Secalonic Acid-F, a Novel Mycotoxin, Represses the Progression of Hepatocellular Carcinoma via MARCH1 Regulation of the PI3K/AKT/β-catenin Signaling Pathway

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
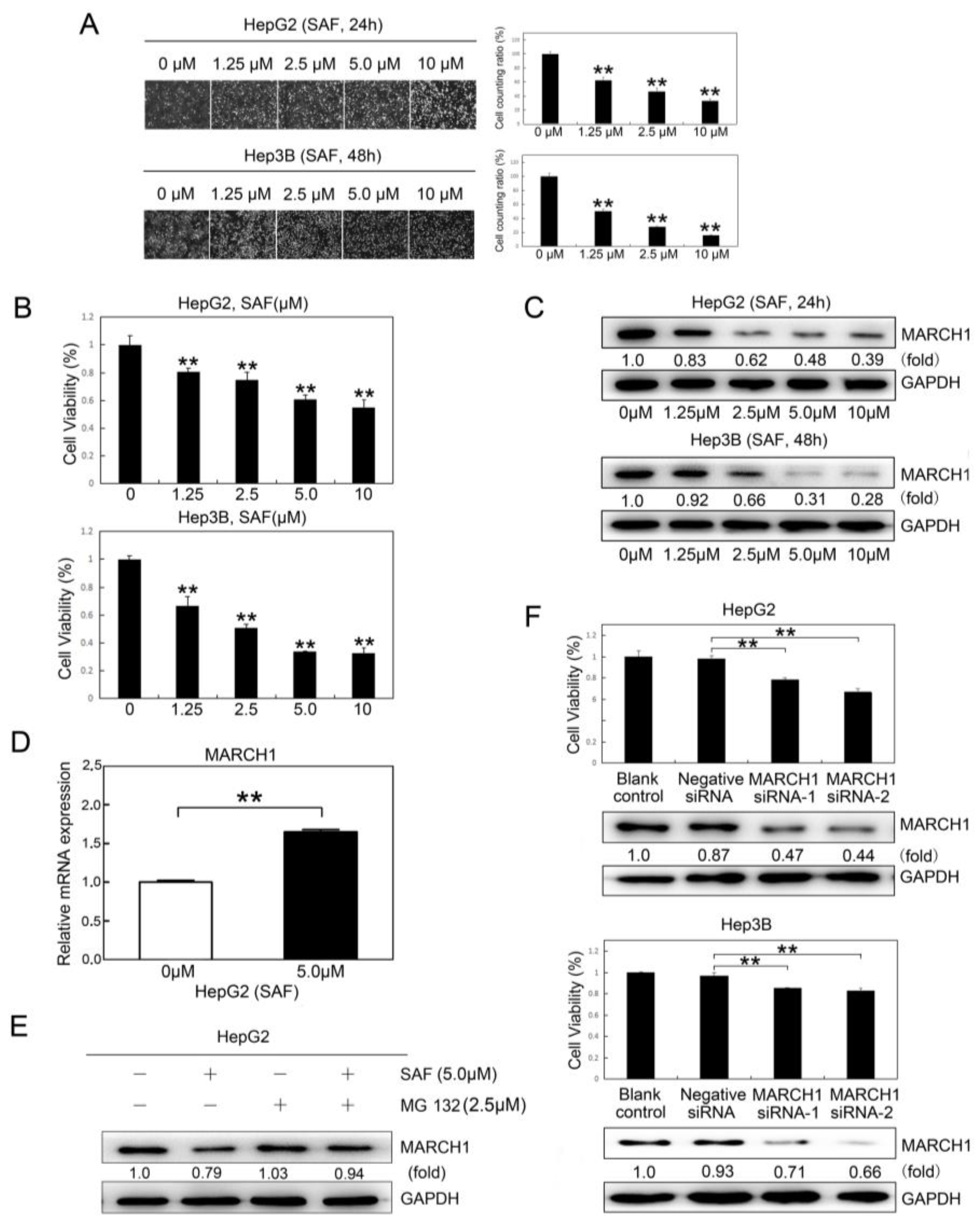
2.1. SAF Inhibited HCC Proliferation and Downregulated MARCH1 Expression in HCC Cells
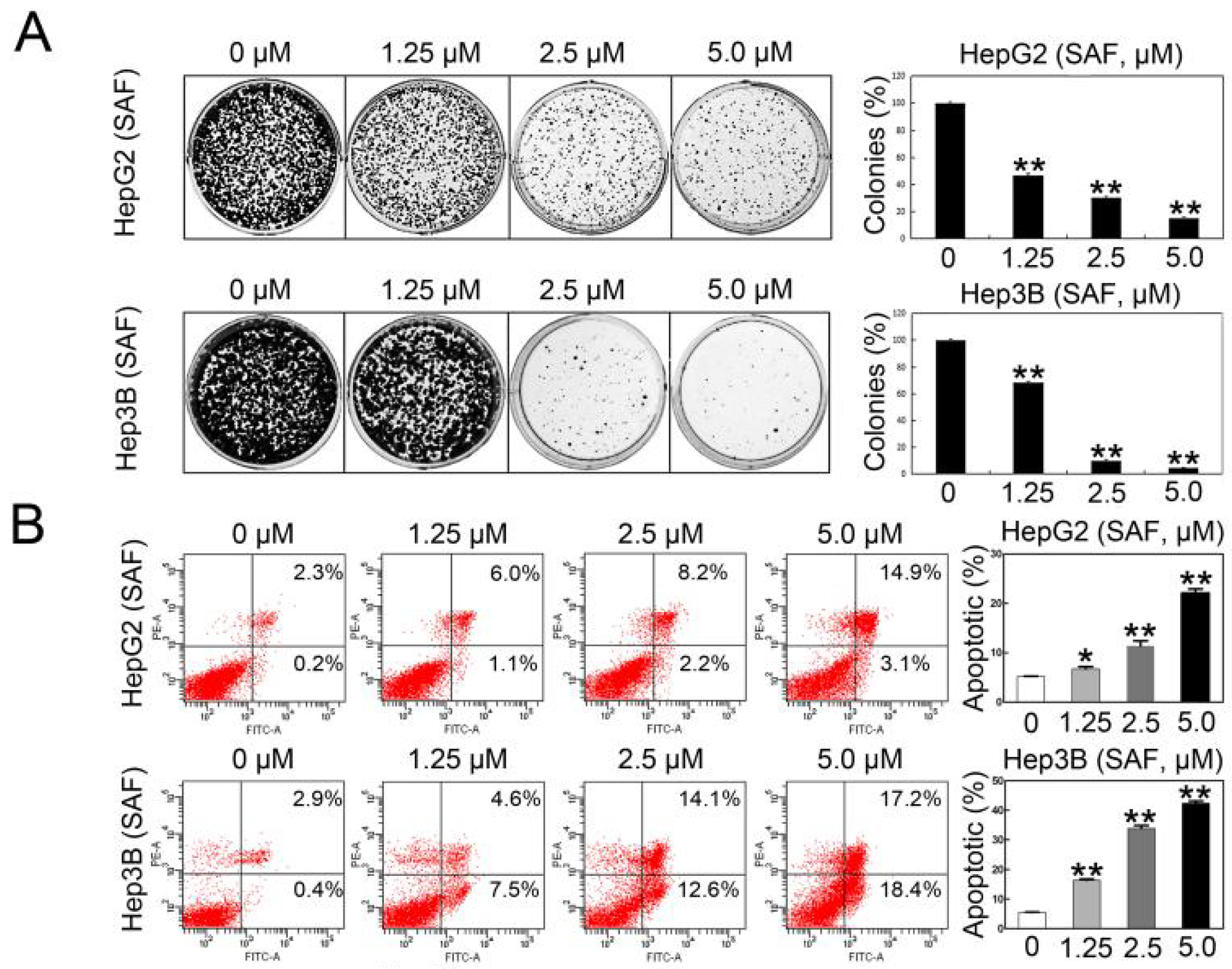
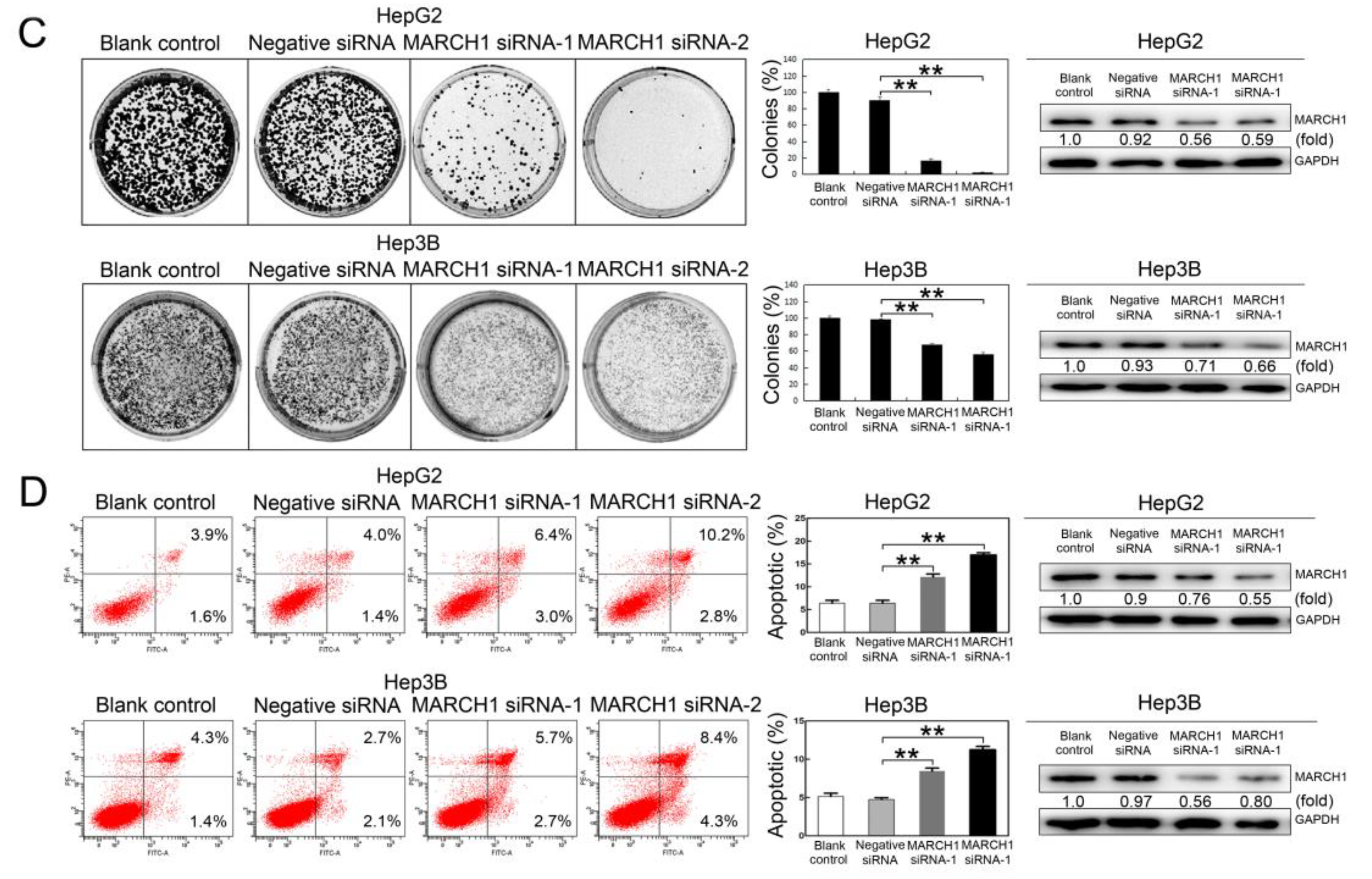
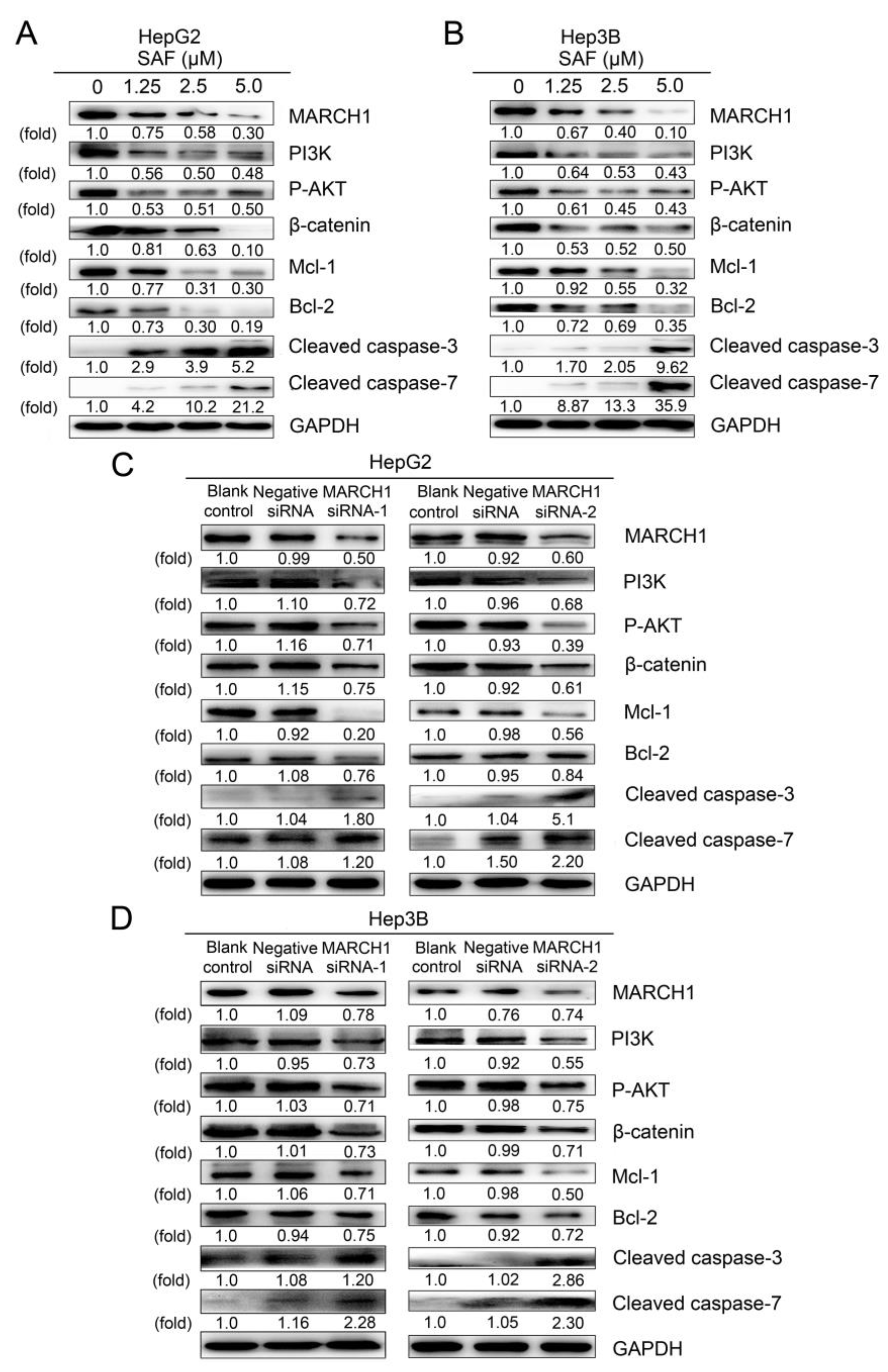
2.2. SAF Induced Apoptosis of HCC Cells by Targeting MARCH1
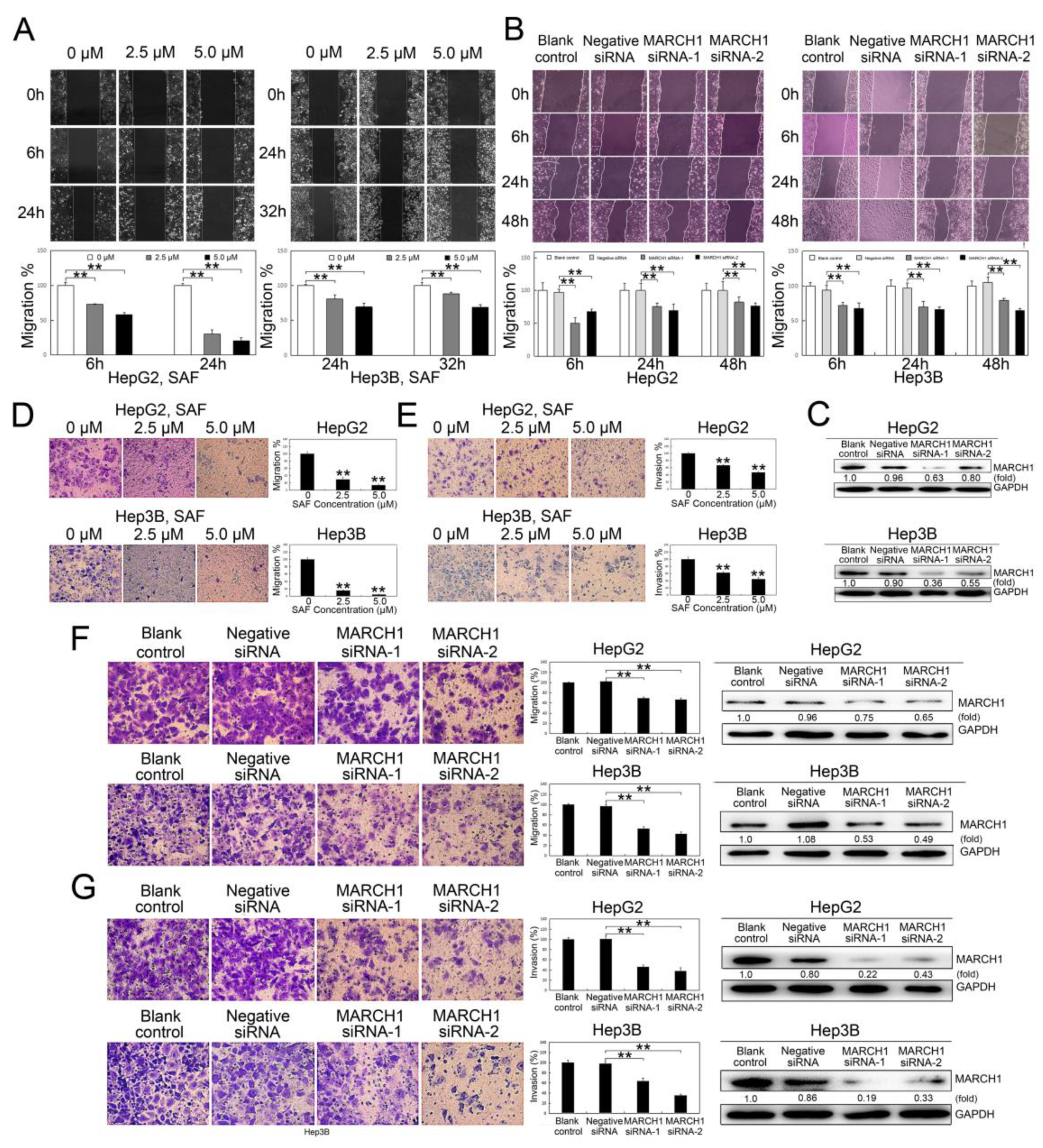
2.3. SAF suppressed HCC Cell Migration and Invasion
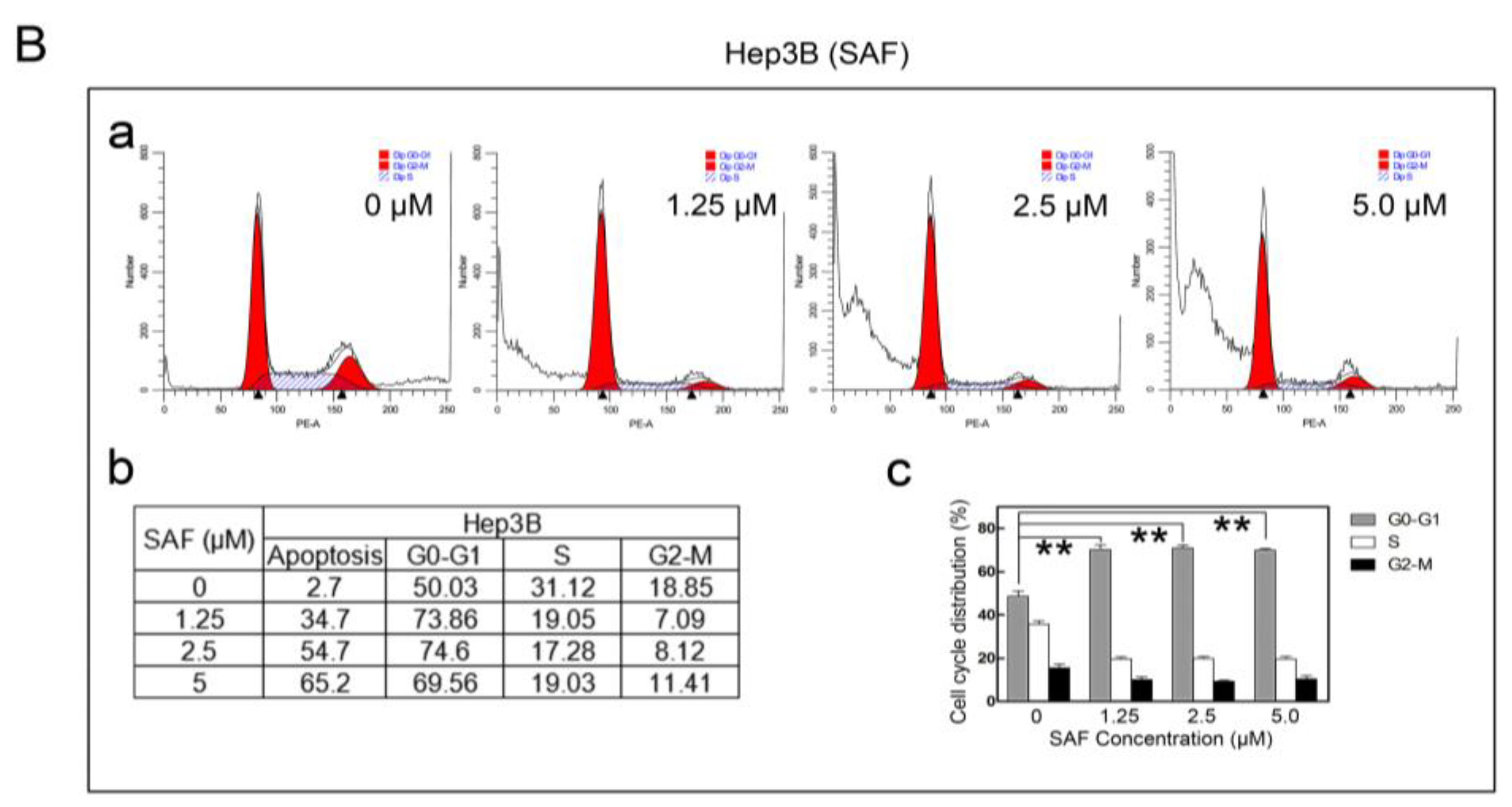
2.4. MARCH1 SAF Induced HCC Cycle Arrest
2.5. PI3K and AKT Mediate the Downregulation of MARCH1 by SAF
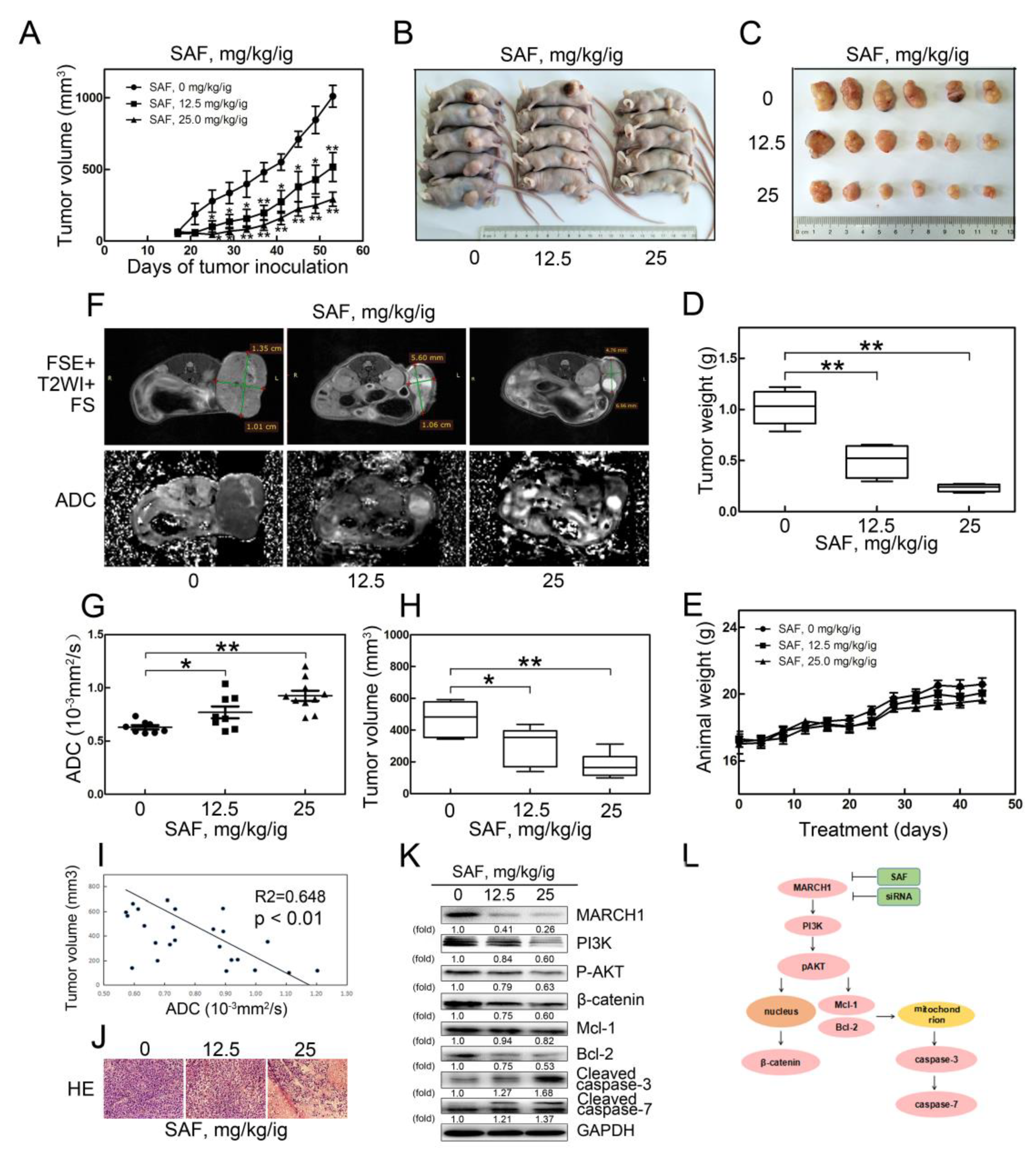
2.6. SAF Significantly Reduced Tumor Growth in a Xenograft Model of HCC
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Reagents and Antibodies
4.3. siRNA Transfection
4.4. Western Blot Analysis
4.5. Cell Proliferation Assay
4.6. Colony Formation Assay
4.7. Flow Cytometric Analysis
4.8. Wound Healing and Transwell Assays
4.9. Xenograft Models and Treatment
4.10. Magnetic Resonance Imaging (MRI)
4.11. Quantitative Real-Time PCR (qRT-PCR)
4.12. Hematoxylin and Eosin (H–E)
4.13. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Forner, A.; Reig, M.; Bruix, J. Hepatocellular carcinoma. Lancet 2018, 391, 1301–1314. [Google Scholar] [CrossRef]
- Fong, Z.V.; Tanabe, K.K. The clinical management of hepatocellular carcinoma in the United States, Europe, and Asia: A comprehensive and evidence-based comparison and review. Cancer 2014, 120, 2824–2838. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, A.; Hoshida, Y.; Battiston, C.; Tovar, V.; Sia, D.; Alsinet, C.; Cornella, H.; Liberzon, A.; Kobayashi, M.; Kumada, H. Combining clinical, pathology, and gene expression data to predict recurrence of hepatocellular carcinoma. Gastroenterology 2011, 140, 1501–1512. [Google Scholar] [CrossRef] [PubMed]
- Lencioni, R.; de Baere, T.; Soulen, M.C.; Rilling, W.S.; Geschwind, J.F. Lipiodol transarterial chemoembolization for hepatocellular carcinoma: A systematic review of efficacy and safety data. Hepatology 2016, 64, 106–116. [Google Scholar] [CrossRef] [Green Version]
