2.1. Mapping Nucleotides according to Sequence Similarity
A variety of methods have been used to identify the network of arbitrary elements [
22]. Hamming distance or edit distance is used to measure the relative distance for various kinds of sequence information, including nucleic acid sequences. Hamming distances used as representative indices define the distance between sequences by quantifying the differences. Also, there are algorithms for the efficient alignment of randomly given strings. The most representative examples are the Needleman—Wunsch algorithm and the Smith—Waterman algorithm [
23,
24]. Each algorithm generates a global and local alignment of the strings, respectively. Both algorithms consider the alignment depending on the match, mismatch, and gap between the strings. The reward or penalty for each match, mismatch, and gap can be reflected at the user’s convenience. In the case of the previously developed miRanda algorithm, the nearest-neighbor parameters are applied to each reward and penalty value to obtain a result in accordance with the hybridization thermodynamic principle [
21]. The distance between the sequences can also be determined by measuring the hybridization profile between sequences. We applied the sociogram to effectively show the relation between the sequence and the hybridization profile.
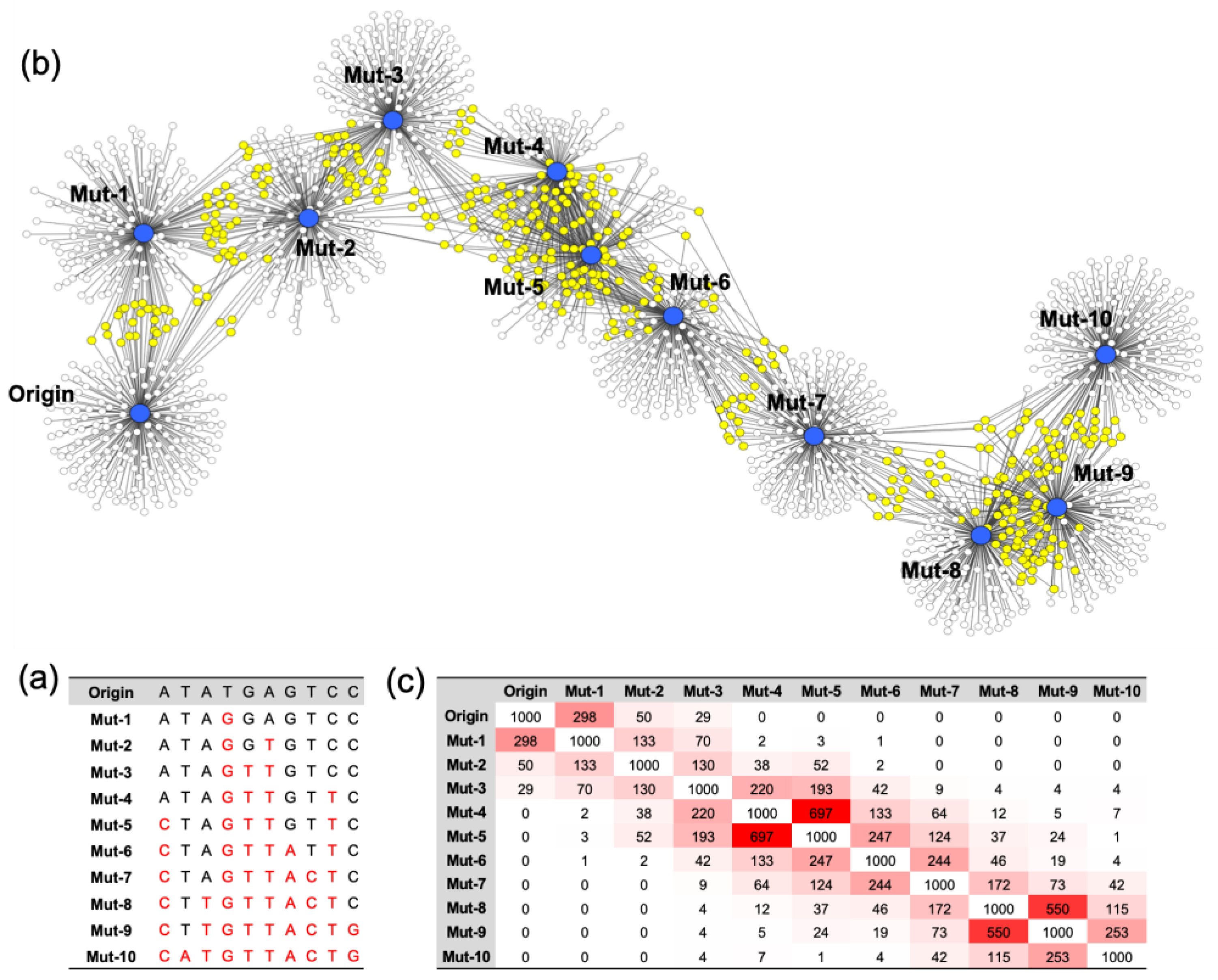
A model nucleotide sequence of 10 random bases (Origin) was generated, and the analog sequences (Mutants) were synthesized by changing the base of the origin in a cumulative manner. Mut-1 was generated by a single base random mutation of the origin, and the mutated base was transferred to the next mutant, Mut-2. Thus, Mut-2 possessed two mismatched sequences from the origin, one of Mut-1 and one of its own. In this way, a total of 10 mutants was generated. When the mutation number becomes higher, the sequence difference between the origin and the mutant is greater. The sequence information of the origin and mutants is noted in
