several C–C Bonds per Chemical Step. J. Am. Chem. Soc. 2014, 136, 2682–2694. [Google Scholar] [CrossRef]
- Das, J.P.; Chechik, H.; Marek, I. A unique approach to aldol products for the creation of all-carbon quaternary stereocentres. Nat. Chem. 2009, 1, 128–132. [Google Scholar] [CrossRef]
- Umezu, S.; Gomes, G.; Yoshinaga, T.; Sakae, M.; Matsumoto, K.; Iwata, T.; Alabugin, I.V.; Shindo, M. Regioselective One-Pot Synthesis of Triptycenes via Triple-Cycloadditions of Arynes to Ynolates. Angew. Chem. Int. Ed. 2016, 56, 1298–1302. [Google Scholar] [CrossRef]
- DeKorver, K.A.; Li, H.; Lohse, A.G.; Hayashi, R.; Lu, Z.; Zhang, Y.; Hsung, R.P. Ynamides: A Modern Functional Group for the New Millennium. Chem. Rev. 2010, 110, 5064–5106. [Google Scholar] [CrossRef]
- Beletskaya, I.P.; Nenajdenko, V.G. Towards the 150th anniversary of the Markovnikov’s rule. Angew. Chem. 2018. [Google Scholar] [CrossRef]
- Beletskaya, I.P.; Ananikov, V.P. Transition-Metal-Catalyzed C−S, C−Se, and C−Te Bond Formation via Cross-Coupling and Atom-Economic Addition Reactions. Chem. Rev. 2011, 111, 1596–1636. [Google Scholar] [CrossRef]
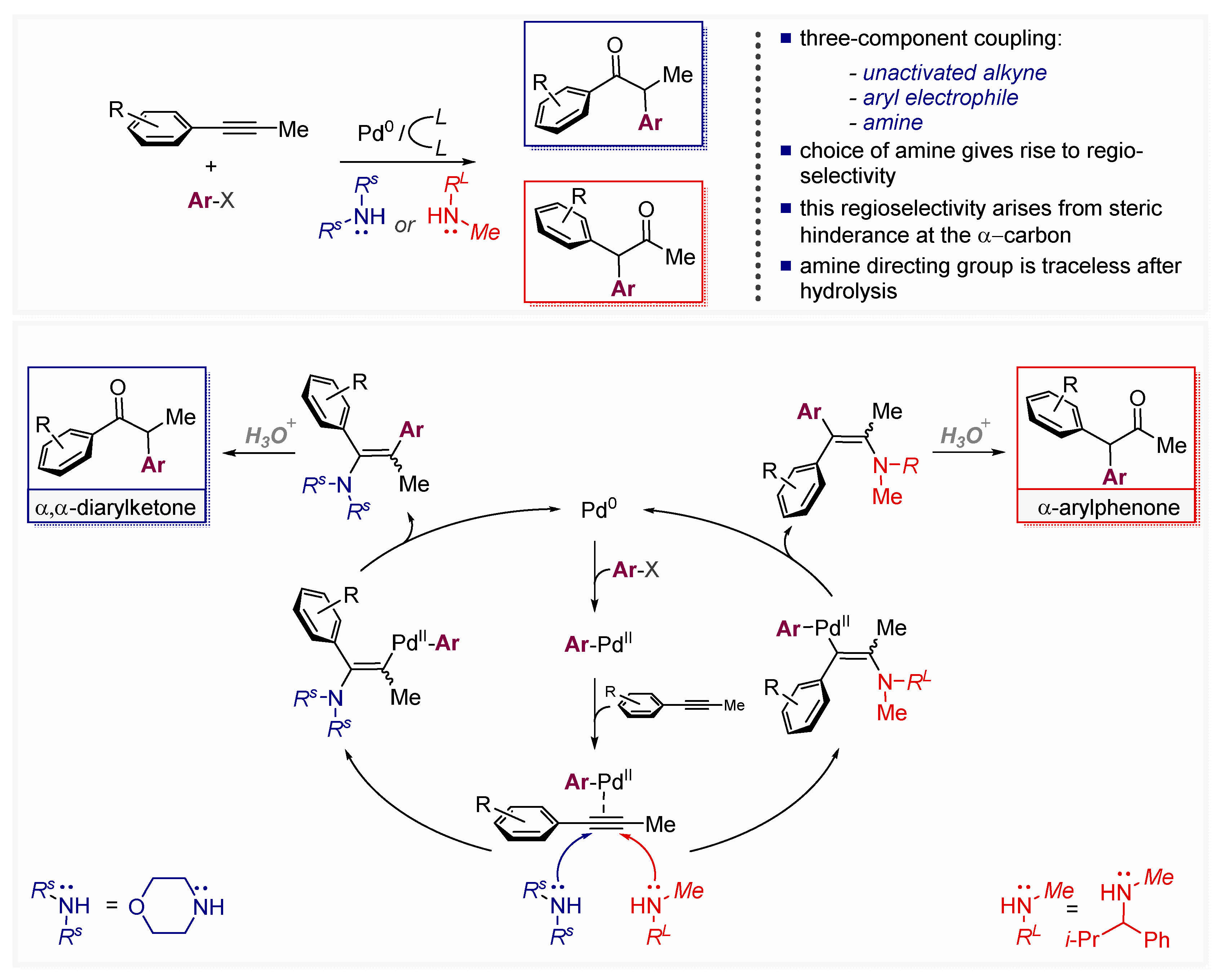
- Park, J.-W.; Kang, B.; Dong, V.M. Catalytic Alkyne Arylation Using Traceless Directing Groups. Angew. Chem. Int. Ed. 2018, 57, 13598–13602. [Google Scholar] [CrossRef]
- Gilmore, K.; Alabugin, I.V. Cyclizations of Alkynes: Revisiting Baldwin’s Rules for Ring Closure. Chem. Rev. 2011, 111, 6513–6556. [Google Scholar] [CrossRef]
- Hashmi, A.S.K. Gold-Catalyzed Organic Reactions. Chem. Rev. 2007, 10, 3180–3211. [Google Scholar] [CrossRef]
- Dorel, R.; Echavarren, A.M. Gold(I)-Catalyzed Activation of Alkynes for the Construction of Molecular Complexity. Chem. Rev. 2015, 115, 9028–9072. [Google Scholar] [CrossRef]
- Arcadi, A. Alternative Synthetic Methods Through New Developments in Catalysis by Gold. Chem. Rev. 2008, 108, 3266–3325. [Google Scholar] [CrossRef]
- Gorin, D.J.; Sherry, B.D.; Toste, F.D. Ligand Effects in Homogeneous Au Catalysis. Chem. Rev. 2008, 108, 3351–3378. [Google Scholar] [CrossRef] [Green Version]
- Fürstner, A. Gold and platinum catalysis-a convenient tool for generating molecular complexity. Chem. Soc. Rev. 2009, 38, 3208–3221. [Google Scholar] [CrossRef]
- Dudnik, A.; Chernyak, N.; Gevorgyan, V. Copper-, Silver-, and Gold Catalyzed Migratory Cycloisomerization Leading to Heterocyclic Five-Membered Rings. Aldrichim. Acta 2010, 43, 37–46. [Google Scholar] [CrossRef]
- Perron, F.; Albizati, K.F. Chemistry of spiroketals. Chem. Rev. 1989, 89, 1617–1661. [Google Scholar] [CrossRef]
- Teles, J.H. Hydration and Hydroalkoxylation of CC Multiple Bonds, in Modern Gold Catalyzed Synthesis; Hashmi, A.S.K., Toste, F.D., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2012; pp. 201–235. [Google Scholar]
- Goodwin, J.A.; Aponick, A. Regioselectivity in the Au-catalyzed hydration and hydroalkoxylation of alkynes. Chem. Commun. 2015, 51, 8730–8741. [Google Scholar] [CrossRef]
- Antoniotti, S.; Genin, E.; Michelet, V.; Genêt, J.-P. Highly efficient access to strained bicyclic ketals via gold-catalyzed cycloisomerization of bis-homopropargylic diols. J. Am. Chem. Soc. 2005, 127, 9976–9977. [Google Scholar] [CrossRef]
- Fukuda, Y.; Utimoto, K. Effective transformation of unactivated alkynes into ketones or acetals with a gold(III) catalyst. J. Org. Chem. 1991, 56, 3729–3731. [Google Scholar] [CrossRef]