- Shen, Q.; Eun, J.W.; Lee, K.; Kim, H.S.; Yang, H.D.; Kim, S.Y.; Lee, E.K.; Kim, T.; Kang, K.; Kim, S.; et al. Barrier to autointegration factor 1, procollagen-lysine, 2-oxoglutarate 5-dioxygenase 3, and splicing factor 3b subunit 4 as early-stage cancer decision markers and drivers of hepatocellular carcinoma. Hepatology 2018, 67, 1360–1377. [Google Scholar] [CrossRef]
- Ganapathy, D.; Reiner, J.R.; Löffler, L.E.; Ma, L.; Gnanaprakasam, B.; Niepötter, B.; Koehne, I.; Tietze, L.F. Enantioselective Total Synthesis of Secalonic Acid E. Chemistry 2015, 21, 16807–16810. [Google Scholar] [CrossRef]
- Zhang, W.; Krohn, K.; Flörke, U.; Pescitelli, G.; Di Bari, L.; Antus, S.; Kurtán, T.; Rheinheimer, J.; Draeger, S.; Schulz, B. New mono- and dimeric members of the secalonic acid family: Blennolides A-G isolated from the fungus Blennoria sp. Chemistry 2008, 14, 4913–4923. [Google Scholar] [CrossRef]
- Guru, S.K.; Pathania, A.S.; Kumar, S.; Ramesh, D.; Kumar, M.; Rana, S.; Kumar, A.; Malik, F.; Sharma, P.R.; Chandan, B.K.; et al. Secalonic Acid-D Represses HIF1α/VEGF-Mediated Angiogenesis by Regulating the Akt/mTOR/p70S6K Signaling Cascade. Cancer Res. 2015, 75, 2886–2896. [Google Scholar] [CrossRef]
- Gao, X.; Sun, H.L.; Liu, D.S.; Zhang, J.R.; Zhang, J.; Yan, M.M.; Pan, X.H. Secalonic acid-F inhibited cell growth more effectively than 5-fluorouracil on hepatocellular carcinoma in vitro and in vivo. Neoplasma 2017, 64, 344–350. [Google Scholar] [CrossRef]
- Llovet, J.M.; Hernandez-Gea, V. Hepatocellular carcinoma: Reasons for phase III failure and novel perspectives on trial design. Clin. Cancer Res. 2014, 20, 2072–2079. [Google Scholar] [CrossRef] [PubMed]
- Bruix, J.; Gores, G.J.; Mazzaferro, V. Hepatocellular carcinoma: Clinical frontiers and perspectives. Gut 2014, 63, 844–855. [Google Scholar] [CrossRef] [PubMed]
- Schulze, K.; Imbeaud, S.; Letouzé, E.; Alexandrov, L.B.; Calderaro, J.; Rebouissou, S.; Couchy, G.; Meiller, C.; Shinde, J.; Soysouvanh, F. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat. Genet. 2015, 47, 505–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forner, A.; Llovet, J.M.; Bruix, J. Hepatocellular carcinoma. Lancet 2012, 379, 1245–1255. [Google Scholar] [CrossRef]