Figure 1a. The Gibbs free energy of all the possible complementary strands against the origin and mutants was calculated. Since the sequence consisted of 10 bases, a total of 4
10 complementary sequences was present. Among all the possible complementary strands, 100 sequences with the lowest Gibbs free energy were selected and connected to each origin or mutant to draw a nondirectional sociogram (
Figure 1b). It was shown that the greater the accumulation of mutations in the model nucleotide sequence, the lower the number of shared complementary sequences. The origin shares most of the complementary sequences with one base mismatched nucleotide, Mut-1. Also, the mismatched nucleotides shared most of the complementary sequences with their most similar analogs. This network of the origin and the mutants with the top 1000 rated complementary sequences was quantified, and is presented in
Figure 1c. A higher number of shared complementary sequences is indicated with a reddish color. It was clear that every nucleotide had the highest connection with those most similar to it. Also, this result indicates that it is possible to map the nucleotides based on their hybridization profiles.
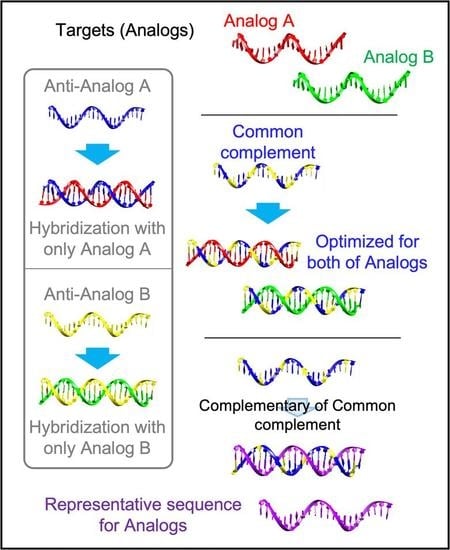
2.2. Generation of an RS from Two Analogs
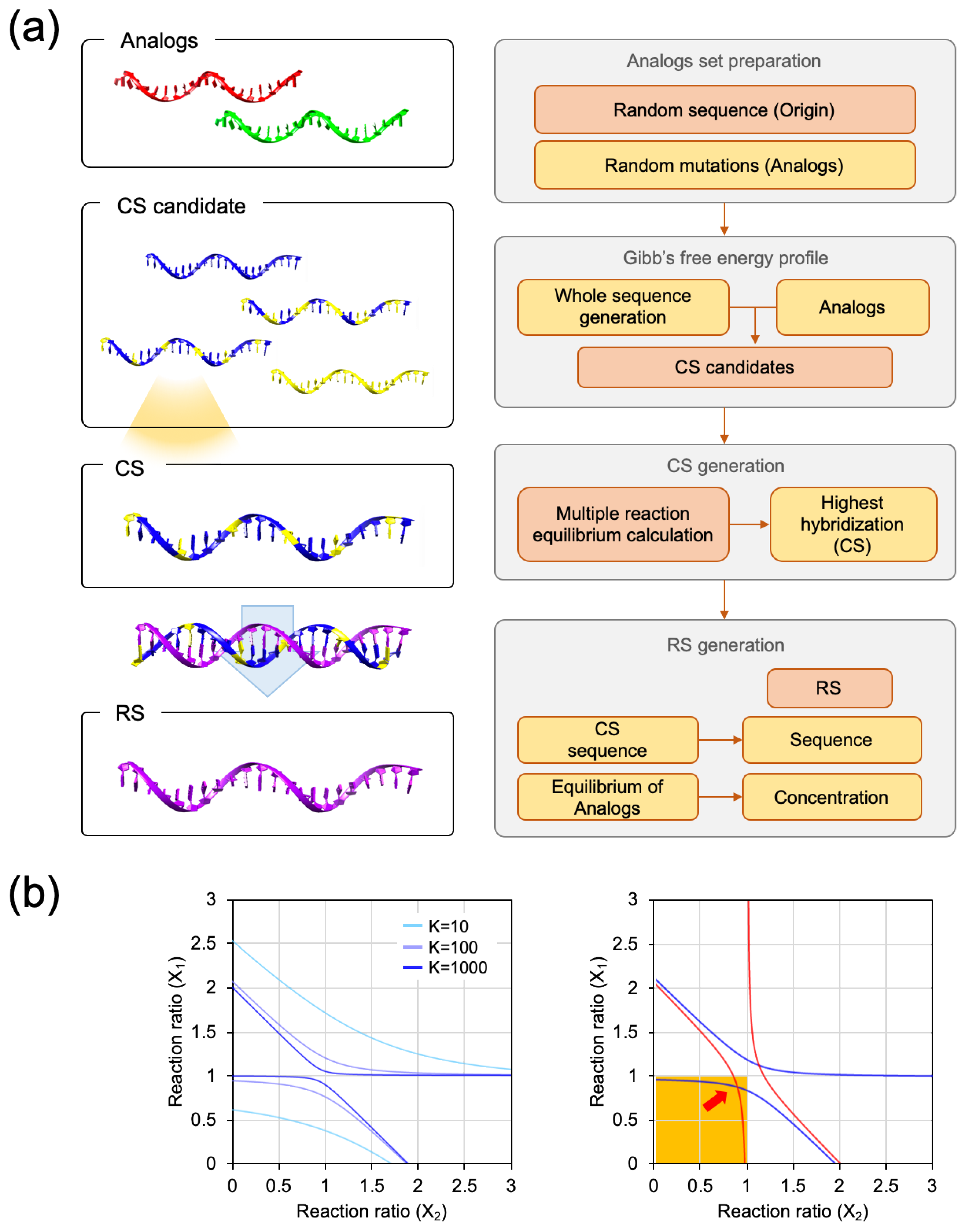
Two analogs having mismatches were used as a model to prove the presence of the RS. The calculation procedure is briefly described below and presented in