- Teles, J.H.; Brode, S.; Chabanas, M. Cationic gold(I) complexes: Highly efficient catalysts for the addition of alcohols to alkynes. Angew. Chem. Int. Ed. 1998, 37, 1415–1418. [Google Scholar] [CrossRef]
- Trost, B.M.; O’Boyle, B.M.; Hund, D. Total Synthesis and Stereochemical Assignment of (−)-Ushikulide, A. J. Am. Chem. Soc. 2009, 131, 15061–15074. [Google Scholar] [CrossRef]
- Benson, S.; Collin, M.-P.; Arlt, A.; Gabor, B.; Goddard, R.; Fürstner, A. Second-generation total synthesis of Spirastrellolide F methyl ester: The alkyne route. Angew. Chem. Int. Ed. 2011, 50, 8739–8744. [Google Scholar] [CrossRef] [PubMed]
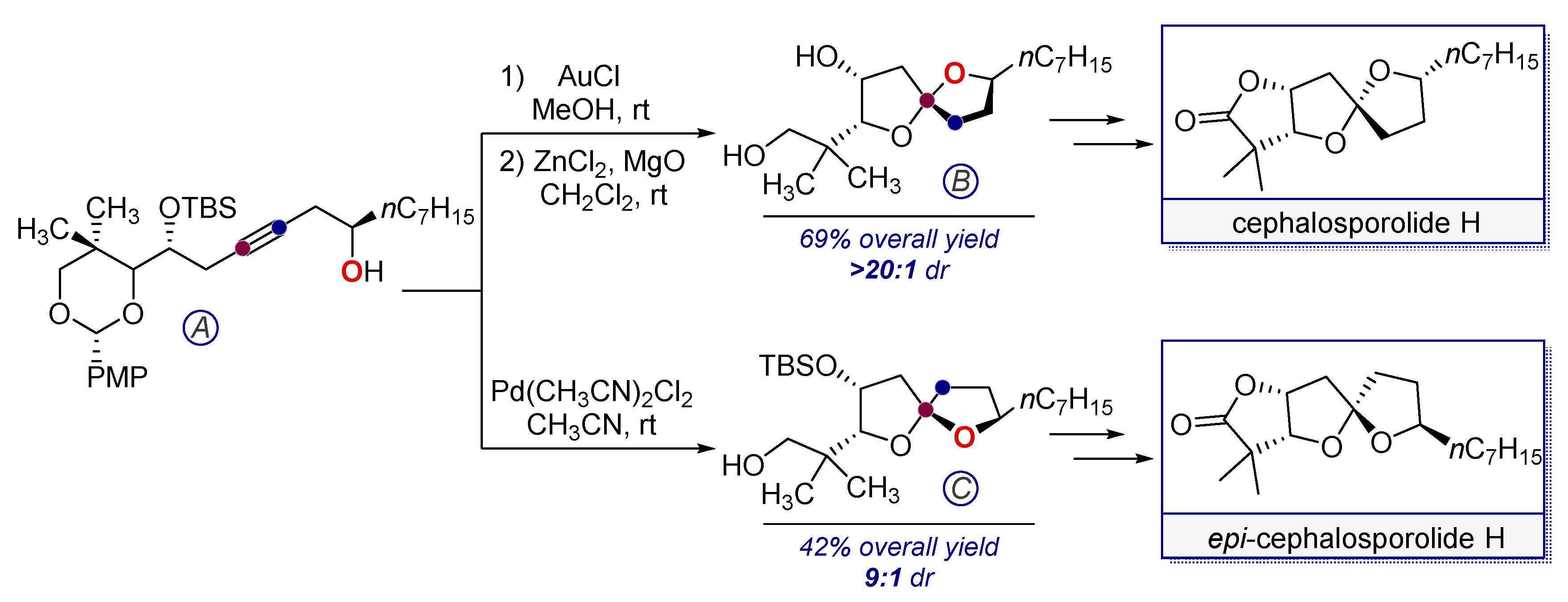
- Tlais, S.F.; Dudley, G.B. Stereocontrol of 5,5-spiroketals in the synthesis of Cephalosporolide H epimers. Org. Lett. 2010, 12, 4698–4701. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhou, F.; Forsyth, C.J. Gold(I)-catalyzed bis-spiroketalization: Synthesis of the trioxadispiroketal-containing A–D rings of Azaspiracid. Angew. Chem. Int. Ed. 2007, 46, 279–282. [Google Scholar] [CrossRef] [PubMed]
- Alabugin, I.V.; Gilmore, K. Finding the right path: Baldwin “Rules for Ring Closure” and stereoelectronic control of cyclizations. (Invited “Viewpoint”). Chem. Commun. 2013, 49, 11246–11250. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, K.; Mohamed, R.K.; Alabugin, I.V. The Baldwin Rules: Revised and Extended. WIREs Comput. Mol. Sci. 2016, 6, 487–514. [Google Scholar] [CrossRef]
- Baldwin, J.E.; Kruse, L.I. Rules for ring closure. Stereoelectronic control in the endocyclic alkylation of ketone enolates. J. Chem. Soc. Chem. Commun. 1977, 233–235. [Google Scholar] [CrossRef]
- Baldwin, J.E.; Lusch, M.J. Rules for ring closure: Application to intramolecular aldol condensations in polyketonic substrates. Tetrahedron 1982, 38, 2939–2947. [Google Scholar] [CrossRef]
- Alabugin, I.V.; Gilmore, K.; Manoharan, M. Rules for Anionic and Radical Ring Closure of Alkynes. J. Am. Chem. Soc. 2011, 133, 12608–12623. [Google Scholar] [CrossRef]
- Gilmore, K.; Manoharan, M.; Wu, J.; Schleyer, P.v.R.; Alabugin, I.V. Aromatic Transition States in Non-Pericyclic Reactions: Anionic 5-Endo Cyclizations are Aborted Sigmatropic Shifts. J. Amer. Chem. Soc. 2012, 134, 10584–10594. [Google Scholar] [CrossRef]
- Alabugin, I.V.; Manoharan, M.; Breiner, B.; Lewis, F. Control of Kinetics and Thermodynamics of [1,5]-Shifts by Aromaticity: A View Through the Prism of Marcus Theory. J. Am. Chem. Soc. 2003, 125, 9329–9342. [Google Scholar] [CrossRef] [PubMed]
- Alabugin, I.V.; Manoharan, M. Thermodynamic and Strain Effects in the Competition Between 5-Exo-dig and 6-Endo-Dig Cyclizations of Vinyl and Aryl Radicals. J. Am. Chem. Soc. 2005, 127, 12583–12594. [Google Scholar] [CrossRef]
- Marvell, E.N.; Titterington, D. A novel synthesis of 4-cycloheptenones. Tetrahedron Lett. 1980, 21, 2123–2124. [Google Scholar] [CrossRef]
- Eglinton, G.; Jones, E.R.H.; Whiting, M.C. Researches on acetylenic compounds. Part XXXVIII. A new method for the introduction of the acetylenic linkage. J. Chem. Soc. 1952, 2873–2882. [Google Scholar] [CrossRef]
- Paul, R.; Tchelitcheff, S. Derivatives of 4-pentyn-1-ol. C. R. Acad. Sci. 1950, 230, 1872–1873. [Google Scholar]
- Padwa, A.; Krumpe, K.E.; Weingarten, M.D. An Unusual Example of a 6-Endo-Dig Addition to an Unactivated Carbon-Carbon Triple Bond. J. Org. Chem. 1995, 60, 5595–5603. [Google Scholar] [CrossRef]
