Synthesis, Structural and Thermal Studies of 3-(1-Benzyl-1,2,3,6-tetrahydropyridin-4-yl)-5-ethoxy-1H-indole (D2AAK1_3) as Dopamine D2 Receptor Ligand

 , , , and
, , , and
Abstract
:
1. Introduction
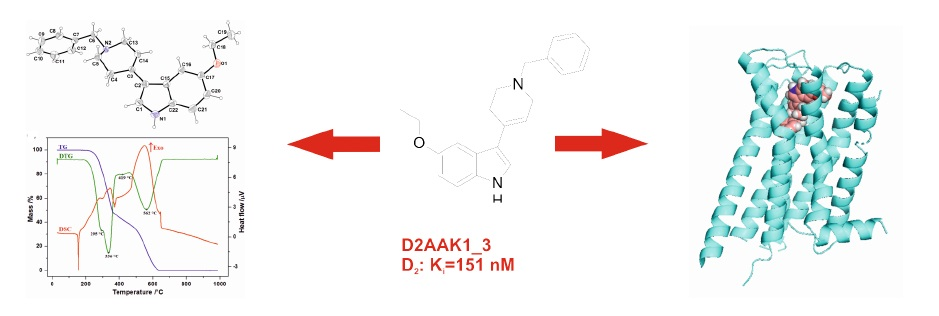
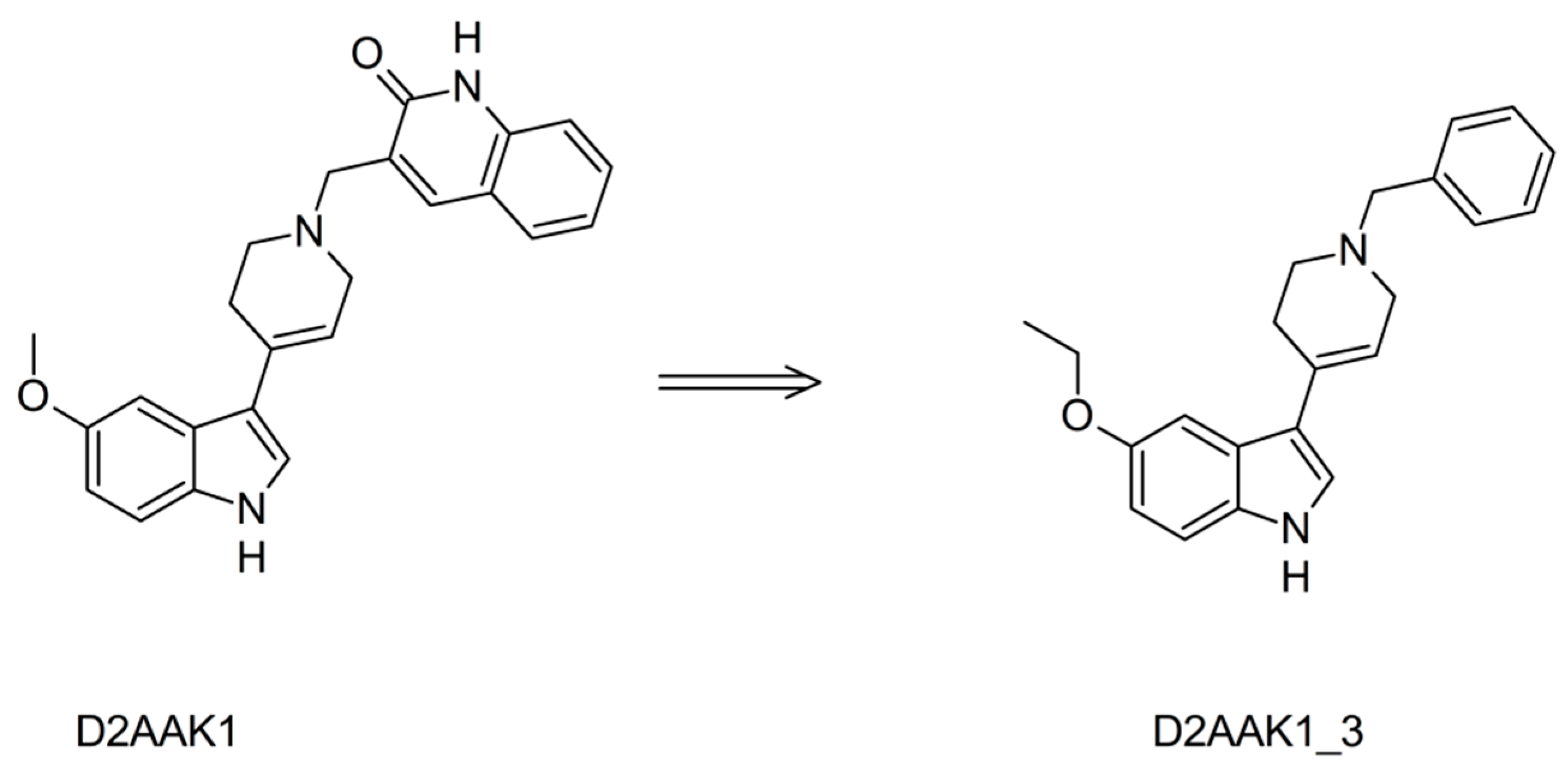
2. Results and Discussion
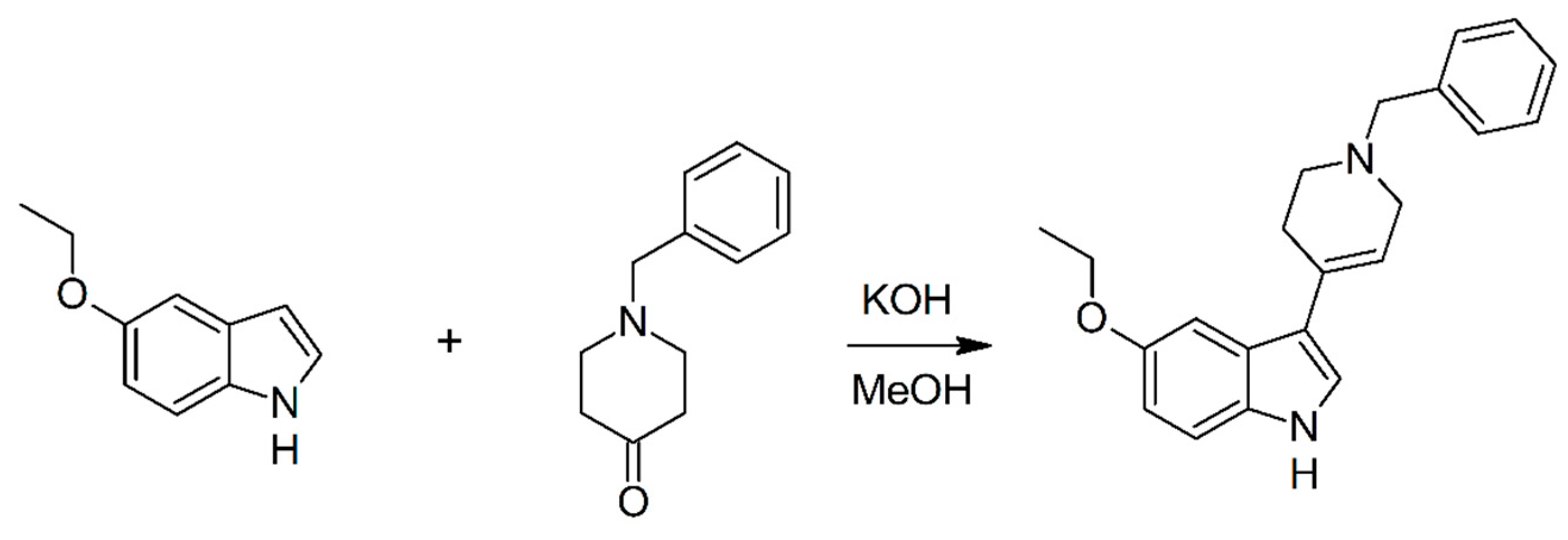
2.1. Chemistry
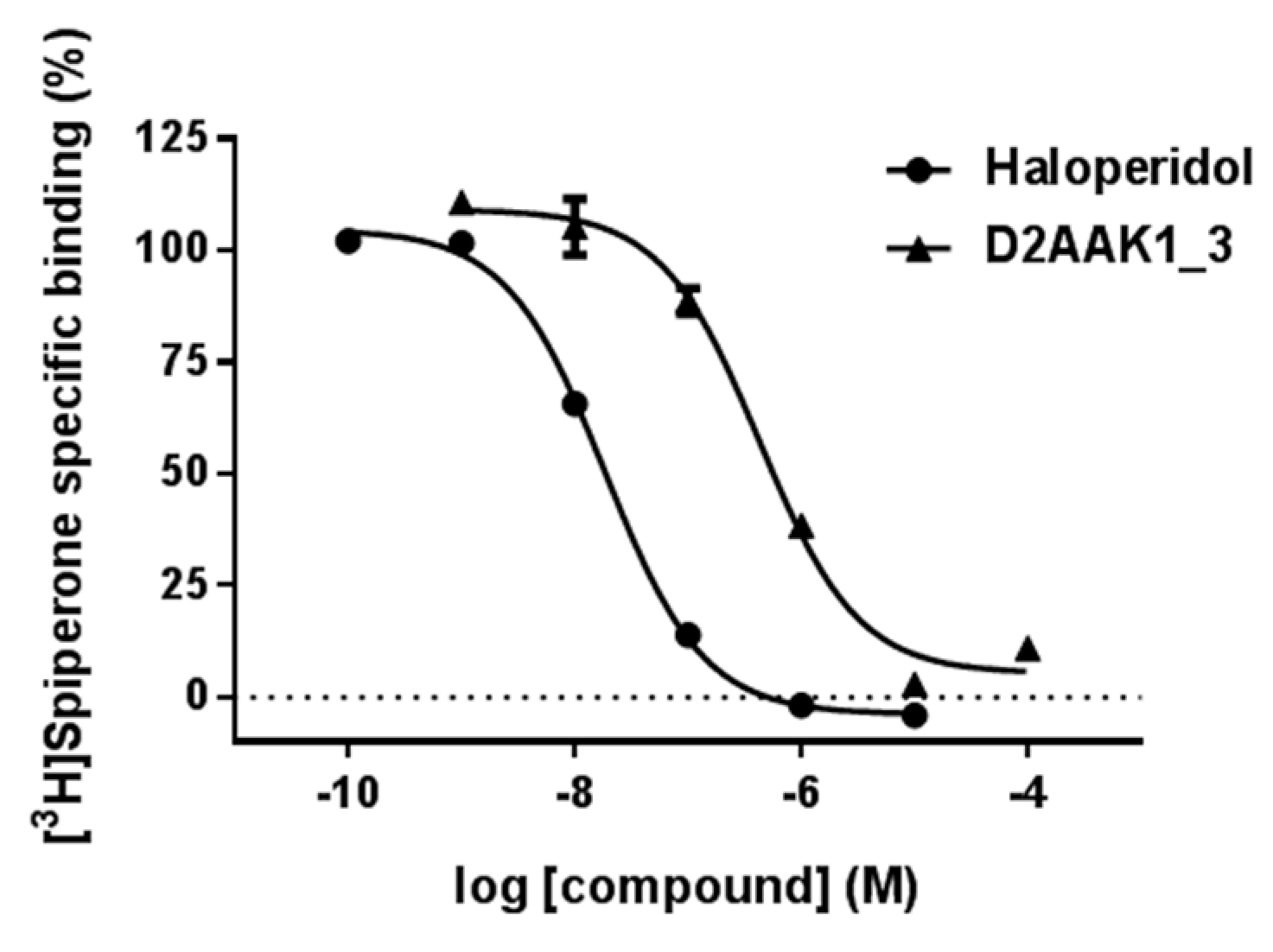
2.2. Affinity to the Dopamine D2 Receptor
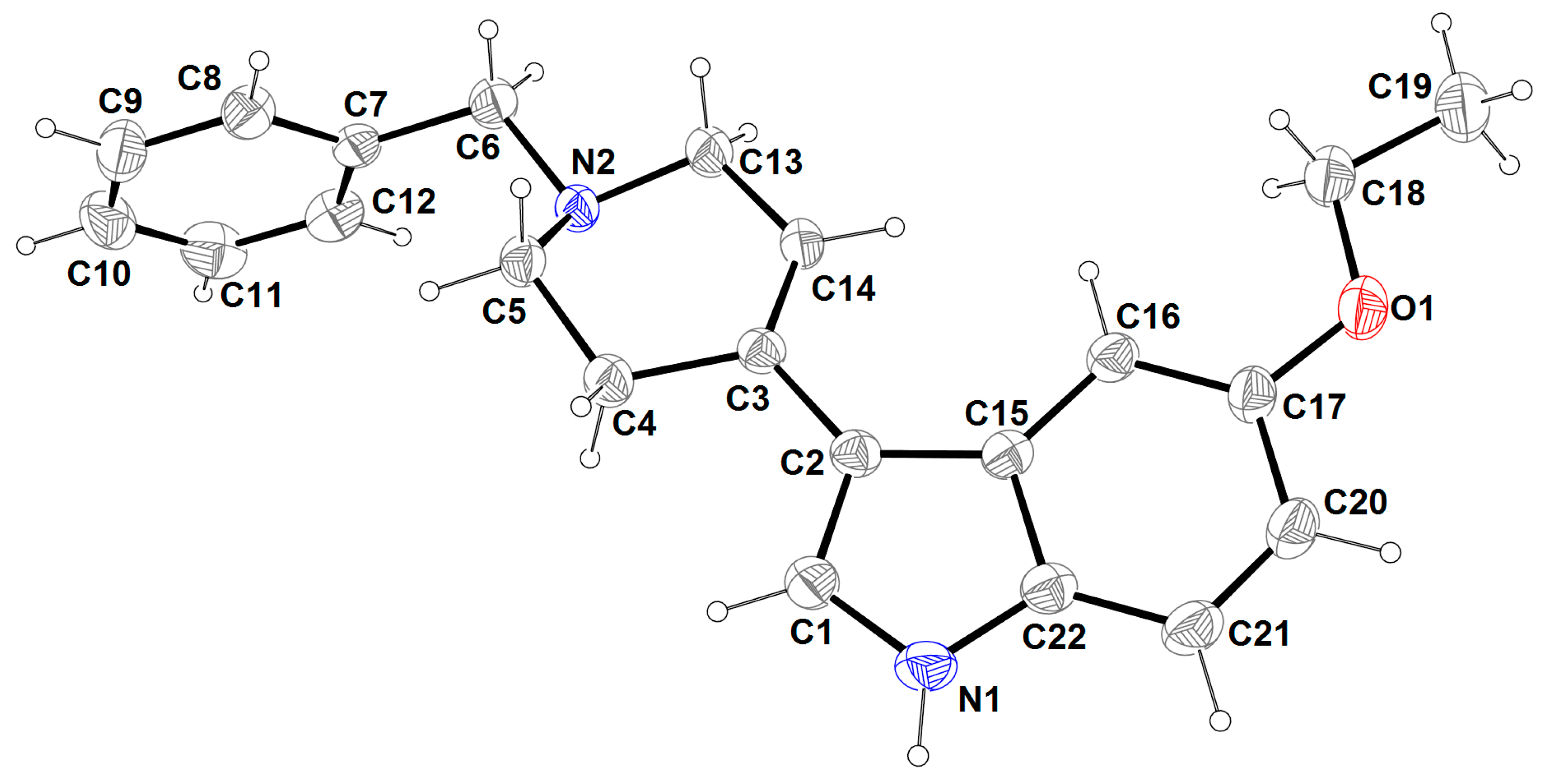
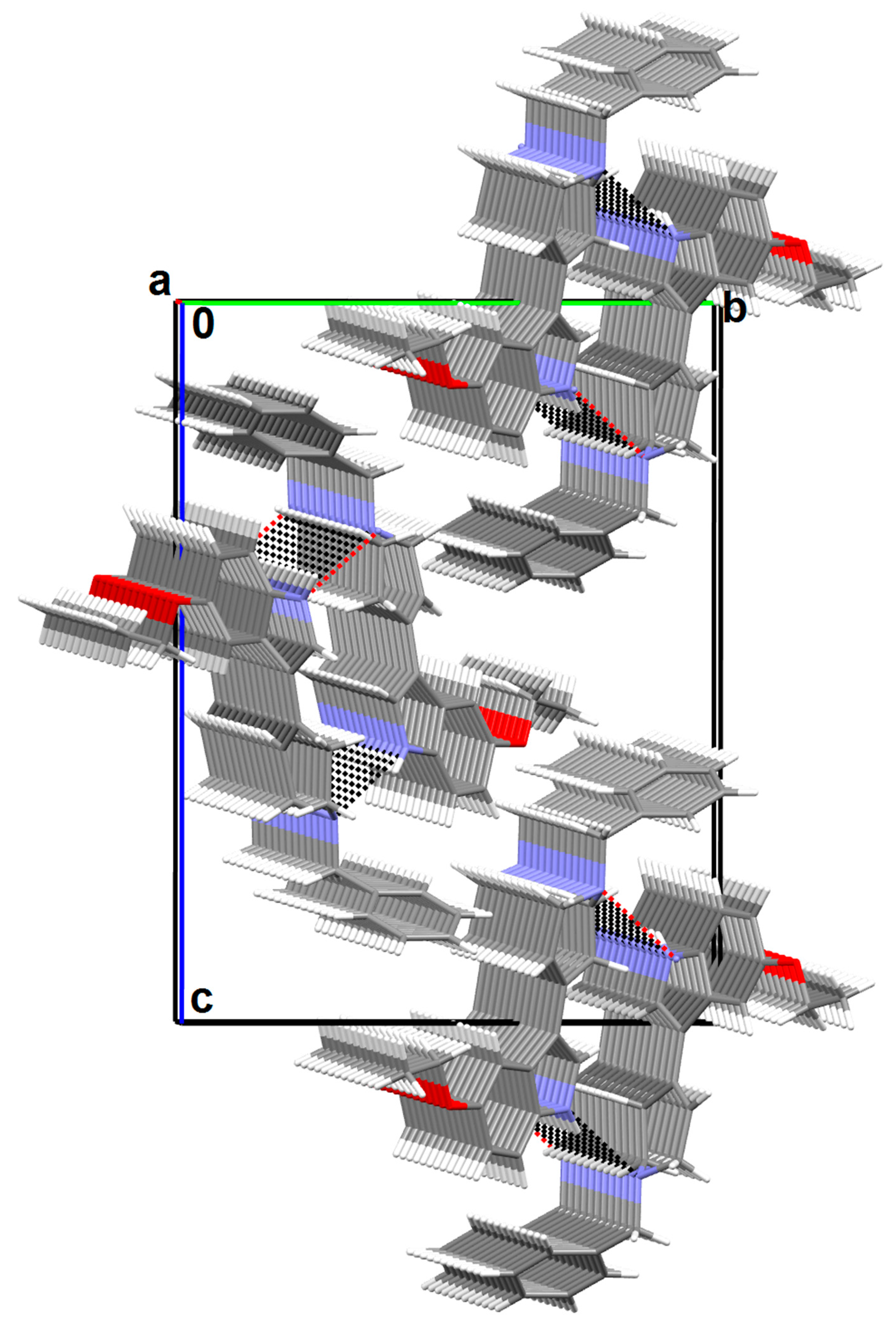
2.3. X-ray Analysis
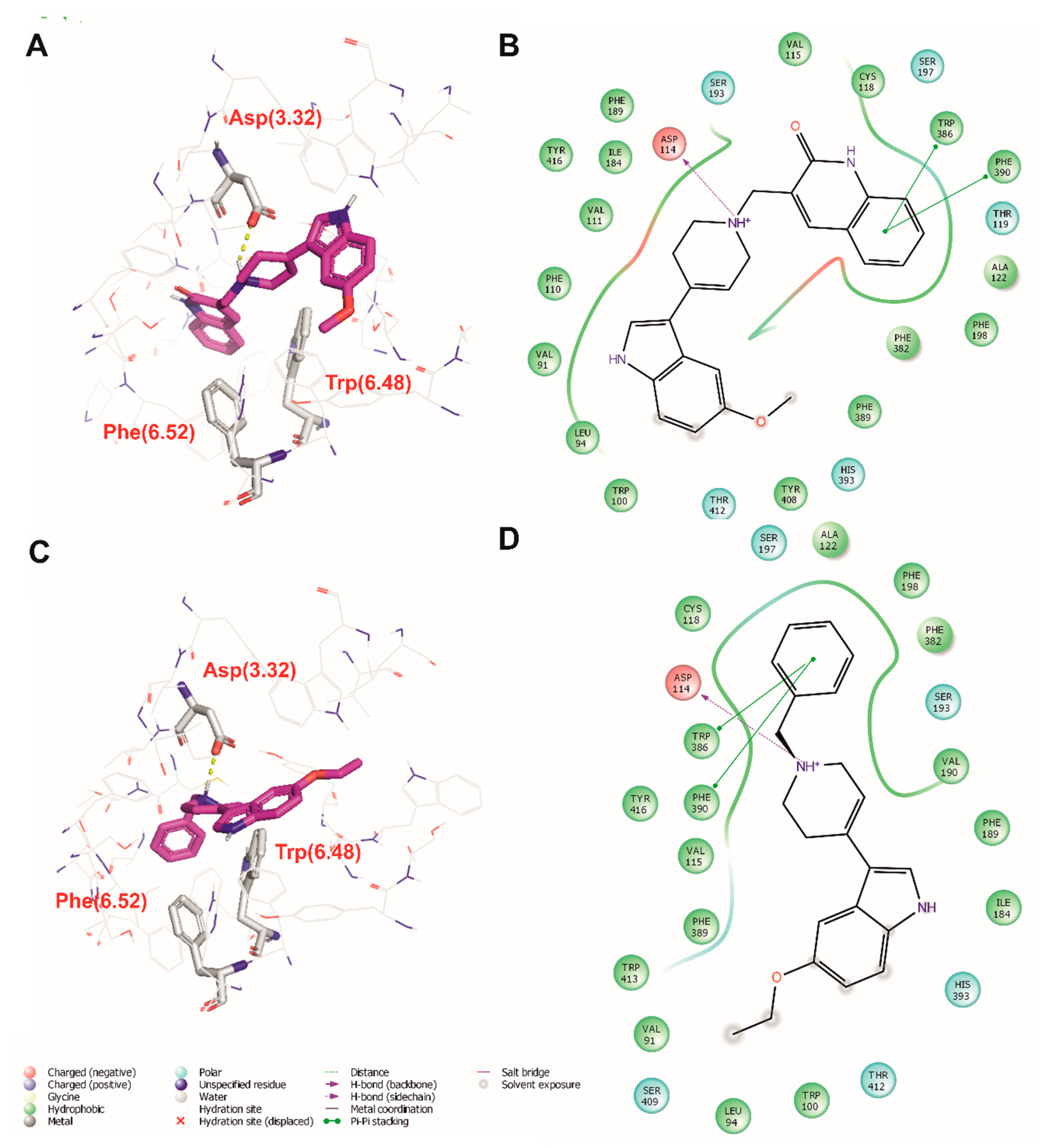
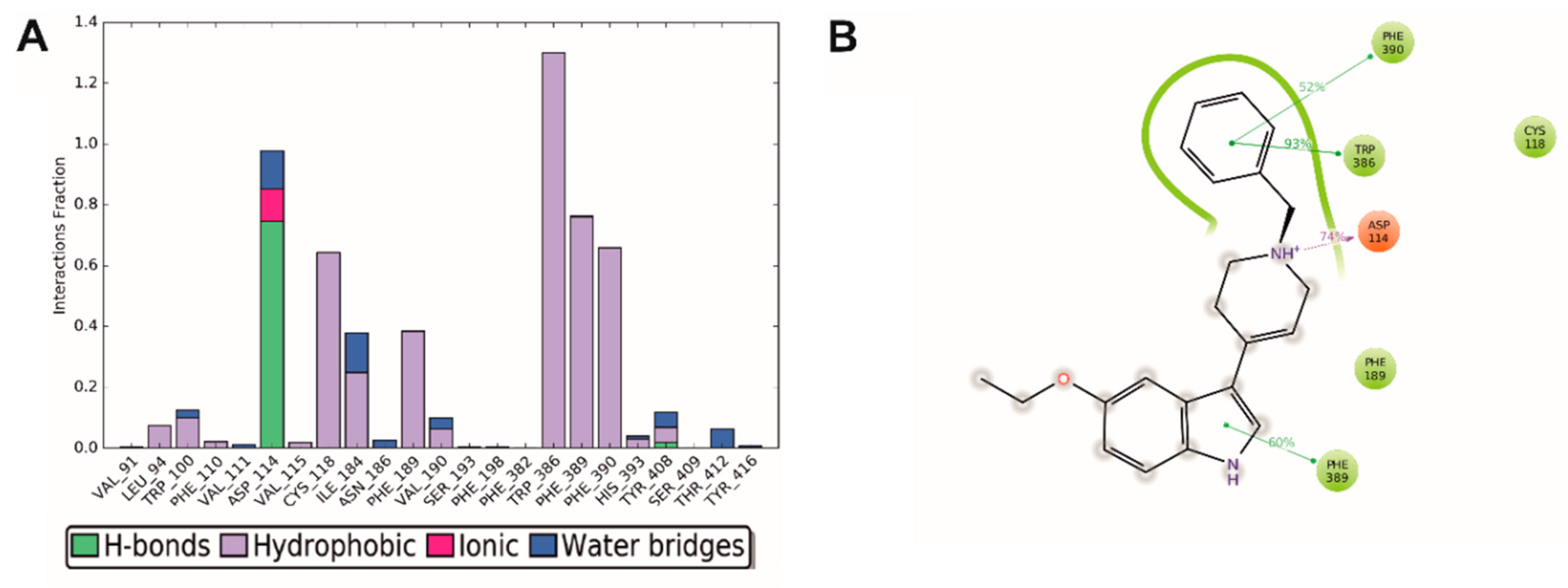
2.4. Molecular Modeling
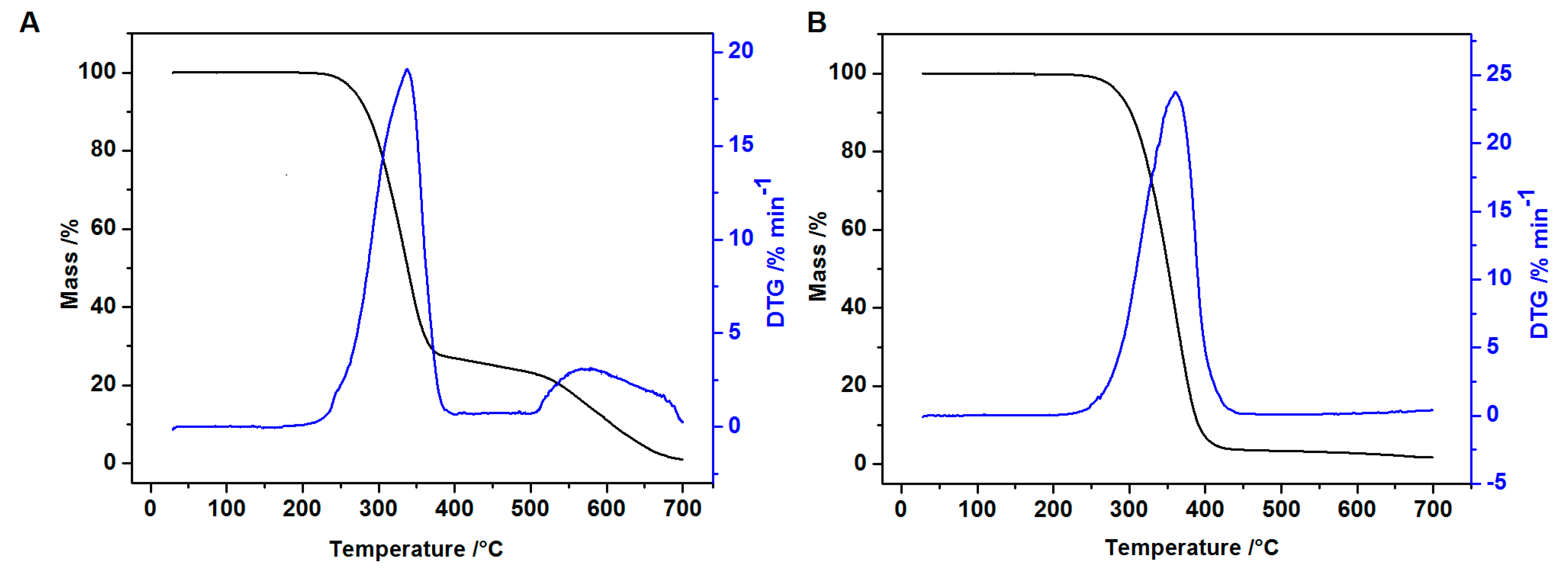
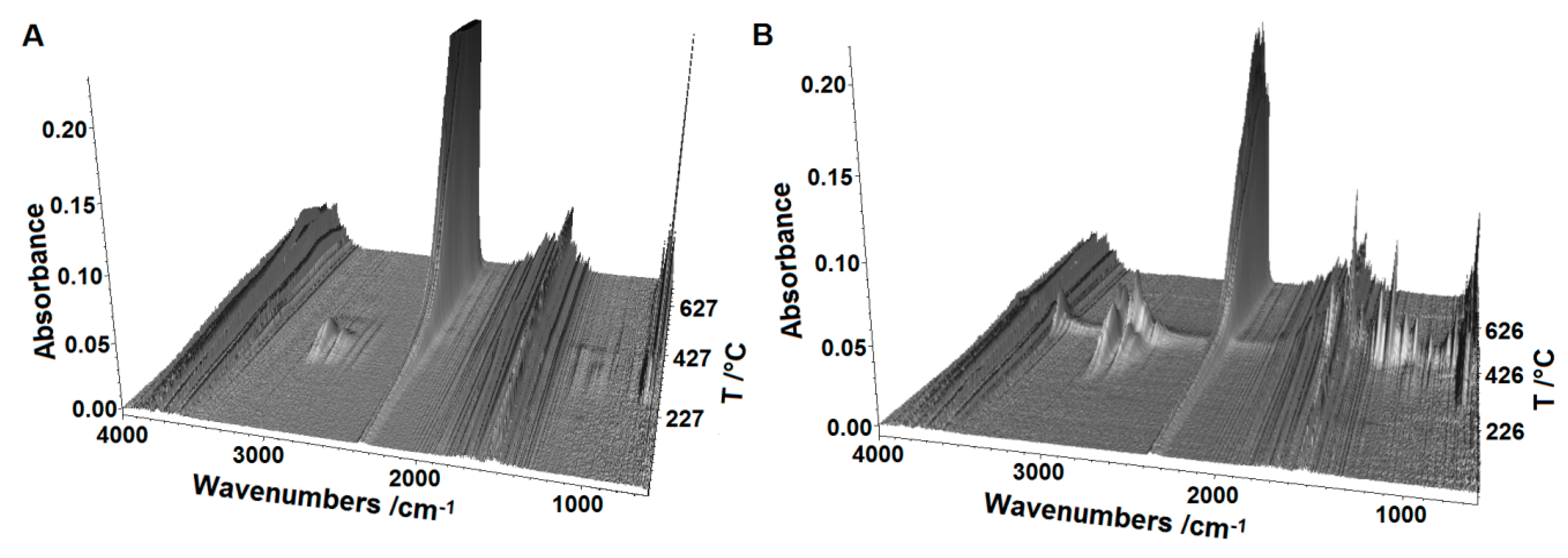
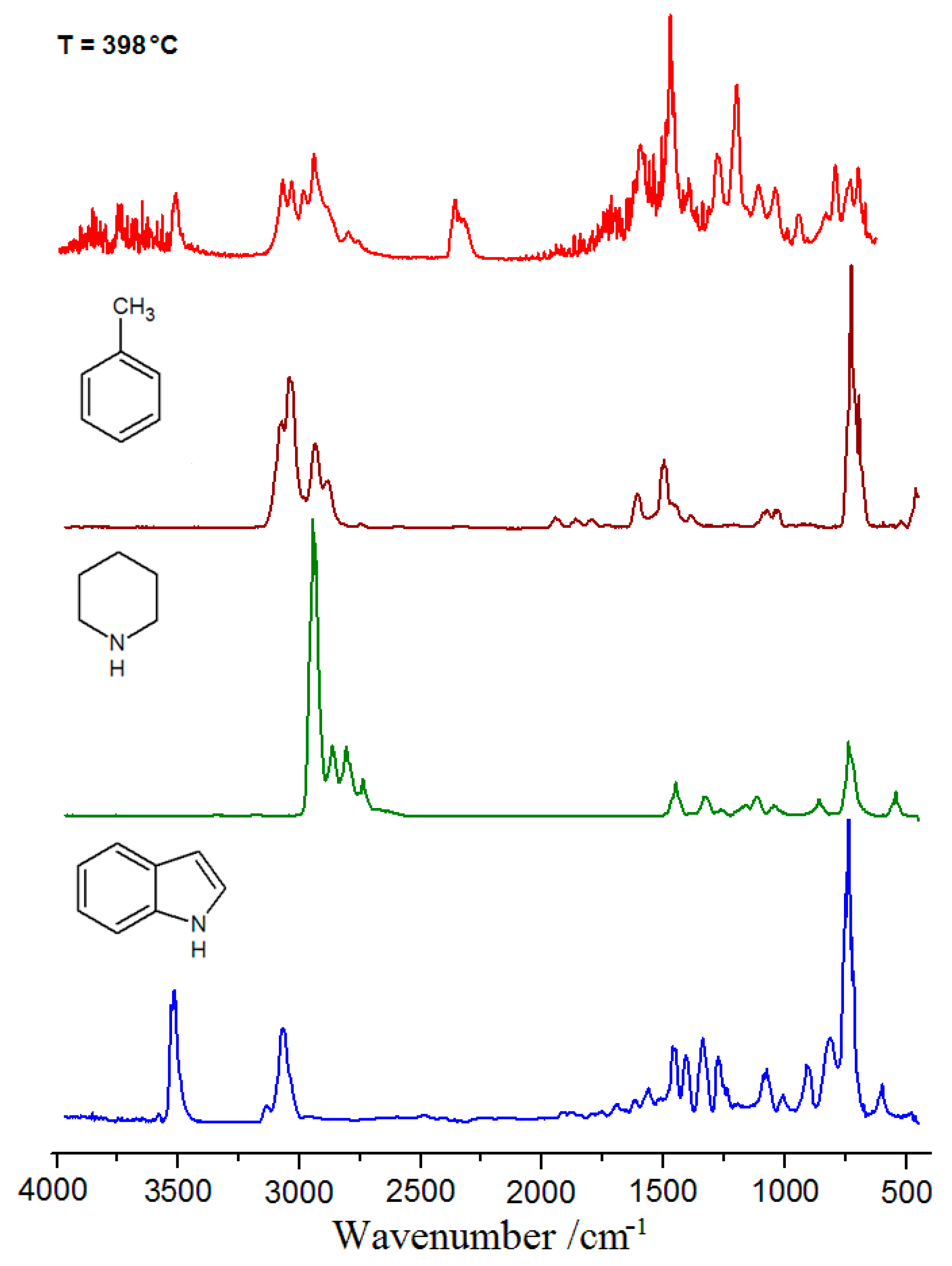
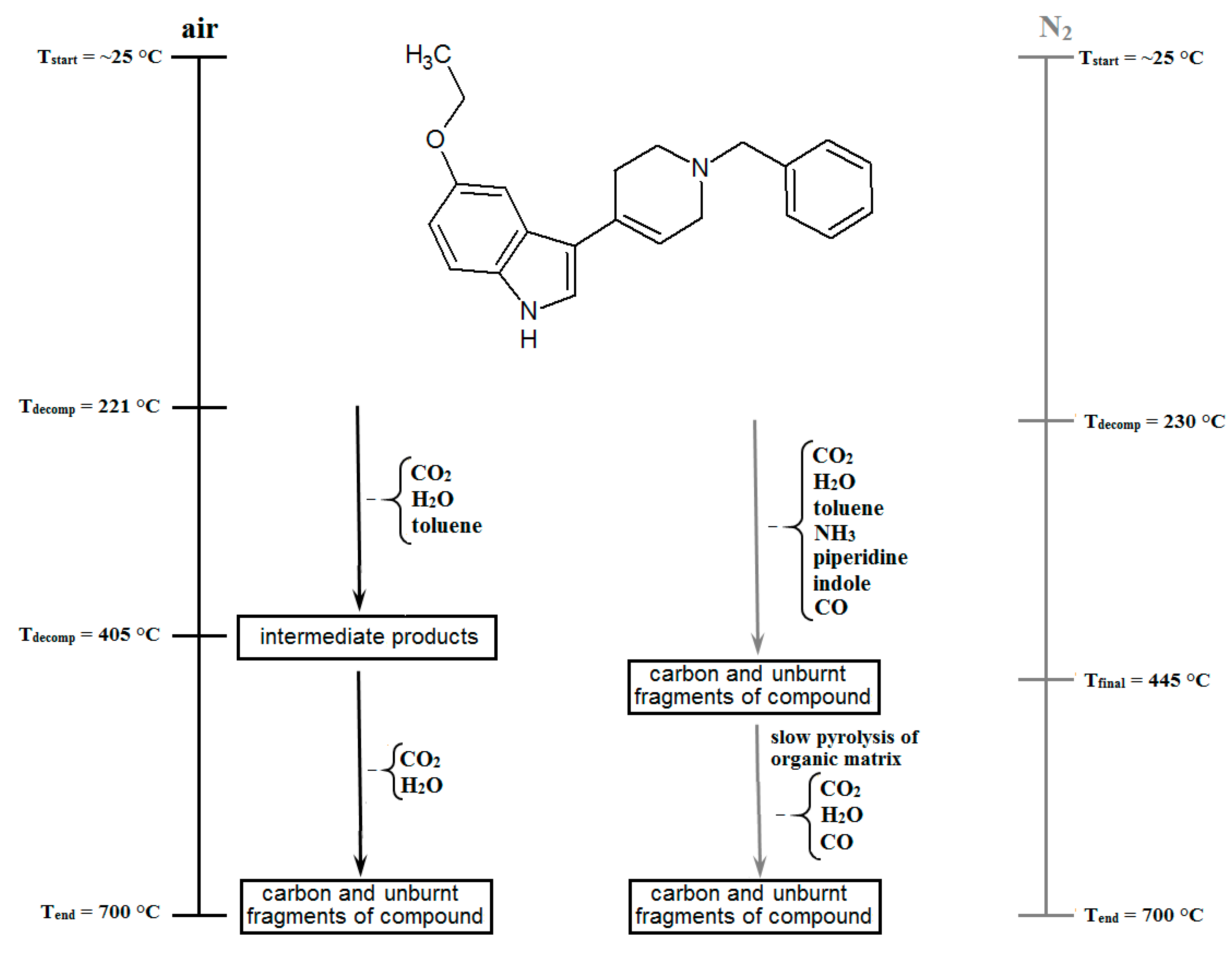
2.5. Thermal Analysis
3. Materials and Methods
3.1. Chemistry
3.2. In Vitro D2 Receptor Binding Assay
3.3. X-ray Structure Analysis
3.4. Molecular Modeling
3.4.1. Compound Preparation
3.4.2. Molecular Docking
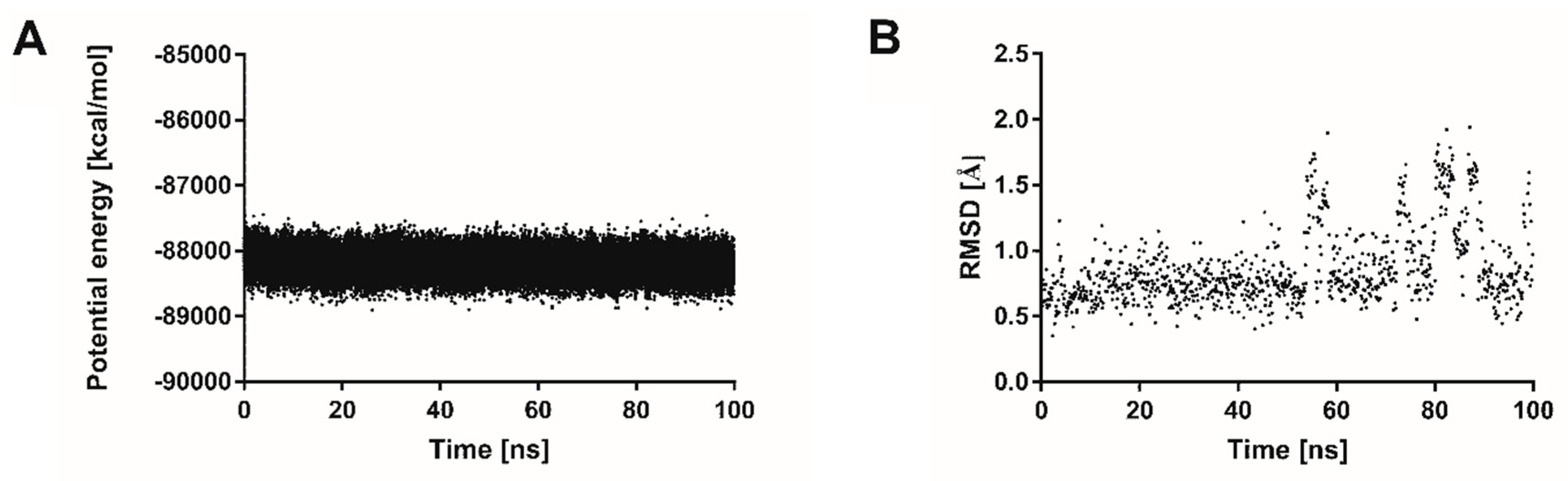
3.4.3. Molecular Dynamics
3.5. Thermal Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Beaulieu, J.-M.; Espinoza, S.; Gainetdinov, R.R. Dopamine receptors—IUPHAR Review 13. Br. J. Pharmacol. 2015, 172, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Singh, S.; Shukla, S. Physiological and Functional Basis of Dopamine Receptors and Their Role in Neurogenesis: Possible Implication for Parkinson’s disease. J. Exp. Neurosci. 2018, 12, 1179069518779829. [Google Scholar] [CrossRef] [PubMed]
- Seeman, P. Schizophrenia and dopamine receptors. Eur. Neuropsychopharmacol. J. Eur. Coll. Neuropsychopharmacol. 2013, 23, 999–1009. [Google Scholar] [CrossRef] [PubMed]