- Galbas, T.; Raymond, M.; Sabourin, A.; Bourgeois-Daigneault, M.C.; Guimont-Desrochers, F.; Yun, T.J.; Cailhier, J.F.; Ishido, S. MARCH1 E3 Ubiquitin Ligase Dampens the Innate Inflammatory Response by Modulating Monocyte Functions in Mice. J. Immunol. 2017, 198, 852–861. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Sheedlo, M.J.; Yu, K.; Tan, Y.; Nakayasu, E.S.; Das, C.; Liu, X.; Luo, Z.Q. Ubiquitination independent of E1 and E2 enzymes by bacterial effectors. Nature 2016, 533, 120–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goru, S.K.; Pandey, A.; Gaikwad, A.B. E3 ubiquitin ligases as novel targets for inflammatory diseases. Pharmacol. Res. 2016, 106, 1–9. [Google Scholar] [CrossRef]
- Oh, J.; Perry, J.S.A.; Pua, H.; Irgens-Möller, N.; Ishido, S.; Hsieh, C.S.; Shin, J.S. MARCH1 protects the lipid raft and tetraspanin web from MHCII proteotoxicity in dendritic cells. J. Cell Biol. 2018, 217, 1395–1410. [Google Scholar] [CrossRef]
- Meng, Y.; Hu, J.; Chen, Y.; Yu, T.; Hu, L. Silencing MARCH1 suppresses proliferation, migration and invasion of ovarian cancer SKOV3 cells via downregulation of NF-κB and Wnt/β-catenin pathways. Oncol. Rep. 2016, 36, 2463–2470. [Google Scholar] [CrossRef] [Green Version]
- Jabbour, M.; Campbell, E.M.; Fares, H.; Lybarger, L. Discrete domains of MARCH1 mediate its localization, functional interactions, and posttranscriptional control of expression. J. Immunol. 2009, 183, 6500–6512. [Google Scholar] [CrossRef]
- Fu, G.; Zhu, L.; Yang, K.; Zhuang, R.; Xie, J.; Zhang, F. Diffusion-Weighted Magnetic Resonance Imaging for Therapy Response Monitoring and Early Treatment Prediction of Photothermal Therapy. ACS Appl. Mater. Interfaces 2016, 8, 5137–5147. [Google Scholar] [CrossRef] [PubMed]
- Sia, D.; Villanueva, A.; Friedman, S.L.; Llovet, J.M. Liver Cancer Cell of Origin, Molecular Class, and Effects on Patient Prognosis. Gastroenterology 2017, 152, 745–761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulze, K.; Nault, J.C.; Villanueva, A. Genetic profiling of hepatocellular carcinoma using next-generation sequencing. J. Hepatol. 2016, 65, 1031–1042. [Google Scholar] [CrossRef]