Figure 2a. First, in the procedure for the Gibbs free energy calculation, the nearest-neighbor model was used with some modifications. In the general usage, the nearest-neighbor parameter of the nucleic acid duplex and the terminal base pairs parameters should be included to calculate the enthalpy and entropy of hybridization. Here, we considered the nearest-neighbor parameter in the complementary base pairing for a facile calculation. The nearest-neighbor parameters were referenced from a previous study [
4]. The details of the calculation procedure are presented in
Figure S1. After the calculation, the Gibbs free energy values of each complementary strand against two analogs were added, and 1000 Strands with the lowest Gibbs free energy were selected as the CS candidates for the next equilibria calculation step. Since the Gibbs free energy values between the analog and the CS candidates were calculated for a single reaction condition, we should convert the Gibbs free energy values to the reaction constants in a multiple reaction, which contains both of the analogs and the CS candidate in a single reaction. The sum of the hybridization yield of the two analogs indicated the involvement of the amount of the CS candidate in the hybridization. Then, the CS was selected from the CS candidates with the highest sum of hybridization yield. The details of the process of the multiple reaction equilibrium are provided in the
Figure S2. The formula of the reaction constant (K) was plotted in hyperbolic graphs; as shown in
Figure 2b, the hyperbolic graph becomes stiffer with the increment of K. Additionally, the points of intersection in a reasonable range represent the multiple reaction equilibrium state (
). Since one unit of Analogs was involved in the reaction, the (1,1) coordinate indicates perfect hybridization for both Analog 1 and Analog 2. Thus, the reasonable range is the area marked in yellow in
Figure 2b. From the CS, an RS and its concentration were calculated. The details of the process for the calculation of the concentration can be found in
Figure S3.