- Missale, C.; Fiorentini, C.; Collo, G.; Spano, P. The neurobiology of dopamine receptors: Evolution from the dual concept to heterodimer complexes. J. Recept. Signal Transduct. Res. 2010, 30, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-M.; Han, C.; Lee, S.-J.; Jun, T.-Y.; Patkar, A.A.; Masand, P.S.; Pae, C.-U. Investigational dopamine antagonists for the treatment of schizophrenia. Expert Opin. Investig. Drugs 2017, 26, 687–698. [Google Scholar] [CrossRef] [PubMed]
- Bielenica, A.; Kozioł, A.E.; Struga, M. Binding modes of chain arylpiperazines to 5-HT1a, 5-HT2a and 5-HT7 receptors. Mini Rev. Med. Chem. 2013, 13, 1516–1539. [Google Scholar] [CrossRef] [PubMed]
- Soskic, V.; Sukalovic, V.; Kostic-Rajacic, S. Exploration of N-arylpiperazine Binding Sites of D2 Dopaminergic Receptor. Mini Rev. Med. Chem. 2015, 15, 988–1001. [Google Scholar] [CrossRef] [PubMed]
- Marciniec, K.; Kurczab, R.; Książek, M.; Bębenek, E.; Chrobak, E.; Satała, G.; Bojarski, A.J.; Kusz, J.; Zajdel, P. Structural determinants influencing halogen bonding: A case study on azinesulfonamide analogs of aripiprazole as 5-HT1A, 5-HT7, and D2 receptor ligands. Chem. Cent. J. 2018, 12, 55. [Google Scholar] [CrossRef] [PubMed]
- Conway, R.J.; Valant, C.; Christopoulos, A.; Robertson, A.D.; Capuano, B.; Crosby, I.T. Synthesis and SAR study of 4-arylpiperidines and 4-aryl-1,2,3,6-tetrahydropyridines as 5-HT2C agonists. Bioorg. Med. Chem. Lett. 2012, 22, 2560–2564. [Google Scholar] [CrossRef] [PubMed]