- Calderaro, J.; Couchy, G.; Imbeaud, S.; Amaddeo, G.; Letouzé, E.; Blanc, J.F.; Laurent, C.; Hajji, Y.; Azoulay, D.; Bioulac-Sage, P. Histological subtypes of hepatocellular carcinoma are related to gene mutations and molecular tumour classification. J. Hepatol. 2017, 67, 727–738. [Google Scholar] [CrossRef]
- Oh, J.; Wu, N.; Baravalle, G.; Cohn, B.; Ma, J.; Lo, B.; Mellman, I.; Ishido, S.; Anderson, M.; Shin, J.S. MARCH1-mediated MHCII ubiquitination promotes dendritic cell selection of natural regulatory T cells. J. Exp. Med. 2013, 210, 1069–1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alissafi, T.; Hatzioannou, A.; Mintzas, K.; Barouni, R.M.; Banos, A.; Sormendi, S.; Polyzos, A.; Xilouri, M.; Wielockx, B.; Gogas, H. Autophagy orchestrates the regulatory program of tumor-associated myeloid-derived suppressor cells. J. Clin. Investig. 2018, 128, 3840–3852. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.J.; Walseng, E.; Ishido, S.; Roche, P.A. Ubiquitination by March-I prevents MHC class II recycling and promotes MHC class II turnover in antigen-presenting cells. Proc. Natl. Acad. Sci. USA 2015, 112, 10449–10454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hindupur, S.K.; Colombi, M.; Fuhs, S.R.; Matter, M.S.; Guri, Y.; Adam, K.; Cornu, M.; Piscuoglio, S.; Ng, C.K.Y.; Betz, C.; et al. The protein histidine phosphatase LHPP is a tumour suppressor. Nature 2018, 555, 678–682. [Google Scholar] [CrossRef] [PubMed]
- Dhar, D.; Antonucci, L.; Nakagawa, H.; Kim, J.Y.; Glitzner, E.; Caruso, S.; Shalapour, S.; Yang, L.; Valasek, M.A.; Lee, S.; et al. Liver Cancer Initiation Requires p53 Inhibition by CD44-Enhanced Growth Factor Signaling. Cancer Cell 2018, 33, 1061–1077.e6. [Google Scholar] [CrossRef] [PubMed]
- Kan, Z.; Zheng, H.; Liu, X.; Li, S.; Barber, T.D.; Gong, Z.; Gao, H.; Hao, K.; Willard, M.D.; Xu, J.; et al. β-catenin (CTNNB1) mutations are not associated with prognosis in advanced hepatocellular carcinoma. Genome Res. 2013, 23, 1422–1433. [Google Scholar] [CrossRef]
- Mao, J.; Wang, D.; Wang, Z.; Tian, W.; Li, X.; Duan, J.; Wang, Y.; Yang, H.; You, L.; Cheng, Y.; et al. Combretastatin A-1 phosphate, a microtubule inhibitor, acts on both hepatocellular carcinoma cells and tumor-associated macrophages by inhibiting the Wnt/β-catenin pathway. Cancer Lett. 2016, 380, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Li, S.; Liu, P.; Sideras, K.; van de Werken, H.J.G.; van