- Trost, B.M.; Runge, T.A. Palladium-catalyzed 1,3-oxygen-to-carbon alkyl shifts. A cyclopentanone synthesis. J. Am. Chem. Soc. 1981, 103, 7559–7572. [Google Scholar] [CrossRef]
- García, H.; Iborra, S.; Primo, J.; Miranda, M.A. 6-Endo-Dig vs. 5-Exo-Dig ring closure in o-hydroxyaryl phenylethynyl ketones. A new approach to the synthesis of flavones and aurones. J. Org. Chem. 1986, 51, 4432–4436. [Google Scholar] [CrossRef]
- Castro, C.E.; Gaughan, E.G.; Owsley, D.C. Indoles, Benzofurans, Phthalides, and Tolanes via Copper(I) Acetylides. J. Org. Chem. 1968, 31, 4071–4078. [Google Scholar] [CrossRef]
- Castro, C.E.; Havlin, R.; Honwad, V.K.; Malte, A.; Moje, S. Copper(I) substitutions. Scope and mechanism of cuprous acetylide substitutions. J. Am. Chem. Soc. 1969, 91, 6464–6470. [Google Scholar] [CrossRef]
- Shvartsberg, M.S.; Vasilevsky, S.F.; Anisimova, T.V.; Gerasimov, V.A. Cyclization of acetylenylpyrazolecarboxylic acids. Russ. Chem. Bull. 1981, 30, 1071–1076. [Google Scholar] [CrossRef]
- Vasilevsky, S.F.; Rubinshtein, E.M.; Shvartsberg, M.S. Condensation of N-methyl-4-iodopyra zolecarboxylic acids with copper acetylides. Russ. Chem. Bull. 1978, 30, 1021–1023. [Google Scholar] [CrossRef]
- Vasilevsky, S.F.; Gerasimov, V.A.; Shvartsberg, M.S. Condensation of iodo-N-methylpyrazole-4-carboxylic acids with copper acetylides. Russ. Chem. Bull. 1981, 30, 683–685. [Google Scholar] [CrossRef]
- Prikhodko, T.A.; Kurilenko, V.M.; Vasilevsky, S.F.; Shvartsberg, M.S. Synthesis and certain properties of acetylenylindoles. Russ. Chem. Bull. 1990, 39, 120–127. [Google Scholar] [CrossRef]
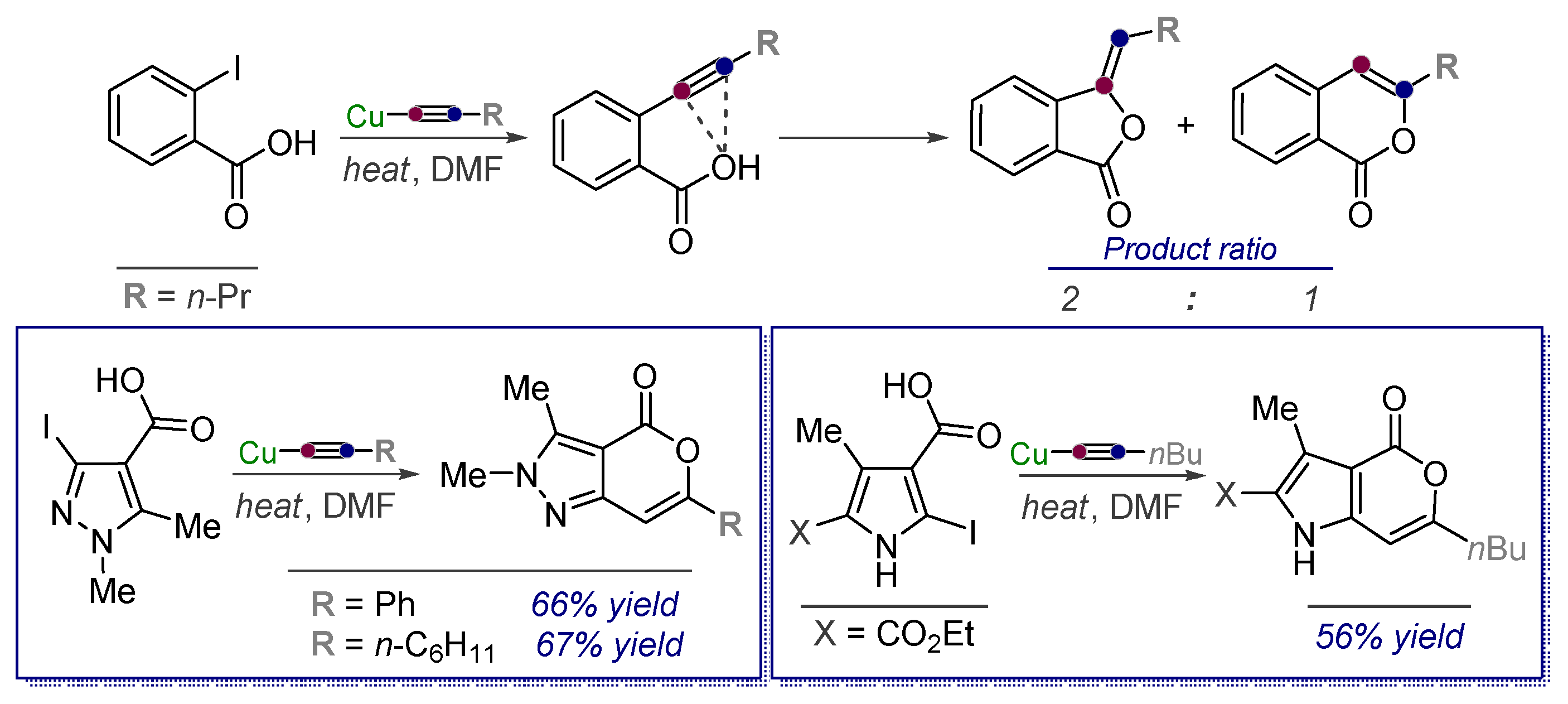
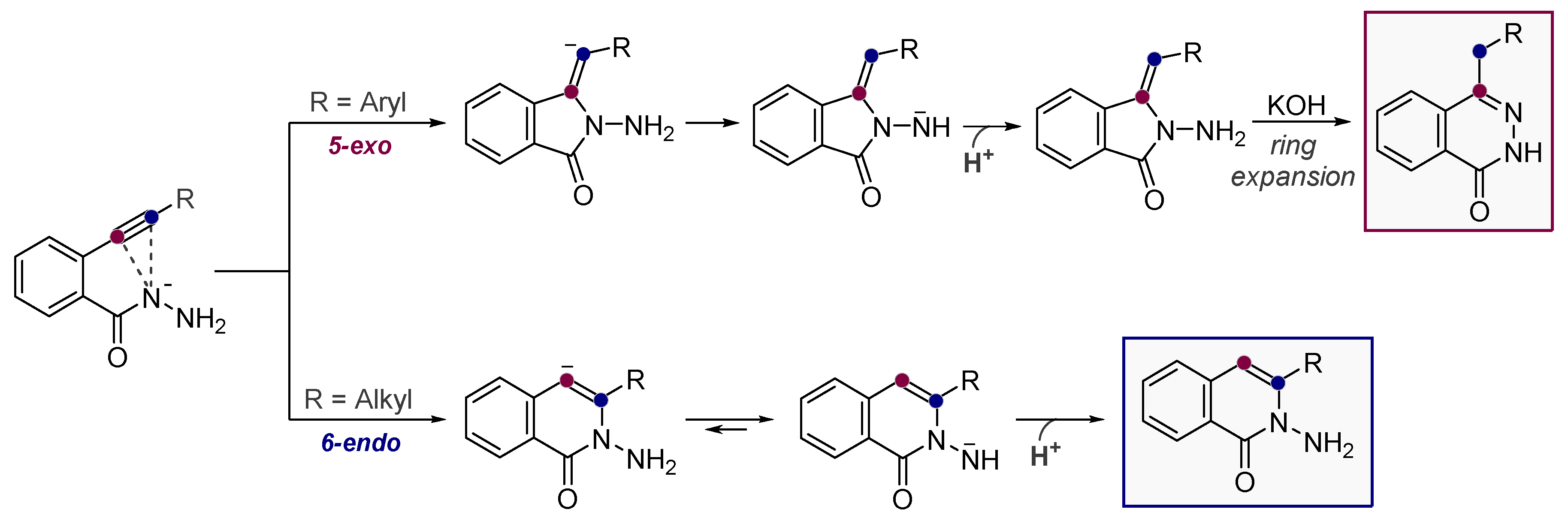
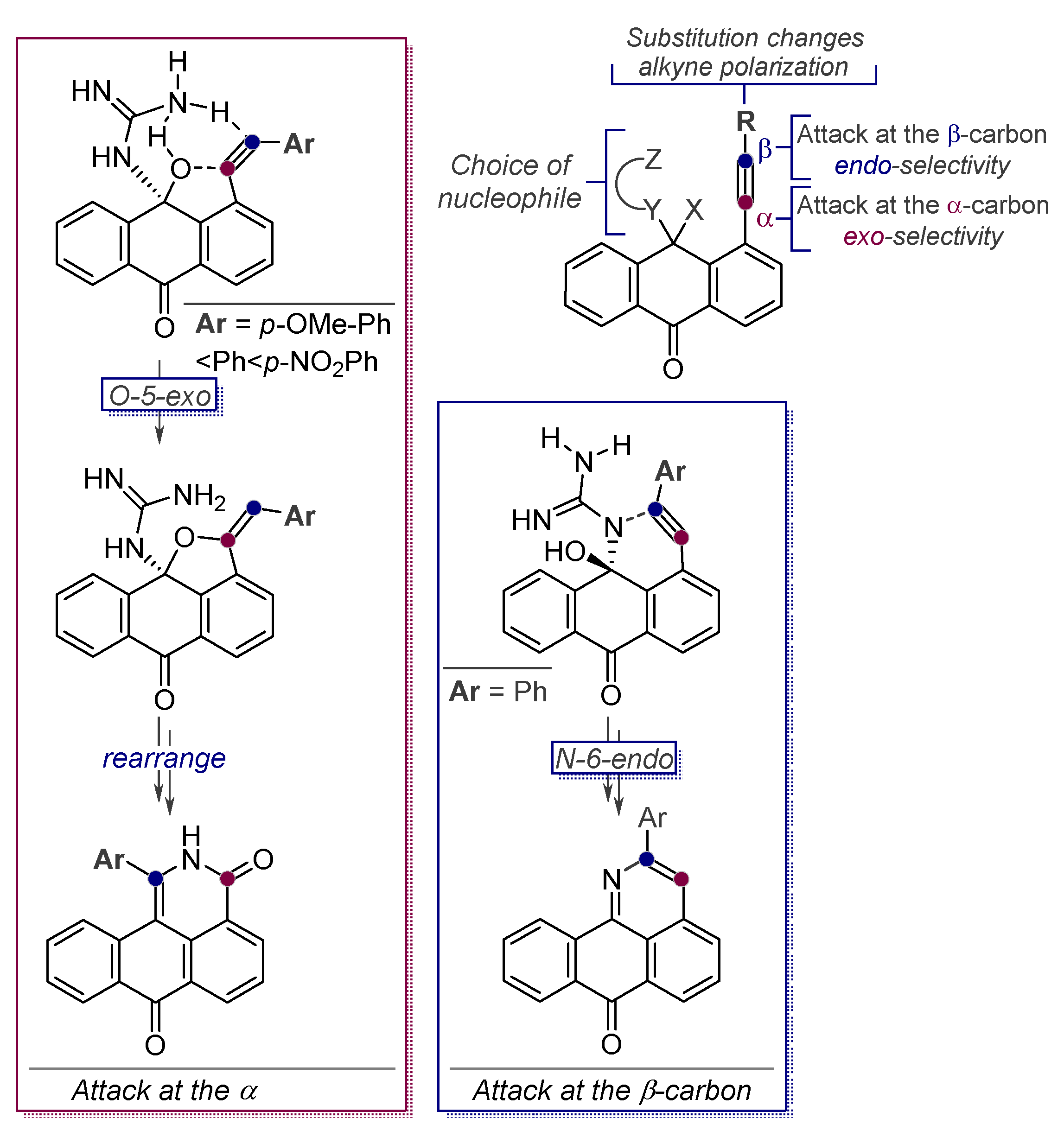
- Vasilevsky, S.F.; Gold, B.; Mikhailovskaya, T.F.; Alabugin, I.V. Strain Control in Nucleophilic Cyclizations: Reversal of exo-Selectivity in Cyclizations of Hydrazides of Acetylenyl Carboxylic Acids by Annealing to a Pyrazole Scaffold. J. Phys. Org. Chem. 2012, 25, 998–1005. [Google Scholar] [CrossRef]
- Vasilevsky, S.F.; Mikhailovskaya, T.F.; Mamatyuk, V.I.; Bogdanchikov, G.A.; Manoharan, M.; Alabugin, I.V. Tuning Selectivity of Anionic Cyclizations: Competition between 5-Exo- and 6-Endo-dig Closures of Hydrazides of o-Acetylenyl Benzoic Acids and Based-catalyzed Fragmentation/Recyclization of the Initial 5-Exo-Dig Products. J. Org. Chem. 2009, 74, 8106–8117. [Google Scholar] [CrossRef] [PubMed]
- Vasilevsky, S.F.; Baranov, D.S.; Mamatyuk, V.I.; Gatilov, Y.V.; Alabugin, I.V. An Unexpected Rearrangement which Disassembles Alkyne Moiety Through Formal Nitrogen Atom Insertion between Two Acetylenic Carbons and Related Cascade Transformations: New Approach to Sampagine Derivatives and Polycyclic Aromatic Amides. J. Org. Chem. 2009, 74, 6143–6150. [Google Scholar] [CrossRef]
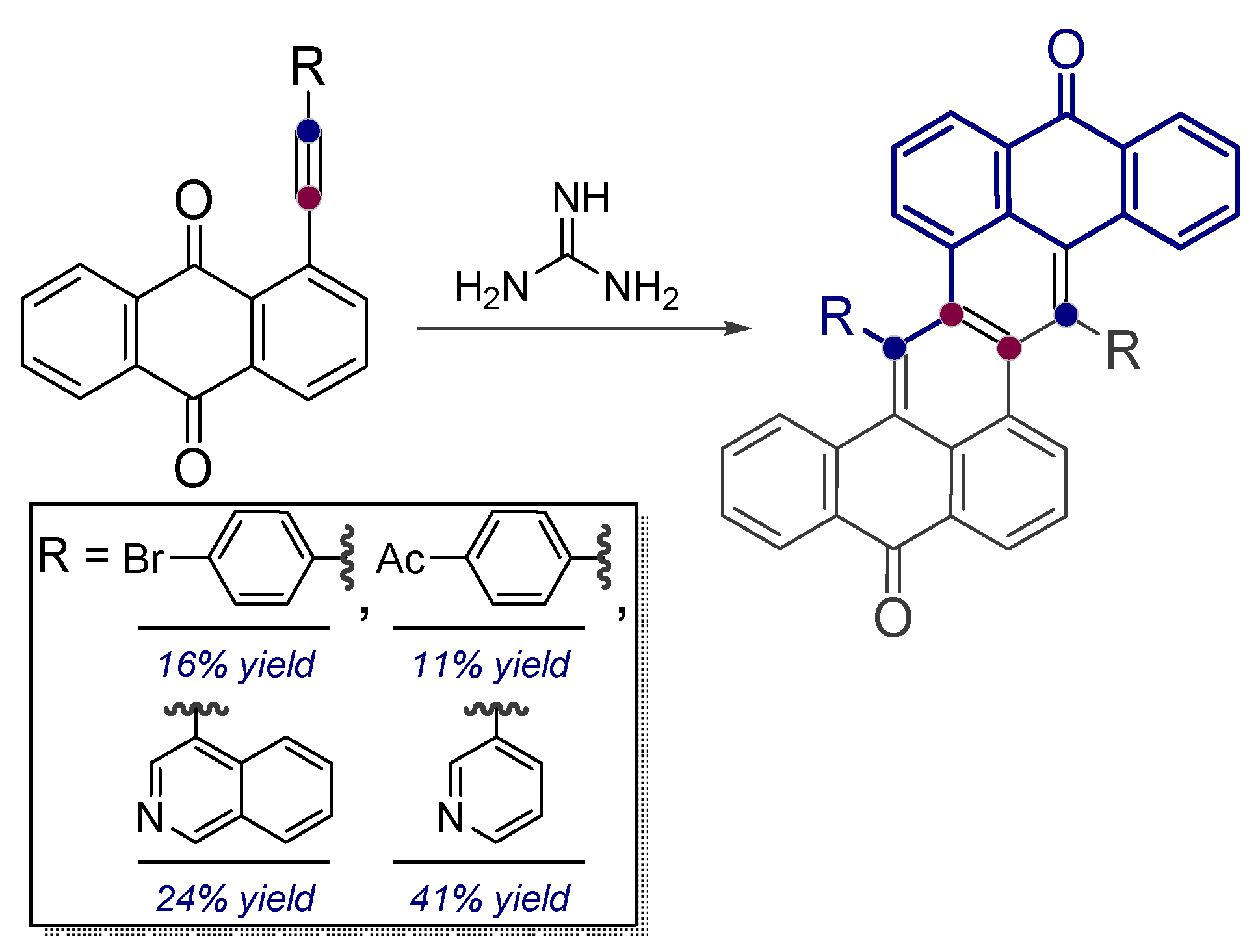
- Baranov, D.S.; Vasilevsky, S.F. Reaction of guanidine with peri-substituted (R-ethynyl)-9,10-anthraquinones bearing electron-donating substituents. Russ. Chem. Bull. 2010, 59, 1031–1034. [Google Scholar] [CrossRef]
- Vasilevsky, S.F.; Baranov, D.S.; Mamatyuk, V.I.; Fadeev, D.S.; Gatilov, Y.V.; Stepanov, A.A.; Vasilieva, N.V.; Alabugin, I.V. Conformational Flexibility of Fused Tetracenedione Propellers Obtained from One-Pot Reductive Dimerization of Acetylenic Quinones. J. Org. Chem. 2015, 80, 1618–1631. [Google Scholar] [CrossRef]