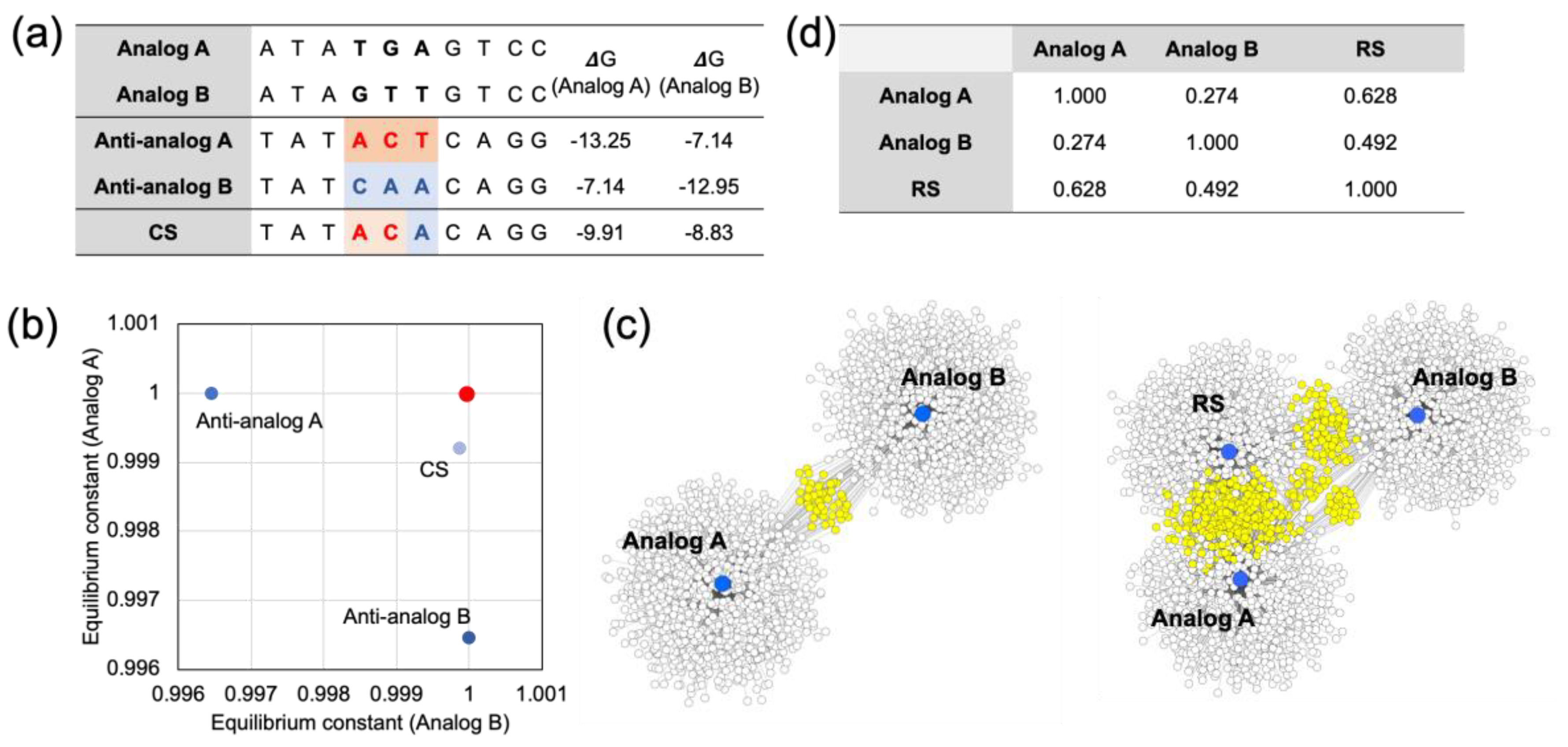
The sequence information of two model analogs with three base mismatches is presented in
Figure 3a. When we compared the Gibbs free energy values of the CS and the perfect complement of the analogs (Anti-analogs), it was shown that the CS did not have a minimized Gibbs free energy; the sum of the hybridization yield was higher than for the Anti-analogs. In addition, the calculated multiple reaction equilibrium coordinates showed a closer distance to the (1,1) coordinates than the coordinates of the Anti-analogs (
Figure 3b). In the case of Anti-analog A, the hybridization yield against Analog B was less than 0.0035 μM to the perfect hybridization (1,1). Meanwhile, the CS was only less than 0.00013 μM and 0.00089 μM for Analog A and Analog B, respectively. The sum of shorts was lower in the CS compared with the Anti-analogs. Furthermore, the RS was obtained from the CS with complementary sequences. Additionally, the RS and Analogs were mapped with 1000 complementary sequences, and are presented in
Figure 3c. The shared complementary sequences are marked with a yellow color. As expected, the RS shared a significant amount of complementary sequences with both Analog A and Analog B. The Pearson’s correlation coefficient of all the Gibbs free energy values calculated from the Analogs was also used to show the similarity of the hybridization profile, and the RS had a higher Pearson’s correlation than the Analogs compared to the value between the Analogs (
Figure 3d). The details of the Pearson’s correlation coefficient calculations are presented in
Figure S4.