der Heide, M.; Cao, W.; Lavrijsen, M.; Peppelenbosch, M.P.; Bruno, M. Oncogenic STRAP Supports Hepatocellular Carcinoma Growth by Enhancing Wnt/β-catenin Signaling. Mol. Cancer Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Hafeez, B.B.; Ganju, A.; Sikander, M.; Kashyap, V.K.; Hafeez, Z.B.; Chauhan, N.; Malik, S.; Massey, A.E.; Tripathi, M.K.; Halaweish, F.T. Ormeloxifene Suppresses Prostate Tumor Growth and Metastatic Phenotypes via Inhibition of Oncogenic β-catenin Signaling and EMT Progression. Mol. Cancer Ther. 2017, 16, 2267–2280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, C.; Xie, D.; Yu, S.C.; Yang, X.J.; He, L.R.; Yang, J.; Ping, Y.F.; Wang, B.; Yang, L.; Xu, S.L.; et al. β-Catenin/POU5F1/SOX2 transcription factor complex mediates IGF-I receptor signaling and predicts poor prognosis in lung adenocarcinoma. Cancer Res. 2013, 73, 3181–3189. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.H.; Hwang-Verslues, W.W.; Chang, Y.C.; Chen, C.C.; Hsiao, M.; Jeng, Y.M.; Chang, K.J.; Lee, E.Y.; Shew, J.Y.; Lee, W.H. Activation of Robo1 signaling of breast cancer cells by Slit2 from stromal fibroblast restrains tumorigenesis via blocking PI3K/Akt/β-catenin pathway. Cancer Res. 2012, 72, 4652–4661. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Ji, Z.; Hong, Y.; Song, Z.; Hu, N.; Zhuang, M.; Bian, B.; Liu, Y.; Wu, F. Sh-MARCH8 Inhibits Tumorigenesis via PI3K Pathway in Gastric Cancer. Cell. Physiol. Biochem. 2018, 49, 306–321. [Google Scholar] [CrossRef]
- Michels, J.; Obrist, F.; Vitale, I.; Lissa, D.; Garcia, P.; Behnam-Motlagh, P.; Kohno, K.; Wu, G.S.; Brenner, C.; Castedo, M.; et al. MCL-1 dependency of cisplatin-resistant cancer cells. Biochem. Pharmacol. 2014, 92, 55–61. [Google Scholar] [CrossRef]
- Levy, M.A.; Claxton, D.F. Therapeutic inhibition of BCL-2 and related family members. Expert Opin. Investig. Drugs 2017, 26, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Zhong, N.; Liu, G.; Chen, K.; Liu, X.; Su, L.; Singhal, S. Usp9x- and Noxa-mediated Mcl-1 downregulation contributes to pemetrexed-induced apoptosis in human non-small-cell lung cancer cells. Cell Death Dis. 2014, 5, e1316. [Google Scholar] [CrossRef] [PubMed]