- Godoi, B.; Schumacher, R.F.; Zeni, G. Synthesis of Heterocycles via Electrophilic Cyclization of Alkynes Containing Heteroatom. Chem. Rev. 2011, 111, 2937–2980. [Google Scholar] [CrossRef]
- Byers, P.M.; Rashid, J.I.; Mohamed, R.K.; Alabugin, I.V. Polyaromatic Ribbon/Benzofuran Fusion via Consecutive Endo Cyclizations of Enediynes. Org. Lett. 2012, 14, 6032–6035. [Google Scholar] [CrossRef] [PubMed]
- Hirano, K.; Inaba, Y.; Takahashi, N.; Shimano, M.; Oishi, S.; Fujii, N.; Ohno, H. Direct Synthesis of Fused Indoles by Gold-Catalyzed Cascade Cyclization of Diynes. J. Org. Chem. 2011, 76, 1212–1227. [Google Scholar] [CrossRef] [PubMed]
- Asiri, A.M.; Hashmi, A.S.K. Gold-catalysed reactions of diynes. Chem. Soc. Rev. 2016, 45, 4471–4503. [Google Scholar] [CrossRef] [PubMed]
- Gomes, G.P.; Alabugin, I.V. Drawing Catalytic Power from Charge Separation: Stereoelectronic and Zwitterionic Assistance in the Au(I)-Catalyzed Bergman Cyclization. J. Am. Chem. Soc. 2017, 139, 3406–3416. [Google Scholar] [CrossRef] [PubMed]
- Cai, T.; Yang, Y.; Li, W.-W.; Qin, W.-B.; Wen, T.-B. Efficient endo Cycloisomerization of Terminal Alkynols Catalyzed by a New Ruthenium Complex with 8-(Diphenylphosphino)quinoline Ligand and Mechanistic Investigation. Chem. Eur. J. 2018, 24, 1606–1618. [Google Scholar] [CrossRef]
- Ferrier, R.J. Unsaturated carbohydrates. Part 21. A carbocyclic ring closure of a hex-5-enopyranoside derivative. J. Chem. Soc. Perkin Trans. 1 1979, 1455–1458. [Google Scholar] [CrossRef]
- Petasis, N.A.; Lu, S.-P. STEREOCONTROLLED SYNTHESIS OF SUBSTITUTED TETRAHYDROPYRANS FROM 1,3-DIOXAN-4-ONES. Tetrahedron Lett. 1996, 37, 141–144. [Google Scholar] [CrossRef]
- Smith III, A.B.; Fox, R.J.; Razler, T.M. Evolution of the Petasis−Ferrier Union/Rearrangement Tactic: Construction of Architecturally Complex Natural Products Possessing the Ubiquitous cis-2,6-Substituted Tetrahydropyran Structural Element. Acc. Chem. Res. 2008, 41, 675–687. [Google Scholar] [CrossRef]
- For selected applications of the Petasis–Ferrier rearrangement in the total synthesis of natural products, see: Smith, A.B., III; Bosanac, T.; Basu, K. Evolution of the Total Synthesis of (−)-Okilactomycin Exploiting a Tandem Oxy-Cope Rearrangement/Oxidation, a Petasis−Ferrier Union/Rearrangement, and Ring-Closing Metathesis. J. Am. Chem. Soc. 2009, 131, 2348–2358. [Google Scholar] [CrossRef]
- Smith, A.B., III; Simov, V. Total Synthesis of the Marine Natural Product (−)-Clavosolide, A. A Showcase for the Petasis—Ferrier Union/Rearrangement Tactic. Org. Lett. 2006, 8, 3315–3318. [Google Scholar] [CrossRef]
- Smith III, A.B.; Mesaros, E.F.; Meyer, E.A. Evolution of a Total Synthesis of (−)-Kendomycin Exploiting a Petasis−Ferrier Rearrangement/Ring-Closing Olefin Metathesis Strategy. J. Am. Chem. Soc. 2006, 128, 5292–5299. [Google Scholar] [CrossRef]
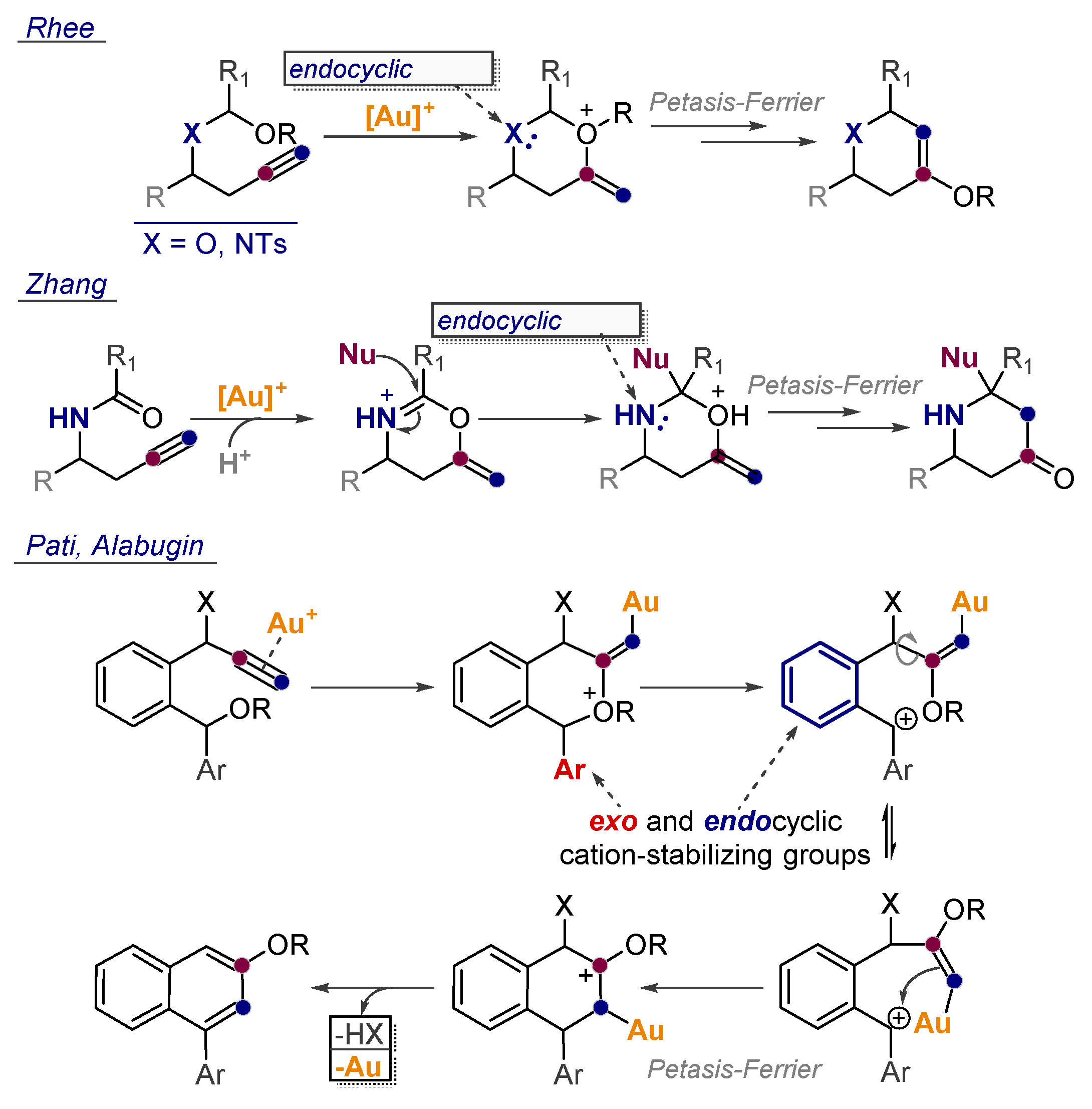
- Bae, H.J.; Jeong, W.; Lee, J.H.; Rhee, Y.H. Gold(I)-Catalyzed Access to Tetrahydropyran-4-ones from 4 (Alkoxyalkyl)oxy-1-butynes: Formal Catalytic Petasis–Ferrier Rearrangement. Chem. Eur. J. 2011, 17, 1433–1436. [Google Scholar] [CrossRef]
- Kim, C.; Bae, H.J.; Lee, J.H.; Jeong, W.; Kim, H.; Sampath, V.; Rhee, Y.H. Formal Alkyne Aza-Prins Cyclization: Gold(I)-Catalyzed Cycloisomerization of Mixed N,O-Acetals Generated from Homopropargylic Amines to Highly Substituted Piperidines. J. Am. Chem. Soc. 2009, 131, 14660–14661. [Google Scholar] [CrossRef]