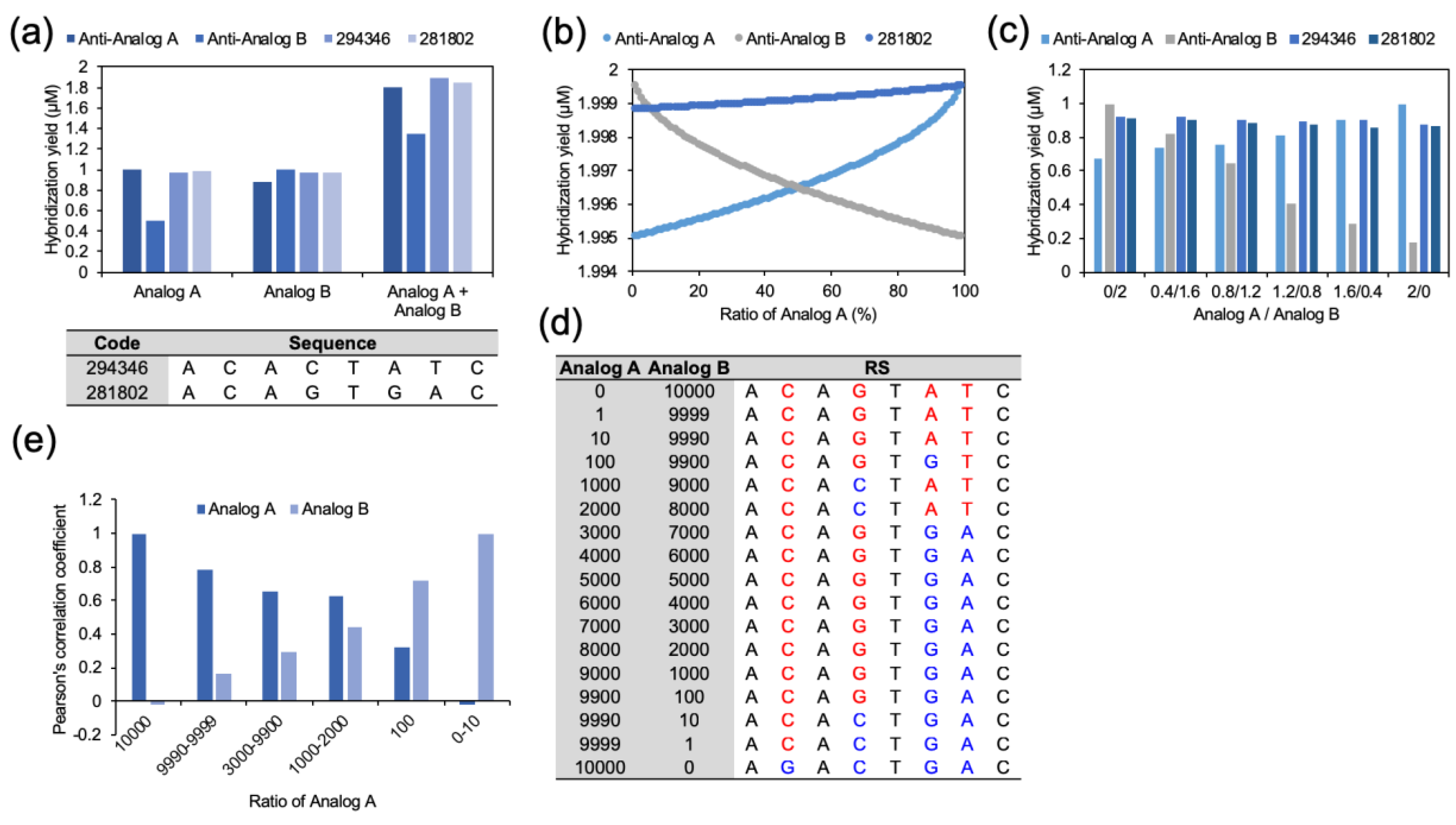
The multiple reactions and hybridization between the analogs and the CS were also proved experimentally. To provide a clear demonstration, we selected two CSs (code numbers: 294346 and 281802). A code number was assigned to every possible complementary sequence, and the sequences of 294,346 and 281,802 are noted in
Figure 4a. As shown in
Figure 4a, the analogs were reacted with CSs, and their hybridization reactions were measured. For the measurement, analogs were labeled with fluorescent dyes (Cy3 and Cy5), and CSs were labeled with non-fluorescent quencher, IOWA Black. When hybridization between the analogs and the CSs occurs, the fluorescence intensities become weaker. First, 1 μM of an analog (Analog A or Analog B) was combined with 2 μM of its Anti-analogs separately. As expected, the Anti-analogs showed the highest hybridization efficiency with their own analog. However, the hybridization to the other analog was not effective. In the case of anti-Analog B, perfect hybridization was shown with Analog B. Meanwhile, anti-Analog B hybridized to Analog A with only 50.4% efficiency. Even though the CSs showed a lower hybridization efficiency compared to the perfect anti-analogs, the hybridization with both analogs was better. Moreover, in the reaction with the mixed Analogs (1 μM each), CSs showed the highest yield in total hybridization.