- Annoura, H.; Nakanishi, K.; Uesugi, M.; Fukunaga, A.; Imajo, S.; Miyajima, A.; Tamura-Horikawa, Y.; Tamura, S. Synthesis and biological evaluation of new 4-arylpiperidines and 4-aryl-4-piperidinols: Dual Na(+) and Ca(2+) channel blockers with reduced affinity for dopamine D(2) receptors. Bioorg. Med. Chem. 2002, 10, 371–383. [Google Scholar] [CrossRef]
- Schuster, D.I.; Pan, Y.P.; Singh, G.; Stoupakis, G.; Cai, B.; Lem, G.; Ehrlich, G.K.; Frietze, W.; Murphy, R.B. N-(1-arylpropionyl)-4-aryltetrahydropyridines, a new class of high-affinity selective sigma receptor ligands. J. Med. Chem. 1993, 36, 3923–3928. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Castagnoli, N. Synthesis and MAO-B substrate properties of 1-methyl-4-heteroaryl-1,2,3,6-tetrahydropyridines. Bioorg. Med. Chem. 1999, 7, 231–239. [Google Scholar] [CrossRef]
- Yu, J.; Castagnoli, N. Synthesis and monoamine oxidase B substrate properties of 1-methyl-4-heteroaryl-1,2,3,6-tetrahydropyridines. Bioorg. Med. Chem. 1999, 7, 2835–2842. [Google Scholar] [CrossRef]
- Kaczor, A.A.; Silva, A.G.; Loza, M.I.; Kolb, P.; Castro, M.; Poso, A. Structure-Based Virtual Screening for Dopamine D2 Receptor Ligands as Potential Antipsychotics. Chem. Med. Chem. 2016, 11, 718–729. [Google Scholar] [CrossRef] [PubMed]
- Kaczor, A.A.; Targowska-Duda, K.M.; Budzyńska, B.; Biała, G.; Silva, A.G.; Castro, M. In vitro, molecular modeling and behavioral studies of 3-{[4-(5-methoxy-1H-indol-3-yl)-1,2,3,6-tetrahydropyridin-1-yl]methyl}-1,2-dihydroquinolin-2-one (D2AAK1) as a potential antipsychotic. Neurochem. Int. 2016, 96, 84–99. [Google Scholar] [CrossRef] [PubMed]