- Cui, L.; Li, C.; Zhang, L. A Modular, Efficient, and Stereoselective Synthesis of Substituted Piperidin-4-ols. Angew. Chem. Int. Ed. 2010, 49, 9178–9181. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Zhang, L. Access to Electron-Rich Arene-Fused Hexahydroquinolizinones through a Gold-Catalysis-Initiated Cascade Process. Angew. Chem. Int. Ed. 2012, 51, 7301–7304. [Google Scholar] [CrossRef]
- Sze, E.M.L.; Rao, W.; Koh, M.J.; Chan, P.W.H. Gold-Catalyzed Tandem Intramolecular Heterocyclization/Petasis–Ferrier Rearrangement of 2-(Prop-2-ynyloxy)benzaldehydes as an Expedient Route to Benzo[b]oxepin-3(2 H)-ones. Chem. Eur. J. 2011, 17, 1437–1441. [Google Scholar] [CrossRef]
- Gade, A.B.; Patil, N.T. Gold(I)-Catalyzed Hydroaminaloxylation and Petasis–Ferrier Rearrangement Cascade of Aminaloalkynes. Org. Lett. 2016, 18, 1844–1847. [Google Scholar] [CrossRef]
- Aaseng, J.E.; Iqbal, N.; Sperger, C.A.; Fiksdahl, A. 3-Fluorotetrahydropyran-4-one derivatives from homopropargyl acetal. J. Fluorine Chem. 2014, 161, 142–148. [Google Scholar] [CrossRef]
- Jiang, G.J.; Wang, Y.; Yu, X.Z. DFT Study on the Mechanism and Stereochemistry of the Petasis–Ferrier Rearrangements. J. Org. Chem. 2013, 78, 6947–6955. [Google Scholar] [CrossRef]
- Pati, K.; Alabugin, I.V. Synthesis of Substituted Biaryls Through Gold-Catalyzed Petasis-Ferrier Rearrangement of Propargyl Ethers. Eur. J. Org. Chem. 2014, 19, 3986–3990. [Google Scholar] [CrossRef]
- Hassall, C.H. Baeyer-Villiger Oxidation of Aldehydes and Ketones. In Organic Reactions; John Wiley & Sons, Inc.: New York, NY, USA, 1957; Volume 9. [Google Scholar] [CrossRef]
- Renz, M.; Meunier, B. 100 Years of Baeyer–Villiger Oxidations. Eur. J. Org. Chem. 1999, 737–750. [Google Scholar] [CrossRef]
- Strukul, G. Transition Metal Catalysis in the Baeyer–Villiger Oxidation of Ketones. Angew. Chem. Int. Ed. 1998, 37, 1198–1209. [Google Scholar] [CrossRef]
- Yaremenko, I.A.; Vil’, V.A.; Demchuk, D.V.; Terent’ev, A.O. Rearrangements of organic peroxides and related processes. Beilstein J. Org. Chem. 2016, 12, 1647–1748. [Google Scholar] [CrossRef] [Green Version]
- Ten Brink, G.J.; Arends, I.W.C.E.; Sheldon, R.A. The Baeyer−Villiger Reaction: New Developments toward Greener Procedures. Chem. Rev. 2004, 104, 4105–4124. [Google Scholar] [CrossRef]
- Rioz-Martínez, A.; Cuetos, A.; Rodríguez, C.; de Gonzalo, G.; Lavandera, I.; Fraaije, M.W.; Gotor, V. Dynamic Kinetic Resolution of α-Substituted β-Ketoesters Catalyzed by Baeyer–Villiger Monooxygenases: Access to Enantiopure α-Hydroxy Esters. Angew. Chem. Int. Ed. 2011, 50, 8387–8390. [Google Scholar] [CrossRef]
- Criegee, R. Die Umlagerung der Dekalin-peroxydester als Folge von kationischem Sauerstoff. Justus Liebigs Ann. Chem. 1948, 560, 127–135. [Google Scholar] [CrossRef]
- Gomes, G.d.P.; Vil, V.; Terent’ev, A.; Alabugin, I.V. Stereoelectronic source of the anomalous stability of bis-peroxides. Chem. Sci. 2015, 6, 6783–6791. [Google Scholar] [CrossRef]
- Crudden, C.M.; Chen, A.C.; Calhoun, L.A. A Demonstration of the Primary Stereoelectronic Effect in the Baeyer–Villiger Oxidation of α-Fluorocyclohexanones. Angew. Chem. Int. Ed. 2000, 39, 2851–2855. [Google Scholar] [CrossRef]
- Chandrasekhar, S.; Roy, C.D. Conformationally restricted Criegee intermediates: Evidence for formation and stereoelectronically controlled fragmentation. J. Chem. Soc. Perkin Trans. 2 1994, 2141–2143. [Google Scholar] [CrossRef]
- Alabugin, I.V.; Gomes, G.d.P.; Miguel, A.A. Hyperconjugation. WIREs Comput. Mol. Sci. 2018, e1389. [Google Scholar] [CrossRef]