- Maji, S.; Samal, S.K.; Pattanaik, L.; Panda, S.; Quinn, B.A.; Das, S.K.; Sarkar, D.; Pellecchia, M.; Fisher, P.B.; Dash, R. Mcl-1 is an important therapeutic target for oral squamous cell carcinomas. Oncotarget 2015, 6, 16623–16637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacquemin, G.; Granci, V.; Gallouet, A.S.; Lalaoui, N.; Morlé, A.; Iessi, E.; Morizot, A.; Garrido, C.; Guillaudeux, T.; Micheau, O. Quercetin-mediated Mcl-1 and survivin downregulation restores TRAIL-induced apoptosis in non-Hodgkin’s lymphoma B cells. Haematologica 2012, 97, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Sieghart, W.; Losert, D.; Strommer, S.; Cejka, D.; Schmid, K.; Rasoul-Rockenschaub, S.; Bodingbauer, M.; Crevenna, R.; Monia, B.P. Mcl-1 overexpression in hepatocellular carcinoma: A potential target for antisense therapy. J. Hepatol. 2006, 44, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Rahmani, M.; Nkwocha, J.; Hawkins, E.; Pei, X.; Parker, R.E.; Kmieciak, M.; Leverson, J.D.; Sampath, D.; Ferreira-Gonzalez, A.; Grant, S. Cotargeting BCL-2 and PI3K Induces BAX-Dependent Mitochondrial Apoptosis in AML Cells. Cancer Res. 2018, 78, 3075–3086. [Google Scholar] [CrossRef] [PubMed]
- Rahmani, M.; Aust, M.M.; Hawkins, E.; Parker, R.E.; Ross, M.; Kmieciak, M.; Reshko, L.B.; Rizzo, K.A.; Dumur, C.I.; Ferreira-Gonzalez, A. Co-administration of the mTORC1/TORC2 inhibitor INK128 and the Bcl-2/Bcl-xL antagonist ABT-737 kills human myeloid leukemia cells through Mcl-1 down-regulation and AKT inactivation. Haematologica 2015, 100, 1553–1563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, N.; Chen, H.Y.; Harris, I.S.; Stover, D.G.; Selfors, L.M.; Bronson, R.T.; Deraedt, T.; Cichowski, K.; Welm, A.L.; Mori, Y.; et al. Cancer Cells Co-opt the Neuronal Redox-Sensing Channel TRPA1 to Promote Oxidative-Stress Tolerance. Cancer Cell 2018, 33, 985–1003. [Google Scholar] [CrossRef] [PubMed]
- Mignion, L.; Dutta, P.; Martinez, G.V.; Foroutan, P.; Gillies, R.J.; Jordan, B.F. Monitoring chemotherapeutic response by hyperpolarized 13C-fumarate MRS and diffusion MRI. Cancer Res. 2014, 74, 686–694. [Google Scholar] [CrossRef] [PubMed]
- Pisanu, M.E.; Ricci, A.; Paris, L.; Surrentino, E.; Liliac, L.; Bagnoli, M.; Canevari, S.; Mezzanzanica, D.; Podo, F.; Iorio, E.; et al. Monitoring response to cytostatic cisplatin in a HER2(+) ovary cancer model by MRI and in vitro and in vivo MR spectroscopy. Br. J. Cancer 2014, 110, 625–635. [Google Scholar] [CrossRef]