- Warnement, M.R.; Tomlinson, I.D.; Chang, J.C.; Schreuder, M.A.; Luckabaugh, C.M.; Rosenthal, S.J. Controlling the reactivity of ampiphilic quantum dots in biological assays through hydrophobic assembly of custom PEG derivatives. Bioconjug. Chem. 2008, 19, 1404–1413. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.; Pearson, P.P.; Taylor, E.W.; Li, H.B.; Dahlgren, T.; Herslöf, M.; Yang, Y.; Lambert, G.; Nelson, D.L.; Regan, J.W. Three-dimensional quantitative structure-activity relationships of 5-HT receptor binding data for tetrahydropyridinylindole derivatives: A comparison of the Hansch and CoMFA methods. J. Med. Chem. 1993, 36, 4006–4014. [Google Scholar] [CrossRef] [PubMed]
- Cole, D.C.; Ellingboe, J.W.; Lennox, W.J.; Mazandarani, H.; Smith, D.L.; Stock, J.R.; Zhang, G.; Zhou, P.; Schechter, L.E. N1-arylsulfonyl-3-(1,2,3,6-tetrahydropyridin-4-yl)-1H-indole derivatives are potent and selective 5-HT6 receptor antagonists. Bioorg. Med. Chem. Lett. 2005, 15, 379–383. [Google Scholar] [CrossRef] [PubMed]
- Gharagozloo, P.; Lazareno, S.; Miyauchi, M.; Popham, A.; Birdsall, N.J.M. Substituted pentacyclic carbazolones as novel muscarinic allosteric agents: Synthesis and structure-affinity and cooperativity relationships. J. Med. Chem. 2002, 45, 1259–1274. [Google Scholar] [CrossRef] [PubMed]
- Santos, S.A.; Lukens, A.K.; Coelho, L.; Nogueira, F.; Wirth, D.F.; Mazitschek, R.; Moreira, R.; Paulo, A. Exploring the 3-piperidin-4-yl-1H-indole scaffold as a novel antimalarial chemotype. Eur. J. Med. Chem. 2015, 102, 320–333. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, E.S.; van Smeden, M.; Schmidt, A.W.; Sprouse, J.S.; Wikström, H.V.; Grol, C.J. Novel 5-HT7 receptor inverse agonists. Synthesis and molecular modeling of arylpiperazine- and 1,2,3,4-tetrahydroisoquinoline-based arylsulfonamides. J. Med. Chem. 2004, 47, 5451–5466. [Google Scholar] [CrossRef] [PubMed]