- Vil’, V.A.; Gomes, G.d.P.; Bityukov, O.V.; Lyssenko, K.A.; Nikishin, G.I.; Alabugin, I.V.; Terent’ev, A.O. Interrupted Baeyer–Villiger Rearrangement: Building A Stereoelectronic Trap for the Criegee Intermediate. Angew. Chem. Int. Ed. 2018, 57, 3372–3376. [Google Scholar] [CrossRef]
- Noyori, R.; Sato, T.; Kobayashi, H. Remote substituent effects in the Baeyer-Villiger oxidation. I. through-bond γ substituent effect on the regioselectivity. Tetrahedron Lett. 1980, 21, 2569–2572. [Google Scholar] [CrossRef]
- Noyori, R.; Kobayashi, H.; Sato, T. Remote substituent effects in the Baeyer-Villiger oxidation. II. regioselection based on the hydroxyl group orientation in the tetrahedral intermediate. Tetrahedron Lett. 1980, 21, 2573–2576. [Google Scholar] [CrossRef]
- Of course, substituent migrations from an sp2-hybridized carbon (usually, a carbonyl) to a nitrogen with a good leaving group are known as the part of Curtius, Schmidt, and Lossen rearrangements.
- Baranov, D.; Gold, B.; Vasilevsky, S.; Alabugin, I.V. Divergent Cyclizations of 1-R-Ethynyl-9,10-anthraquinones: Use of Thiourea as a “S2-” Equivalent in an “Anchor-Relay” Addition Mediated by Formal C-H Activation. J. Org. Chem. 2013, 78, 2074–2082. [Google Scholar] [CrossRef]
- Baranov, D.S.; Vasilevsky, S.F.; Gold, B.; Alabugin, I.V. Urea as a Solvent and Reagent for the Addition/Cyclization/Fragmentation Cascades Leading to 2-R-7H-dibenzo[de,h]quinolin-7- one Analogues of Aporphinoid Alkaloids. RSC Adv. 2011, 1, 1745–1750. [Google Scholar] [CrossRef]
- Heathcock, C.H. The Aldol Reactions. In Comprehensive Organic Synthesis; Trost, B.M., Fleming, I., Heathcock, C.H., Eds.; Pergamon: Oxford, UK, 1991; Volume 2, pp. 99–181. [Google Scholar]
- Palomo, C.; Oiarbide, M.; García, J.M. The Aldol Addition Reaction: An Old Transformation at Constant Rebirth. Chem. Eur. J. 2002, 8, 36–44. [Google Scholar] [CrossRef]
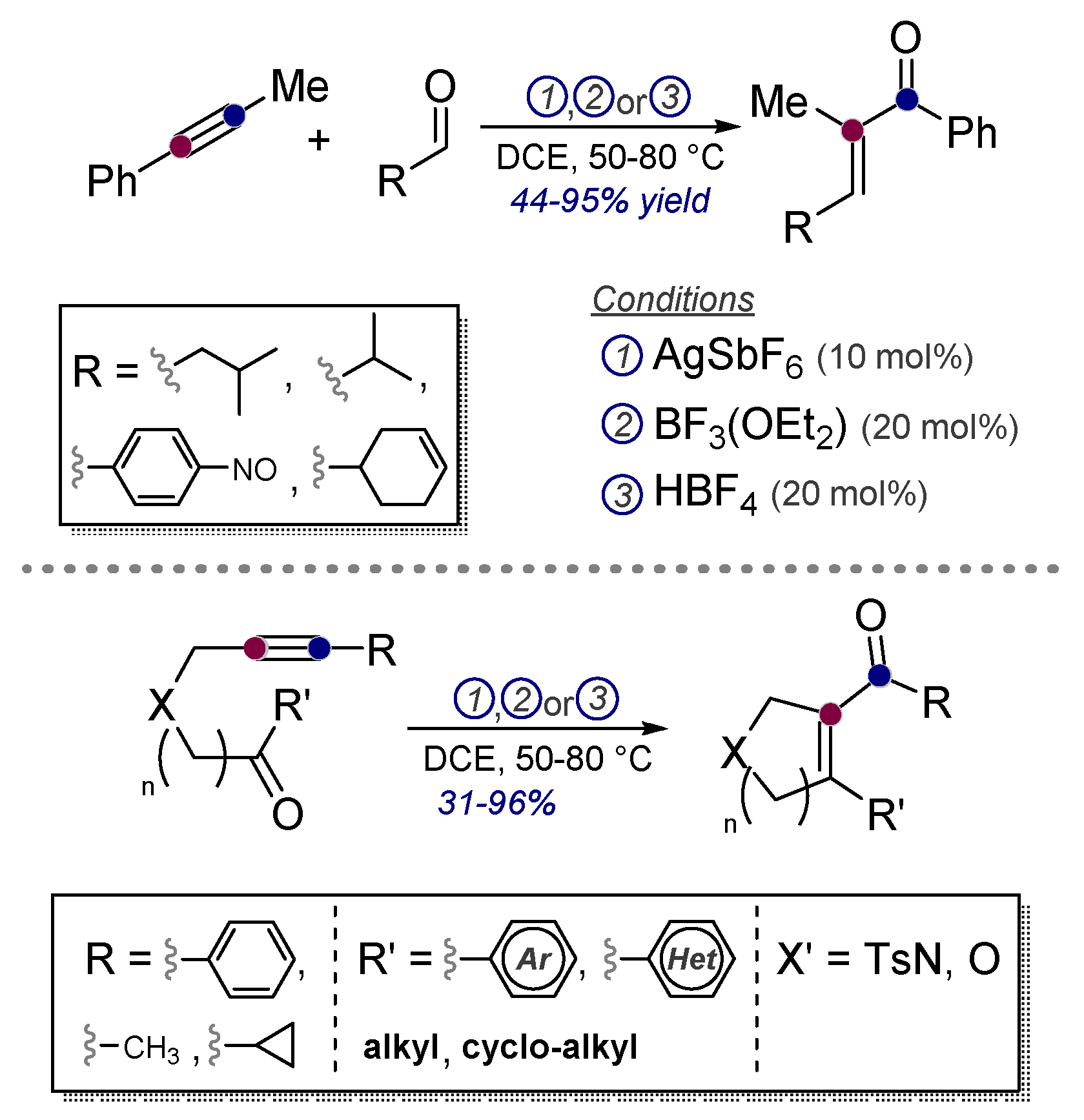
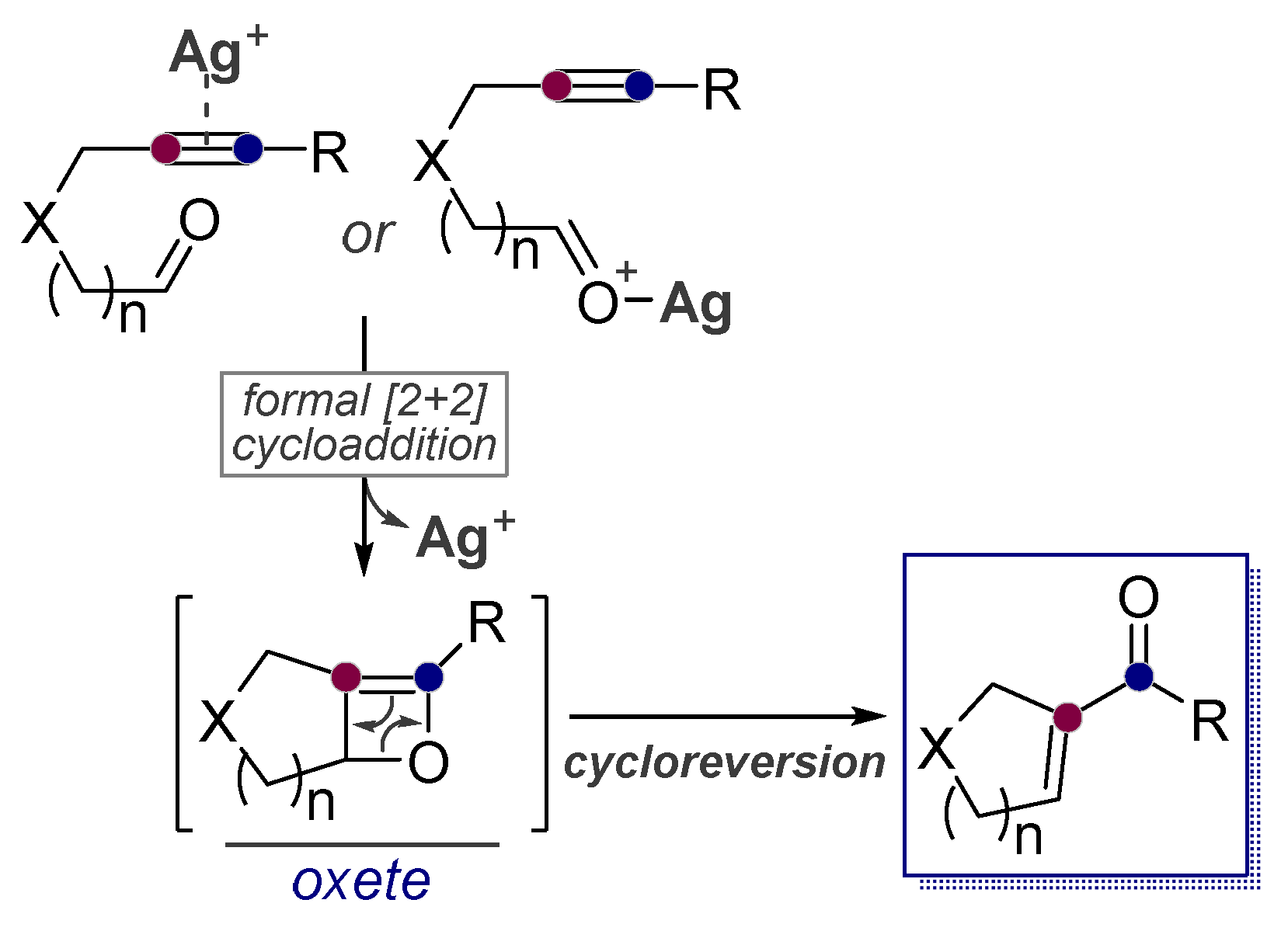
- Rhee, J.U.; Krische, M.J. Alkynes as Synthetic Equivalents to Stabilized Wittig Reagents: Intra- and Intermolecular Carbonyl Olefinations Catalyzed by Ag(I), BF3, and HBF4. Org. Lett. 2005, 7, 2493–2495. [Google Scholar] [CrossRef]
- Jin, T.; Yang, F.; Liu, C.; Yamamoto, Y. TfOH-catalyzed intramolecular alkyne–ketone metathesis leading to highly substituted five-membered cyclic enones. Chem. Commun. 2009, 3533–3535. [Google Scholar] [CrossRef]
- Kurtz, K.C.M.; Hsung, P.R.; Zhang, Y. A Ring-Closing Yne-Carbonyl Metathesis of Ynamides. Org. Lett. 2006, 8, 231–234. [Google Scholar] [CrossRef]
- Balog, A.; Geib, S.J.; Curran, D.P. Additive and Medium Effects on Lewis Acid-Promoted Cationic.pi.-Cyclizations of Alkenyl- and Alkynylcyclopentane-1,3-diones. J. Org. Chem. 1995, 60, 345–352. [Google Scholar] [CrossRef]
- For a detailed review that outlines many mechanistic scenarios, see: Yamamoto, Y.; Gridnev, I.D.; Patil, N.T.; Jin, T. Alkyne activation with Brønsted acids, iodine, or gold complexes, and its fate leading to synthetic application. Chem. Commun. 2009, 5075–5087. [Google Scholar] [CrossRef]
- Curini, M.; Epifano, F.; Maltese, F.; Rosati, O. Ytterbium Triflate Promoted Coupling Reaction Between Aryl Alkynes and Aldehydes. Synlett 2003, 552–554. [Google Scholar] [CrossRef]
- Saito, A.; Umakoshi, M.; Yagyu, N.; Hanazawa, Y. Novel One-Pot Approach to Synthesis of Indanones through Sb(V)-Catalyzed Reaction of Phenylalkynes with Aldehydes. Org. Lett. 2008, 10, 1783–1785. [Google Scholar] [CrossRef]