This phenomenon was also observed when the concentration of the analogs was verified. Before the actual experiment, the hybridization efficiency of the Anti-analogs and CSs in various concentrations of analogs was calculated in silico (
Figure 4b). The concentrations of the analogs were increased from 0 μM to 2 μM, and the sum of the concentration was fixed at 2 μM. At the end-points and nearby, where Analog A or Analog B occupied all of the nucleotides at 2 μM, the perfect complementary sequences showed the highest hybridization efficiencies. However, hybridization of the Anti-analogs significantly decreased with the decrement of their own complements. In contrast, CSs demonstrated sustained hybridization efficiency at all concentrations. The triangle region where CSs showed higher hybridization efficiency well-described the potential of CSs and the RSs. This tendency was also observed in the actual experiments (
Figure 4c). The hybridization efficiency of the perfect complementary sequences became lower with the increment of their less-compatible targets, but the CSs showed better hybridization at an Analog A/Analog B ratio from 0.4:1.6 to 1.2:0.8. To make this result more reliable, 100 random analog sets were used to generate the RS (
Figure S5). Even though there were differences in the hybridization efficiency values, the results showed solid evidence of the same process in RSs. The Gibbs free energy profile of the analogs and the RSs against all possible complementary sequences were compared using Pearson’s correlation coefficient. Between two-base mismatched analogs, the Pearson’s correlation coefficient was 0.636 ± 0.049. In contrast, the average Pearson’s correlation coefficient between the RS and the analogs was 0.805 ± 0.042. This increment of the coefficient indicated that the RS can delegate the hybridization profile of the analogs. In addition, it was possible to calculate the optimized RSs from various concentrations of analogs. As shown in
Figure 4d, two analogs sharing four of a total of eight bases were used for calculating the RS in varied concentrations. Analog concentrations were applied from 0:10,000 to 10,000:0, and optimized RSs were obtained. With the concentration biases, the optimized RSs had greater closeness and a higher Pearson’s correlation coefficient than the dominant analog (
Figure 4e).
2.3. Generation of RSs from Multiple Sequences
In the coordinate system, the K-means clustering algorithm can generate intuitive and rational centroids for clustering [
25,
26]. For clustering with the K-means algorithm, the sum of distances from the centroid to the data objects is measured, and the coordinates of centroids are updated to minimize the sum. The centroid itself has a coordinate just like other data objects, although it is not real. Thus, it can be said that the centroid represents the properties of the data objects in the cluster. This is quite similar to the calculation of the RS from the analog sequences. We tried to apply the K-means clustering algorithm to the generation of the RS with multiple sequences. By equating a map of nucleotides obtained from the sociogram with a coordinate system where K-means clustering is performed, it is possible to obtain the RS representing multiple nucleotides similar to the calculation of the centroid using the K-means clustering method. The sum of distance was replaced with the sum of hybridization yield of the CS to each analog, and the maximized hybridization yield of the CS reflects the coordinates of the centroid in the minimized sum of distance of the K-means clustering algorithm.
Several remarkable approaches have been developed to calculate multiple nucleotide equilibrium states. For instance, Robert Dirt and his colleagues developed methodologies for calculating multistrand interactions and the formation of secondary structures by the combination of graph theory and a partition function [
6,
27,
28,
29]. However, tremendous resources and calculation times are needed to obtain reasonable results for thousands of reactions simultaneously. Therefore, we applied the calculation in a step-by-step manner. It was shown that the calculation of the RSs from two different analogs was achieved by a simple multiple reaction equilibrium calculation. If we repeat this calculation, it would be possible to discover the RS for a number of sequences. To determine the order of stepwise calculation, the closeness was used. Two analogs with the highest closeness were calculated first to generate their own RS, and the RS participated in the next iterative calculation as one of the analogs. Finally, it was possible to obtain the RS information and its equivalent concentration from the cluster of the analogs.