- Bartyzel, A.; Kaczor, A.A.; Głuchowska, H.; Pitucha, M.; Wróbel, T.M.; Matosiuk, D. Thermal and spectroscopic studies of 2, 3, 5-trisubstituted and 1, 2, 3, 5-tetrasubstituted indoles as non-competitive antagonists of GluK1/GluK2 receptors. J. Therm. Anal. Cal. 2018, 133, 935–944. [Google Scholar] [CrossRef]
- Socrates, G. Infrared and Raman Characteristic Group Frequencies Tables and Charts; John Wiley & Sons Ltd.: Chichester, UK, 2001. [Google Scholar]
- Allen, F.H.; Watson, D.G.; Brammer, L.; Orpen, A.G.; Taylor, R. Chapter 9.5 Typical Interatomic Distances: Organic Compounds in International Tables for Crystallography; John Wiley & Sons Ltd.: Chichester, UK, 2006. [Google Scholar]
- Bates, R.B.; Bruck, M.A.; Camou, F.A.; Martin, A.R.; Nikam, S.S.; Nelson, D.L. 3-(1-Methyl-1,2,3,6-tetrahydropyrid-4-yl)indole. Acta Cryst. 1989, C45, 109–111. [Google Scholar] [CrossRef]
- Rasztawicka, M.; Wolska, I.; Maciejewska, D. Solid state structure by X-ray and 13C CP/MAS NMR of new 5,5′-diethoxy-3,3′-methanediyl-bis-indole. J. Mol. Struct. 2007, 831, 174–179. [Google Scholar] [CrossRef]
- Chandrakantha, T.N.; Puttaraja; Kokila, M.K.; Shivaprakash, N.C. Ethyl 5-Ethoxy-3-methyl-1H-indole-2-carboxylate. Acta Cryst. 1998, 54, 1685–1687. [Google Scholar] [CrossRef]
- Cremer, D.; Pople, J.A. General definition of ring puckering coordinates. J. Am. Chem. Soc. 1975, 97, 1354–1358. [Google Scholar] [CrossRef]
- Boeyens, J.C.A. Conformation of 6-Membered Rings. J. Cryst. Mol. Struct. 1978, 8, 317–320. [Google Scholar] [CrossRef]