- Viswanathan, G.S.; Lee, C.-J. A highly stereoselective, novel coupling reaction between alkynes and aldehydes. Tetrahedron Lett. 2002, 43, 1613–1615. [Google Scholar] [CrossRef]
- González-Rodríguez, C.; Escalante, L.; Varela, J.A.; Castedo, L.; Saá, C. Brønsted Acid-Promoted Intramolecular Carbocyclization of Alkynals Leading to Cyclic Enones. Org. Lett. 2009, 11, 1531–1533. [Google Scholar] [CrossRef]
- Sperger, C.; Fiksdahl, A. Gold-Catalyzed Cyclizations of 1,6-Diynes. Org. Lett. 2009, 11, 2449–2452. [Google Scholar] [CrossRef]
- Trost, B.M.; Rudd, M.T. A Mechanistic Dichotomy in Ruthenium-Catalyzed Propargyl Alcohol Reactivity: A Novel Hydrative Diyne Cyclization. J. Am. Chem. Soc. 2003, 125, 11516–11517. [Google Scholar] [CrossRef]
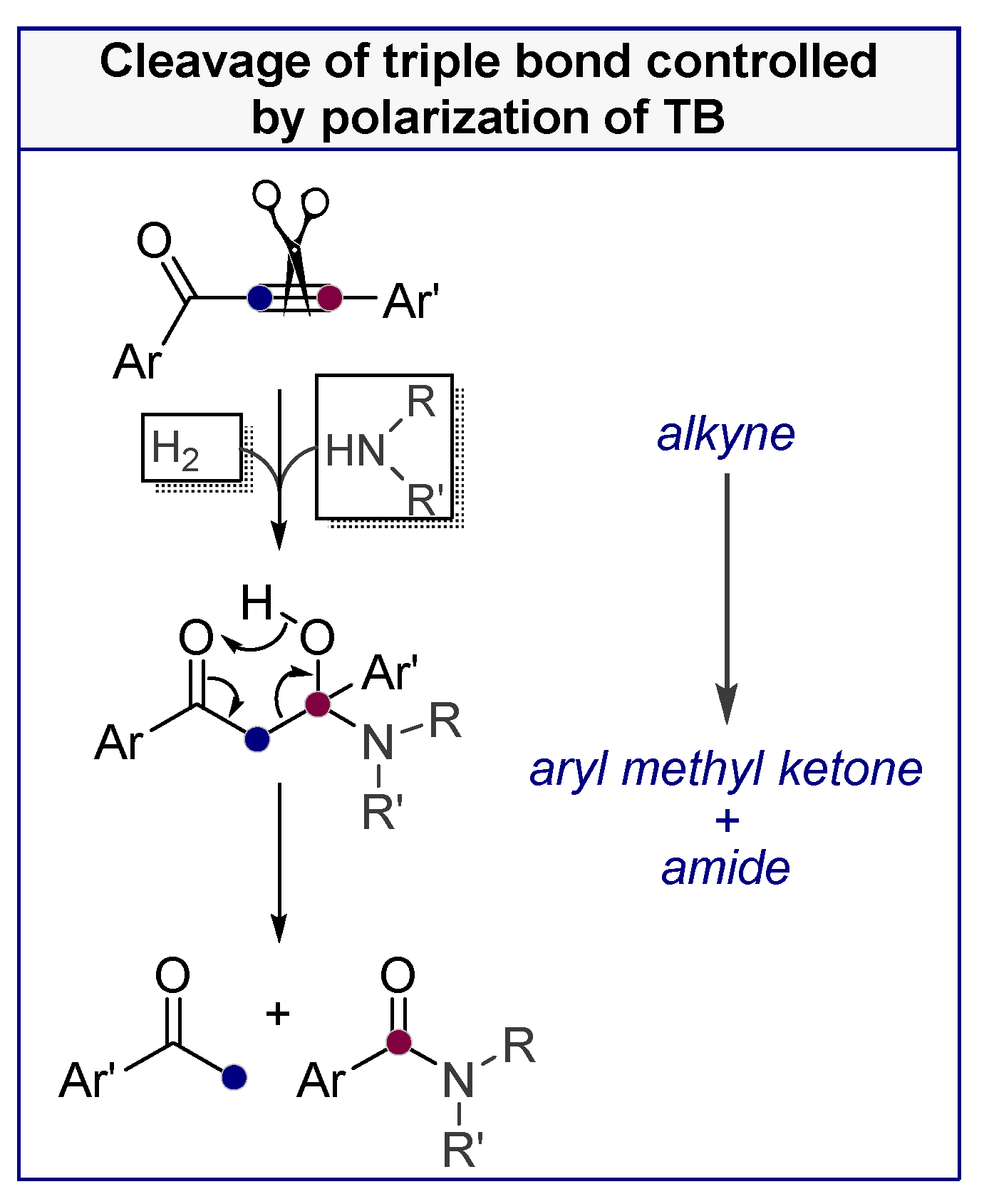
- Roy, S.; Davydova, M.P.; Pal, R.; Gilmore, K.; Tolstikov, G.A.; Vasilevsky, S.F.; Alabugin, I.V. Dissecting Alkynes: Full Cleavage of Polarized C≡C Moiety via Sequential Bis-Michael Addition/Retro-Mannich Cascade. J. Org. Chem. 2011, 76, 7482. [Google Scholar] [CrossRef]
- Baldwin, J.E. Rules for ring closure. J. Chem. Soc. Chem. Commun. 1976, 734–736. [Google Scholar] [CrossRef]
- Alabugin, I.V. Stereoelectronic Effects: A Bridge between Structure and Reactivity; John Wiley & Sons, Ltd.: Chichester, UK, 2016. [Google Scholar] [CrossRef]
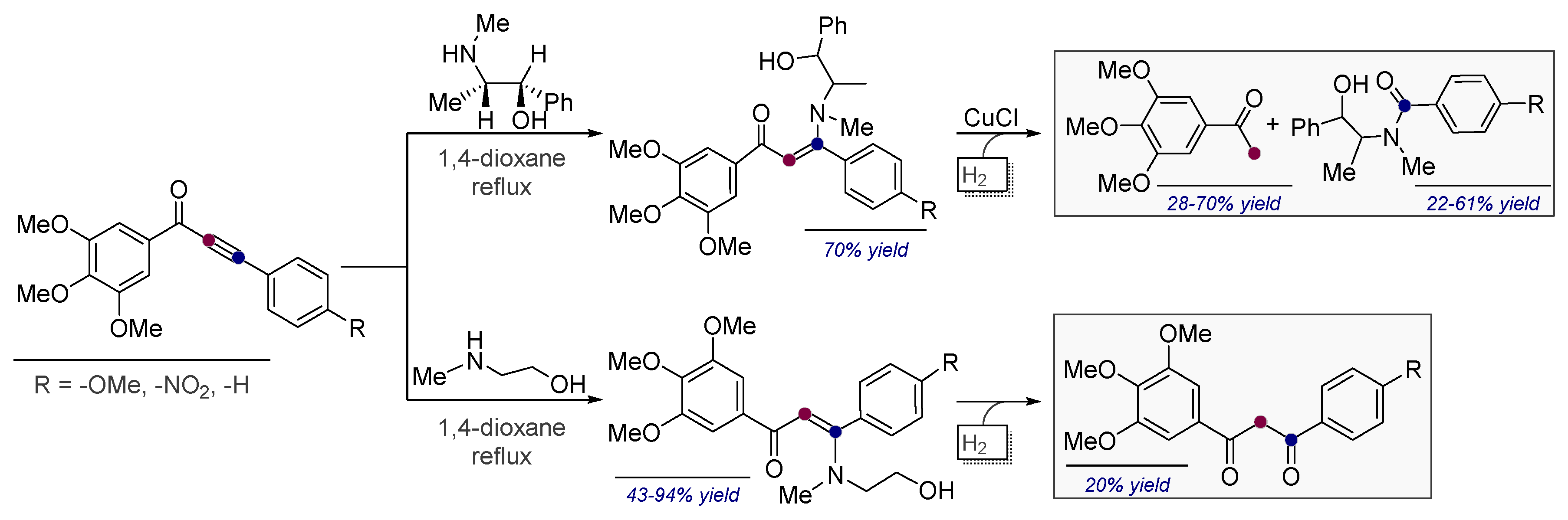
- Vasilevsky, S.F.; Davydova, M.P.; Mamatyuk, V.I.; Pleshkova, N.V.; Fadeev, D.S.; Alabugin, I.V. Reaction of a,b-alkynylketones with b-amino alcohols: Pseudoephedrine assisted cleavage of triple bond via formal internal redox process. Mendeleev Commun. 2015, 25, 377–379. [Google Scholar] [CrossRef]
- Vasilevsky, S.F.; Davydova, M.P.; Mamatyuk, V.I.; Tsvetkov, N.; Hughes, A.; Baranov, D.S.; Alabugin, I.V. Full Cleavage of C≡C Bond in Electron-Deficient Acetylenes via Reaction with Ethylenediamine. Aust. J. Chem. 2017, 70, 421–429. [Google Scholar] [CrossRef]
- Davydova, M.P.; Vasilevsky, S.F.; Nenajdenko, V.G. Reaction of trifluoroacetyl acetylenes with β-amino alcohols. Synthesis of enaminoketones and unusual fragmentation. J. Fluorine Chem. 2016, 190, 61–67. [Google Scholar] [CrossRef]
- Druzhinin, S.V.; Balenkova, E.S.; Nenajdenko, V.G. Recent advances in the chemistry of α,β-unsaturated trifluoromethylketones. Tetrahedron 2007, 63, 7753–7808. [Google Scholar] [CrossRef]
- Doyle, M.P.; McKervey, M.A.; Ye, T. Modern Catalytic Methods for Organic Synthesis with Diazo Compounds: From Cyclopropanes to Ylides; Wiley: New York, NY, USA, 1998. [Google Scholar]
- Barluenga, J.; Rodríguez, F.; Fañanás, F.J.; Flórez, J. Metal Carbenes in Organic Synthesis. In Topics in Organometallic Chemistry; Dötz, K.H., Ed.; Springer: Berlin, Germany, 2004; Volume 13, pp. 59–122. [Google Scholar] [CrossRef]
- Blanco Jaimes, M.C.; Hashmi, A.S.K. Gold-Catalyzed Oxygen- Atom Transfer to Alkynes. In Modern Gold Catalyzed Synthesis; Hashmi, A.S.K., Toste, F.D., Eds.; Wiley-VCH: Weinheim, Germany, 2012; pp. 273–296. [Google Scholar]
- Zheng, Z.; Wang, Z.; Wang, Y.; Zhang, L. Au-Catalysed oxidative cyclisation. Chem. Soc. Rev. 2016, 45, 4448–4458. [Google Scholar] [CrossRef]