- Mignion, L.; Danhier, P.; Magat, J.; Porporato, P.E.; Masquelier, J.; Gregoire, V.; Muccioli, G.G.; Sonveaux, P.; Gallez, B.; Jordan, B.F. Non-invasive in vivo imaging of early metabolic tumor response to therapies targeting choline metabolism. Int. J. Cancer 2016, 138, 2043–2049. [Google Scholar] [CrossRef]
- Papaevangelou, E.; Almeida, G.S.; Jamin, Y.; Robinson, S.P.; deSouza, N.M. Diffusion-weighted MRI for imaging cell death after cytotoxic or apoptosis-inducing therapy. Br. J. Cancer 2015, 112, 1471–1479. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; Hua, H.; Li, M.; Liu, S.; Kong, Q.; Shao, T.; Wang, J.; Luo, Y.; Wang, Q.; Luo, T.; et al. mTORC2 promotes type I insulin-like growth factor receptor and insulin receptor activation through the tyrosine kinase activity of mTOR. Cell Res. 2016, 26, 46–65. [Google Scholar] [CrossRef]
- Xie, N.; Cai, J.B.; Zhang, L.; Zhang, P.F.; Shen, Y.H.; Yang, X.; Lu, J.C.; Gao, D.M.; Kang, Q.; Liu, L.X.; et al. Upregulation of B7-H4 promotes tumor progression of intrahepatic cholangiocarcinoma. Cell Death Dis. 2017, 8, 3205. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Qiu, J.; Xiao, Y.; Hu, X.; Liu, Q.; Chen, L.; Huang, W.; Li, X.; Li, L.; Zhang, J.; et al. AP-2β inhibits hepatocellular carcinoma invasion and metastasis through Slug and Snail to suppress epithelial-mesenchymal transition. Theranostics 2018, 8, 3707–3721. [Google Scholar] [CrossRef] [PubMed]
- Lim, W.; Yang, C.; Bazer, F.W.; Song, G. Chrysophanol Induces Apoptosis of Choriocarcinoma Through Regulation of ROS and the AKT and ERK1/2 Pathways. J. Cell. Physiol. 2017, 232, 331–339. [Google Scholar] [CrossRef]
- Yang, C.; Lim, W.; Bazer, F.W.; Song, G. Myricetin suppresses invasion and promotes cell death in human placental choriocarcinoma cells through induction of oxidative stress. Cancer Lett. 2017, 399, 10–19. [Google Scholar] [CrossRef]