- Pérez, P.J.; Carrascosa, R.; García, L.; Barandika, G.; Calderón-Casado, A.; Pérez, E.; Serrano, J.L.; Santana, M.D. Coordination to metal centers: A tool to fix high energy conformations in organic molecules. Application to 2,4,4-trimethyl-1,5,9-triazacyclododec-1-ene and related macrocycles. Dalton Trans. 2011, 40, 9504–9511. [Google Scholar] [CrossRef] [PubMed]
- Mayes, H.B.; Broadbelt, L.J.; Beckham, G.T. How Sugars Pucker: Electronic Structure Calculations Map the Kinetic Landscape of Five Biologically Paramount Monosaccharides and Their Implications for Enzymatic Catalysis. J. Am. Chem. Soc. 2014, 136, 1008–1022. [Google Scholar] [CrossRef] [PubMed]
- Ballesteros, J.A.; Weinstein, H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci. 1995, 25, 366–428. [Google Scholar] [CrossRef]
- Bartyzel, A.; Sztanke, M.; Sztanke, K. An insight into the thermal behaviour of biologically active 8-aryl-4-oxo-4,6,7,8-tetrahydroimidazo[2,1-c][1,2,4]triazine-3-carbohydrazides. J. Anal. Appl. Pyrol. 2016, 121, 138–145. [Google Scholar] [CrossRef]
- Bartyzel, A.; Kaczor, A.A. The formation of a neutral manganese(III) complex containing a tetradentate Schiff base and a ketone—Synthesis and characterization. J. Coord. Chem. 2015, 68, 3701–3717. [Google Scholar] [CrossRef]
- Bartyzel, A. Synthesis, thermal behaviour and some properties of CuII complexes with N,O-donor Schiff bases. J. Therm. Anal. Calorim. 2018, 131, 1221–1236. [Google Scholar] [CrossRef]
- Bartyzel, A. Synthesis, thermal study and some properties of N2O4—Donor Schiff base and its Mn(III), Co(II), Ni(II), Cu(II) and Zn(II) complexes. J. Therm. Anal. Calorim. 2017, 127, 2133–2147. [Google Scholar] [CrossRef]