- Zhang, L. A non-diazo approach to α-oxo gold carbenes via gold-catalyzed alkyne oxidation. Acc. Chem. Res. 2014, 47, 877–888. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Cui, L.; Zhang, G.; Zhang, L. Alkynes as Equivalents of α-Diazo Ketones in Generating α-Oxo Metal Carbenes: A Gold-Catalyzed Expedient Synthesis of Dihydrofuran-3-ones. J. Am. Chem. Soc. 2010, 132, 3258–3259. [Google Scholar] [CrossRef] [PubMed]
- Benitez, D.; Shapiro, N.D.; Tkatchouk, E.; Wang, Y.; Goddard, W.A., III; Toste, F.D. A Bonding Model for Gold(I) Carbene Complexes. Nat. Chem. 2009, 1, 482–486. [Google Scholar] [CrossRef] [PubMed]
- Santelli-Rouvier, E.J.; Santelli, M. The Nazarov Cyclisation. Synthesis 1983, 6, 429–442. [Google Scholar] [CrossRef]
- Rautenstrauch, V. 2-Cyclopentenones from 1-ethynyl-2-propenyl acetates. J. Org. Chem. 1984, 49, 950–952. [Google Scholar] [CrossRef]
- Mainetti, E.; Mouries, V.; Fensterbank, L.; Malacria, M.; Marco-Contelles, J. The Effect of a Hydroxy Protecting Group on the PtCl2-Catalyzed Cyclization of Dienynes—A Novel, Efficient, and Selective Synthesis of Carbocycles. Angew. Chem. Int. Ed. 2002, 41, 2132–2135. [Google Scholar] [CrossRef]
- Miki, K.; Ohe, K.; Uemura, S. Ruthenium-Catalyzed Cyclopropanation of Alkenes Using Propargylic Carboxylates as Precursors of Vinylcarbenoids. J. Org. Chem. 2003, 68, 8505–8513. [Google Scholar] [CrossRef]
- Nevado, C.; Cardenas, D.J.; Echavarren, A.M. Reaction of Enol Ethers with Alkynes Catalyzed by Transition Metals: 5exo-dig versus 6endo-dig Cyclizations via Cyclopropyl Platinum or Gold Carbene Complexes. Chem. Eur. J. 2003, 9, 2627–2635. [Google Scholar] [CrossRef] [PubMed]
- Mamane, V.; Gress, T.; Krause, H.; Fürstner, A. Platinum- and Gold-Catalyzed Cycloisomerization Reactions of Hydroxylated Enynes. J. Am. Chem. Soc. 2004, 126, 8654–8655. [Google Scholar] [CrossRef]
- Harrak, Y.; Blasykowski, C.; Fensterbank, L.; Malacria, M. PtCl2-Catalyzed Cycloisomerizations of 5-En-1-yn-3-ol Systems. J. Am. Chem. Soc. 2004, 126, 8656–8657. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Gorin, D.J.; Toste, F.D. Synthesis of 2-Cyclopentenones by Gold(I)-Catalyzed Rautenstrauch Rearrangement. J. Am. Chem. Soc. 2005, 127, 5802–5803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
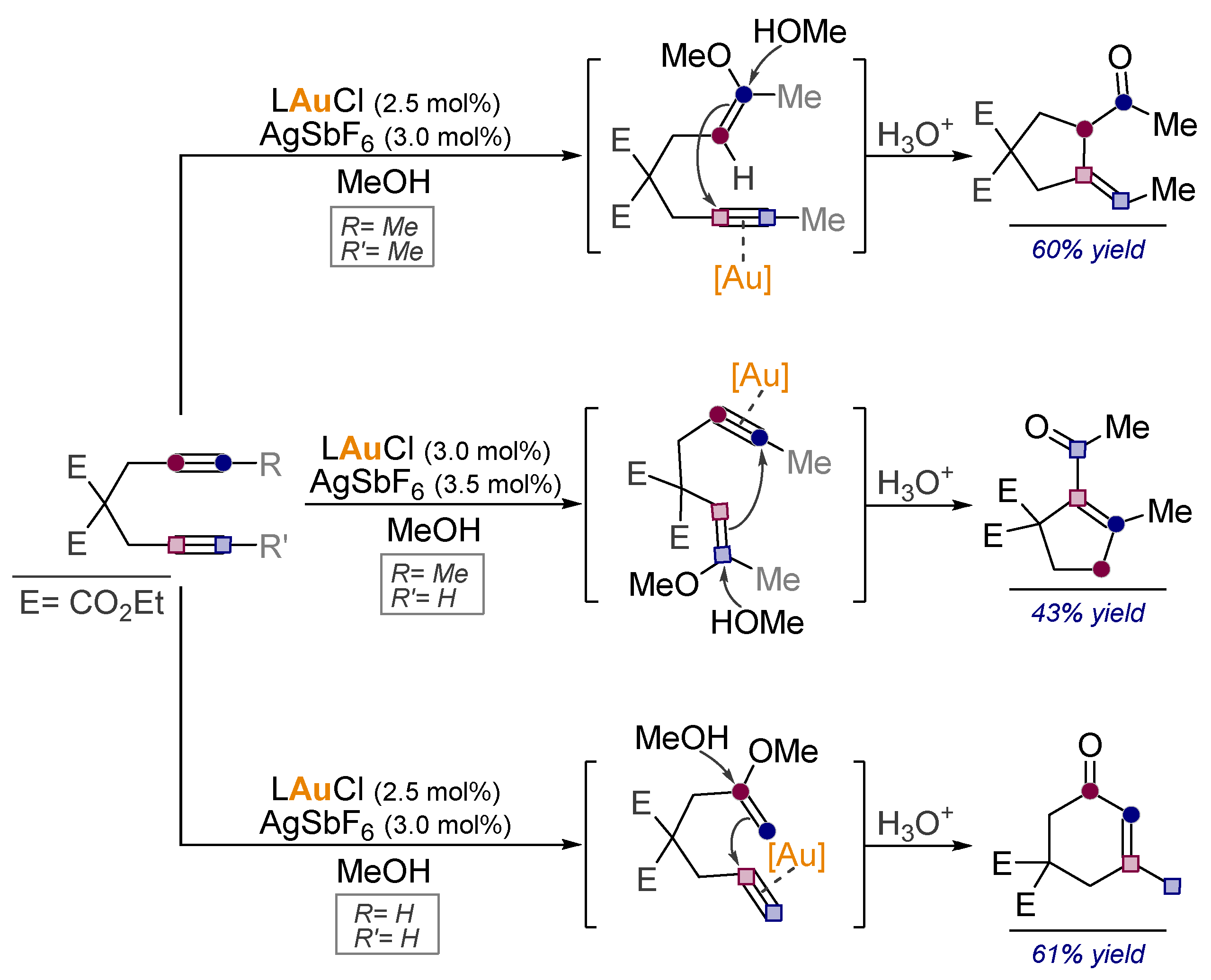
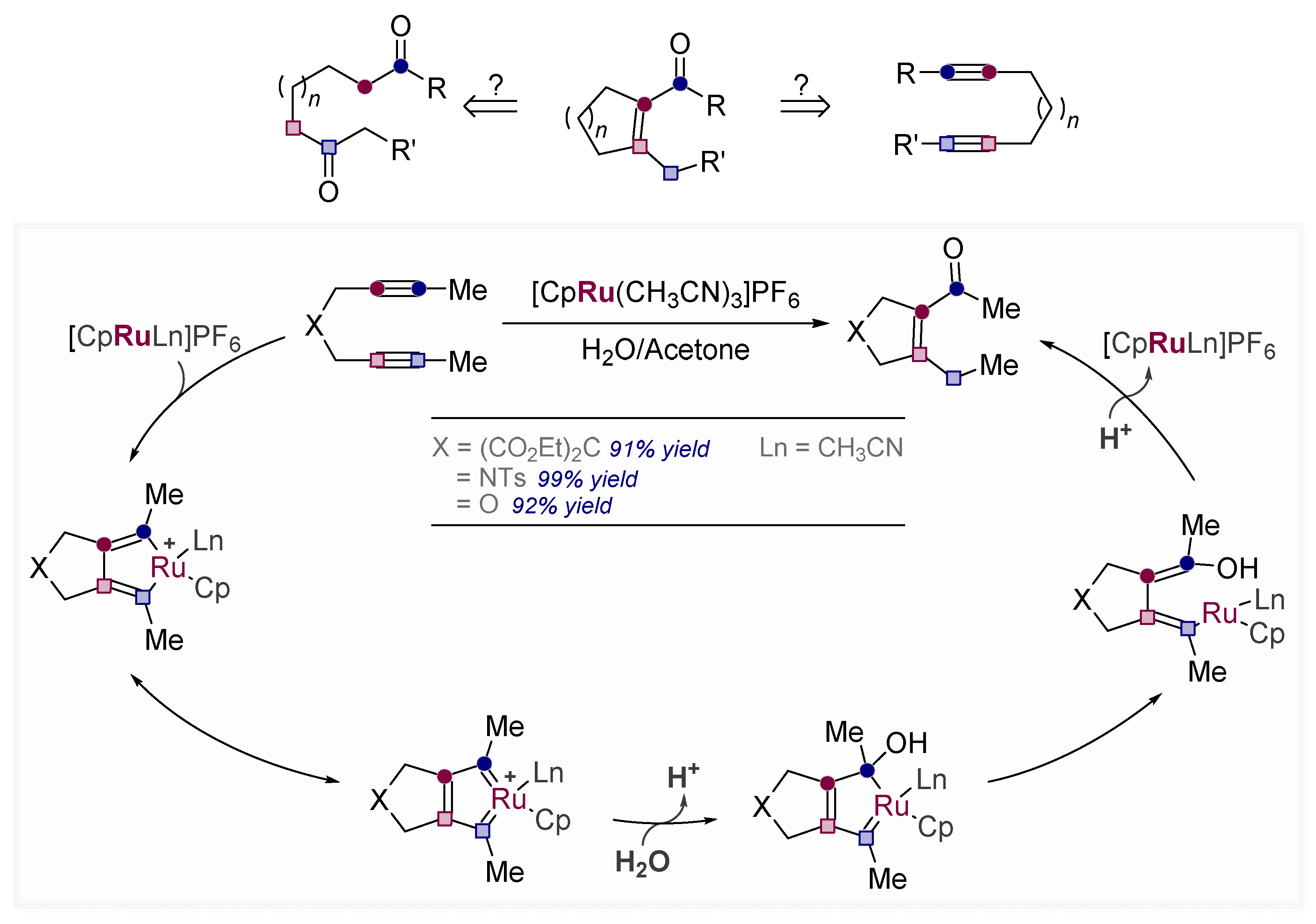
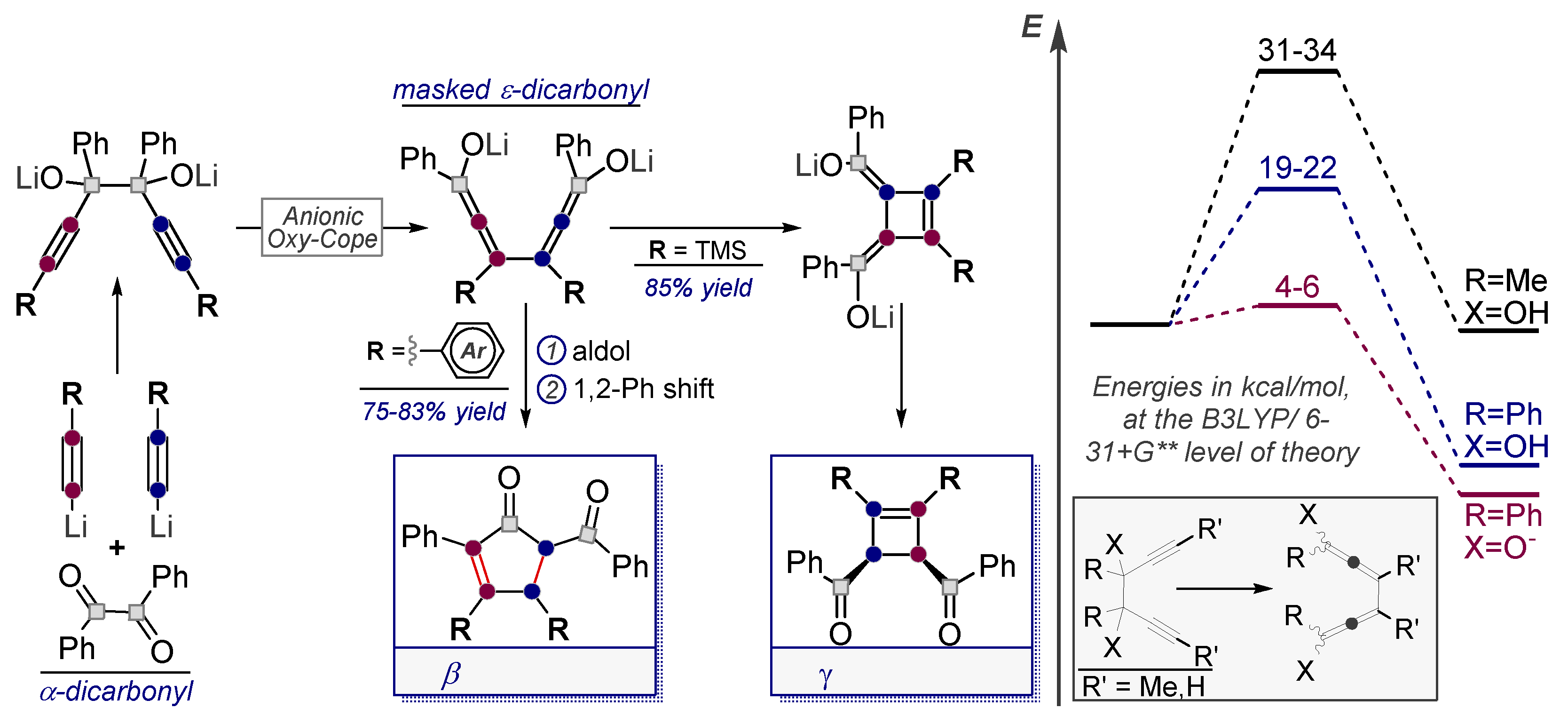
- Pal, R.; Clark, R.J.; Manoharan, M.; Alabugin, I.V. Fast Oxy-Cope Rearrangements of Bis-alkynes: Competition with Central C−C Bond Fragmentation and Incorporation in Tunable Cascades Diverging from a Common Bis-allenic Intermediate. J. Org. Chem. 2010, 75, 8689–8692. [Google Scholar] [CrossRef]















































© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alabugin, I.V.; Gonzalez-Rodriguez, E.; Kawade, R.K.; Stepanov, A.A.; Vasilevsky, S.F. Alkynes as Synthetic Equivalents of Ketones and Aldehydes: A Hidden Entry into Carbonyl Chemistry. Molecules 2019, 24, 1036. https://doi.org/10.3390/molecules24061036
Alabugin IV, Gonzalez-Rodriguez E, Kawade RK, Stepanov AA, Vasilevsky SF. Alkynes as Synthetic Equivalents of Ketones and Aldehydes: A Hidden Entry into Carbonyl Chemistry. Molecules. 2019; 24(6):1036. https://doi.org/10.3390/molecules24061036
Chicago/Turabian StyleAlabugin, Igor V., Edgar Gonzalez-Rodriguez, Rahul Kisan Kawade, Aleksandr A. Stepanov, and Sergei F. Vasilevsky. 2019. "Alkynes as Synthetic Equivalents of Ketones and Aldehydes: A Hidden Entry into Carbonyl Chemistry" Molecules 24, no. 6: 1036. https://doi.org/10.3390/molecules24061036