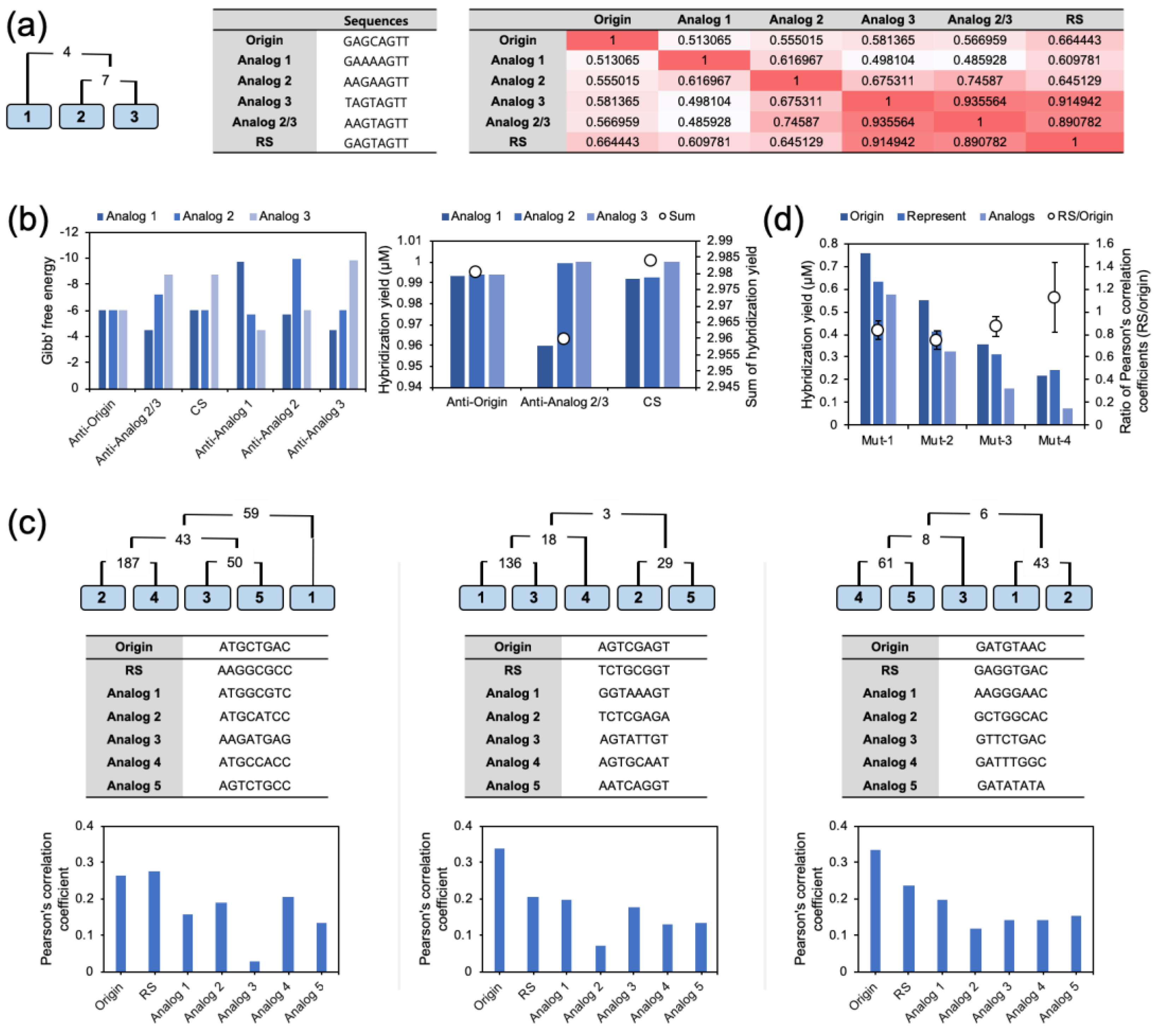
To see how the iterative calculation worked, three analogs were generated from an eight-based random origin. The analogs were prepared with two mutations from the origin. The sequence information of the three analogs (Analog 1, Analog 2, and Analog 3) is presented in
Figure 5a. The initial concentrations of the analogs were assumed to be 1. In the top 1000 rated complementary sequences-based closeness calculation, Analog 2 and Analog 3 showed the highest closeness. Therefore, the RS of Analog 2 and Analog 3 was calculated first, and named Analog 2/3. Subsequently, the concentration of Analog 2/3 was calculated. The sequences of Analog 2 and Analog 3 were replaced with the sequence and concentration of the calculated Analog 2/3. In succession, Analog 1 and Analog 2/3 were used to calculate the final RS. The Pearson’s correlation coefficients of the origin, Analogs, and RS are noted in
Figure 5a. The Pearson’s correlation coefficients of Analog 2/3 against Analog 2 and Analog 3 (0.746 and 0.936, respectively) were higher than the coefficient between Analog 2 and Analog 3 (0.675). In contrast, the Pearson’s correlation coefficient of Analog 2/3 against Analog 1 were not increased. This indicated that the calculated RS was specific to the target analogs. Meanwhile, the RS calculated from Analog 1 and Analog 2/3 showed an increment of the Pearson’s correlation coefficient against the origin and modest Gibbs free energy values. The coefficient value was 0.664, which was higher than the coefficient value of any other Analog. It was remarkable that the Pearson’s correlation of the RS for Analog 2 and Analog 3 was lower than that of Analog 2/3; however, the average coefficient was increased within all the analogs. Moreover, the average Pearson’s correlation coefficient of the initial analogs against the RS was higher than that against the origin. This result not only demonstrates that it was possible to make a RS, but also that its performance might be better than that of its origin.