- Bartyzel, A.; Kaczor, A.A. Synthesis, crystal structure, thermal, spectroscopic and theoretical studies of N3O2-donor Schiff base and its complex with CuII ions. Polyhedron 2018, 139, 271–281. [Google Scholar] [CrossRef]
- Selent, J.; Marti-Solano, M.; Rodríguez, J.; Atanes, P.; Brea, J.; Castro, M.; Sanz, F.; Loza, M.I.; Pastor, M. Novel insights on the structural determinants of clozapine and olanzapine multi-target binding profiles. Eur. J. Med. Chem. 2014, 77, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Brea, J.; Castro, M.; Loza, M.I.; Masaguer, C.F.; Raviña, E.; Dezi, C.; Pastor, M.; Sanz, F.; Cabrero-Castel, A.; Galán-Rodríguez, B.; et al. QF2004B, a potential antipsychotic butyrophenone derivative with similar pharmacological properties to clozapine. Neuropharmacology 2006, 51, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Crysalis Pro; Agilent Technologies: Oxfordshire, UK, 2013.
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Faruggia, L.J. WinGX suite for small-molecule single-crystal crystallography. J. Appl. Crystallogr. 1999, 32, 837–838. [Google Scholar] [CrossRef]
- Spek, L.A. Single-crystal structure validation with the program PLATON. J. Appl. Cryst. 2003, 36, 7–13. [Google Scholar] [CrossRef]
- Flack, H.D. On Enantiomorph-Polarity Estimation. Acta Crystallogr. 1993, A39, 876–881. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Cryst. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; van de Streek, J. Mercury: Visualization and Analysis of Crystal Structures. J. Appl. Cryst. 2006, 39, 453–457. [Google Scholar] [CrossRef]
- Schrödinger Release 2018-2: LigPrep; Schrödinger, LLC: New York, NY, USA, 2018.
- Schrödinger Release 2018-2: Epik; Schrödinger, LLC: New York, NY, USA, 2018.