- Sun, X.; Wang, S.C.; Wei, Y.; Luo, X.; Jia, Y.; Li, L.; Gopal, P.; Zhu, M.; Nassour, I.; Chuang, J.C.; et al. Arid1a Has Context-Dependent Oncogenic and Tumor Suppressor Functions in Liver Cancer. Cancer Cell 2017, 32, 574–589. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.; Zhang, C.; Huang, H.; Yang, B.; Xiao, G.; Kong, D.; Tian, Q.; Song, Q.; Song, Y.; Tan, H.; et al. The Natural Compound Myricetin Effectively Represses the Malignant Progression of Prostate Cancer by Inhibiting PIM1 and Disrupting the PIM1/CXCR4 Interaction. Cell. Physiol. Biochem. 2018, 48, 1230–1244. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Chen, X.; Xie, C.; Wang, Y.; Lin, L.; Zhu, K.; Shuai, X. Co-Delivery of Doxorubicin and Anti-BCL-2 siRNA by pH-Responsive Polymeric Vector to Overcome Drug Resistance in In Vitro and In Vivo HepG2 Hepatoma Model. Biomacromolecules 2018, 19, 2248–2256. [Google Scholar] [CrossRef]
- Duan, L.; Wu, R.; Zhang, X.; Wang, D.; You, Y.; Zhang, Y.; Zhou, L.; Chen, W. HBx-induced S100A9 in NF-κB dependent manner promotes growth and metastasis of hepatocellular carcinoma cells. Cell Death Dis. 2018, 9, 629. [Google Scholar] [CrossRef]
- Arif, T.; Vasilkovsky, L.; Refaely, Y.; Konson, A.; Shoshan-Barmatz, V. Silencing VDAC1 Expression by siRNA Inhibits Cancer Cell Proliferation and Tumor Growth In Vivo. Mol. Ther. Nucleic Acids 2014, 3, e159. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds secalonic acid-F are available from the authors. |








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, L.; Li, M.; Liu, D.; Wang, X.; Wang, P.; Dai, H.; Yang, W.; Liu, W.; Hu, X.; Zhao, M. Secalonic Acid-F, a Novel Mycotoxin, Represses the Progression of Hepatocellular Carcinoma via MARCH1 Regulation of the PI3K/AKT/β-catenin Signaling Pathway. Molecules 2019, 24, 393. https://doi.org/10.3390/molecules24030393
Xie L, Li M, Liu D, Wang X, Wang P, Dai H, Yang W, Liu W, Hu X, Zhao M. Secalonic Acid-F, a Novel Mycotoxin, Represses the Progression of Hepatocellular Carcinoma via MARCH1 Regulation of the PI3K/AKT/β-catenin Signaling Pathway. Molecules. 2019; 24(3):393. https://doi.org/10.3390/molecules24030393
Chicago/Turabian StyleXie, Lulu, Minjing Li, Desheng Liu, Xia Wang, Peiyuan Wang, Hanhan Dai, Wei Yang, Wei Liu, Xuemei Hu, and Mingdong Zhao. 2019. "Secalonic Acid-F, a Novel Mycotoxin, Represses the Progression of Hepatocellular Carcinoma via MARCH1 Regulation of the PI3K/AKT/β-catenin Signaling Pathway" Molecules 24, no. 3: 393. https://doi.org/10.3390/molecules24030393