To validate the RS, the Gibbs free energy and the Pearson’s correlation between the sequences were calculated. As shown in
Figure 5b, the Gibbs free energy values of Analog 2/3 were higher than those of the perfect complementary sequences, Anti-Analog 2 for Analog 2, and Anti-Analog3 for Analog 3; however, they were higher than those of the opposite analogs Anti-Analog 2 for Analog 3 and Anti-Analog 3 for Analog 2. The potential of the RS was also revealed in the hybridization yield calculation in
Figure 5b. The perfect complementary sequences of the origin, Analog 2/3, and the RS were used to measure the hybridization efficiency. The hybridization yields of the anti-sense origin were similar in the three analogs, and the sum of the constant was 2.980. In the case of Anti-analog 2/3, which was calculated from Analog 2 and Analog 3, the hybridization yields of Analog 2 and Analog 3 were increased compared to the Anti-analogs; however, the constant of Analog 1 was significantly decreased. Thus, the sum of hybridization yields was decreased overall (2.960). Finally, the CS showed a recovered hybridization yield of Analog 1, and the sum was the highest (2.984). Through this result, the potential of the generated RS was proved directly.
For the analysis of multiple nucleotides, the RSs were obtained by the same procedure with five analogs and three mismatched bases. In the model sets, the average Pearson’s correlations of the origin and the RS against the initial analogs were calculated and were compared. Interestingly, the Pearson’s correlation coefficient of the RS was not always higher or lower than the coefficient of the origin. As shown in
Figure 5c, the Pearson’s correlation coefficient of the RS was higher or lower than the Pearson’s correlation of the origin in a sequence-dependent manner; however, statistically, it was higher than the coefficients of the analogs. To obtain solid evidence, we created a random set of 100 analogs and measured the averages (
Figure S6). The Pearson’s correlation coefficients of the origin, the RS, and the analogs were 0.359 ± 0.074, 0.312 ± 0.0821, and 0.163 ± 0.0922, respectively. This result indicated that our procedure can generate RSs from multiple nucleotides statistically; however, they are not optimized as in the origin. Also, to clarify the effect of the number of mutations in the analogs, 30 sets of analogs were prepared with 1–4 mutations from the eight-based origin, and the Pearson’s correlation coefficients were measured (
Figure S7). As shown in
Figure 5d, the average Pearson’s correlation coefficient of the origin against the analogs was decreased with the increment of the number of mutations because the difference of the sequences led to less similarity in the hybridization profile. The decrement of the Pearson’s correlation coefficient was also observed in the RS; however, the amount of decrease was smaller than that of the origins. Thus, the ratio of the Pearson’s correlation coefficient (RS/origin) was increased with the number of mutations. When the number of mutations was 4, the average coefficient of the RS was higher than that of the origins.
We believe this shortage was generated from the imprecise calculation of the Gibbs free energy and the equilibrium constant. Especially, since there are several factors to consider in nucleotide hybridization, such as secondary structure, the equilibrium constant calculation with simple thermodynamic principles may not be sufficient for specific optimization for RS generation. To overcome these inaccuracies, more complex and simultaneous calculations could be applied in the Gibbs energy and equilibrium state calculation process. However, it was demonstrated that the RS showed a much higher correlation with the analogs than any of the single analogs.

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