- Wang, S.; Che, T.; Levit, A.; Shoichet, B.K.; Wacker, D.; Roth, B.L. Structure of the D2 dopamine receptor bound to the atypical antipsychotic drug risperidone. Nature 2018, 555, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Bowers, K.J.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossváry, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the ACM/IEEE Conference on Supercomputing (SC06), Tampa, FL, USA, 11–17 November 2006. [Google Scholar]
- Schrödinger Release 2018-2: Desmond Molecular Dynamics System; D.E. Shaw Research; Maestro-Desmond Interoperability Tools; Schrödinger: New York, NY, USA, 2018.
- Kaczor, A.A.; Karczmarzyk, Z.; Fruziński, A.; Pihlaja, K.; Sinkkonen, J.; Wiinämaki, K.; Kronbach, C.; Unverferth, K.; Poso, A.; Matosiuk, D. Structural studies, homology modeling and molecular docking of novel non-competitive antagonists of GluK1/GluK2 receptors. Bioorg. Med. Chem. 2014, 22, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Jozwiak, K.; Targowska-Duda, K.M.; Kaczor, A.A.; Kozak, J.; Ligeza, A.; Szacon, E.; Wrobel, T.M.; Budzynska, B.; Biala, G.; Fornal, E.; et al. Synthesis, in vitro and in vivo studies, and molecular modeling of N-alkylated dextromethorphan derivatives as non-competitive inhibitors of α3β4 nicotinic acetylcholine receptor. Bioorg. Med. Chem. 2014, 22, 6846–6856. [Google Scholar] [CrossRef] [PubMed]
- Arias, H.R.; Feuerbach, D.; Targowska-Duda, K.; Kaczor, A.A.; Poso, A.; Jozwiak, K. Pharmacological and molecular studies on the interaction of varenicline with different nicotinic acetylcholine receptor subtypes. Potential mechanism underlying partial agonism at human α4β2 and α3β4 subtypes. Biochim. Biophys. Acta 2015, 1848, 731–741. [Google Scholar] [CrossRef] [PubMed]
- Kaczor, A.A.; Żuk, J.; Matosiuk, D. Comparative molecular field analysis and molecular dynamics studies of the dopamine D2 receptor antagonists without a protonatable nitrogen atom. Med. Chem. Res. 2018, 27, 1149–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ennis, Z.N.; Damkier, P. Pregnancy exposure to olanzapine, quetiapine, risperidone, aripiprazole and risk of congenital malformations. A systematic review. Basic Clin. Pharmacol. Toxicol. 2015, 116, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Asmal, L.; Flegar, S.J.; Wang, J.; Rummel-Kluge, C.; Komossa, K.; Leucht, S. Quetiapine versus other atypical antipsychotics for schizophrenia. Cochrane Database Syst. Rev. 2013, 11, CD006625. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compound are available from the authors. |














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formula | C22H24N2O |
| Formula weight | 332.43 |
| Temperature K | 293(2) |
| Crystal system | orthorhombic |
| Space group | P212121 |
| a (Å) | 5.9600(4) |
| b (Å) | 15.0986(9) |
| c (Å) | 16.1571(9) |
| Volume (Å3) | 1814.5(2) |
| Z | 4 |
| Calculated density (g cm−3) | 1.217 |
| μ (mm−1) | 0.075 |
| Absorption correction | multi-scan |
| F(000) | 712 |
| Crystal size (mm) | 0.50 × 0.20 × 0.20 |
| θ range (°) | 2.698 to 27.103 |
| Index ranges | −7 ≤ h ≤ 7 −19 ≤ k ≤ 19 −25 ≤ l ≤ 18 |
| Reflections collected/unique | 14,278/3993 |
| Rint | 0.0442 |
| Data/restraints/parameters | 3993/0/231 |
| GooF on F2 | 1.000 |
| Final R indices[I > 2σ(I)] | R1 = 0.0437, wR2 = 0.0887 |
| R indices(all data) | R1 = 0.0745, wR2 = 0.1019 |
| Largest diff. peak/hole, e Å−3 | 0.156/−0.111 |
| Bond Lengths (Å) | |||
| C1-N1 | 1.359(3) | C10-C11 | 1.364(5) |
| C1-C2 | 1.369(3) | C11-C12 | 1.376(4) |
| C2-C15 | 1.441(3) | C13-N2 | 1.462(3) |
| C2-C3 | 1.465(3) | C13-C14 | 1.497(3) |
| C3-C14 | 1.325(3) | C15-C22 | 1.406(3) |
| C3-C4 | 1.506(3) | C15-C16 | 1.409(3) |
| C4-C5 | 1.512(3) | C16-C17 | 1.373(4) |
| C5-N2 | 1.459(3) | C17-O1 | 1.380(3) |
| C6-N2 | 1.472(3) | C17-C20 | 1.392(4) |
| C6-C7 | 1.503(3) | C18-O1 | 1.416(3) |
| C7-C8 | 1.376(3) | C18-C19 | 1.493(4) |
| C7-C12 | 1.383(3) | C20-C21 | 1.366(4) |
| C8-C9 | 1.379(4) | C21-C22 | 1.386(4) |
| C9-C10 | 1.360(4) | C22-N1 | 1.376(3) |
| Bond angles (°) | |||
| C14-C3-C2 | 124.6(2) | C3-C14-C13 | 124.1(2) |
| C14-C3-C4 | 118.6(2) | C16-C17-O1 | 124.3(2) |
| C2-C3-C4 | 116.7(2) | O1-C17-C20 | 114.6(2) |
| C3-C4-C5 | 113.0 (2) | C5-N2-C13 | 108.4(2) |
| N2-C5-C4 | 111.0(2) | C5-N2-C6 | 111.2(2) |
| N2-C6-C7 | 114.1(2) | C13-N2-C6 | 107.7(2) |
| N2-C13-C14 | 113.1(2) | C17-O1-C18 | 118.0(2) |
| Torsion angles (°) | |||
| C4-C5-N2-C13 | −65.0(3) | C4-C3-C14-C13 | 0.6(4) |
| C3-C4-C5-N2 | 47.6(3) | N2-C13-C14-C3 | −18.2(4) |
| C14-C3-C4-C5 | −15.0(3) | C14-C13-N2-C5 | 49.2(3) |
| Hydrogen Bond [Å, °] | ||||
| D-H···A | d(D-H) | d(H···A) | d(D···A) | DHA |
| N1-H1N···N2 i | 0.88(3) | 2.41(3) | 3.217(3) | 154(2) |
| C-H···Cg interactions [Å, °] | ||||
| C-H···Cg | d(D-H) | d(H···Cg) | d(C···Cg) | CHCg |
| C4-H4B···Cg1 ii | 0.97 | 2.94 | 3.567(3) | 124 |
| C19-H19C···Cg4 iii | 0.96 | 3.00 | 3.799(3) | 142 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kondej, M.; Bartyzel, A.; Pitucha, M.; Wróbel, T.M.; Silva, A.G.; Matosiuk, D.; Castro, M.; Kaczor, A.A. Synthesis, Structural and Thermal Studies of 3-(1-Benzyl-1,2,3,6-tetrahydropyridin-4-yl)-5-ethoxy-1H-indole (D2AAK1_3) as Dopamine D2 Receptor Ligand. Molecules 2018, 23, 2249. https://doi.org/10.3390/molecules23092249
Kondej M, Bartyzel A, Pitucha M, Wróbel TM, Silva AG, Matosiuk D, Castro M, Kaczor AA. Synthesis, Structural and Thermal Studies of 3-(1-Benzyl-1,2,3,6-tetrahydropyridin-4-yl)-5-ethoxy-1H-indole (D2AAK1_3) as Dopamine D2 Receptor Ligand. Molecules. 2018; 23(9):2249. https://doi.org/10.3390/molecules23092249
Chicago/Turabian StyleKondej, Magda, Agata Bartyzel, Monika Pitucha, Tomasz M. Wróbel, Andrea G. Silva, Dariusz Matosiuk, Marián Castro, and Agnieszka A. Kaczor. 2018. "Synthesis, Structural and Thermal Studies of 3-(1-Benzyl-1,2,3,6-tetrahydropyridin-4-yl)-5-ethoxy-1H-indole (D2AAK1_3) as Dopamine D2 Receptor Ligand" Molecules 23, no. 9: 2249. https://doi.org/10.3390/molecules23092249