A Simple, Sensitive, and Reliable Method for the Simultaneous Determination of Multiple Antibiotics in Vegetables through SPE-HPLC-MS/MS

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
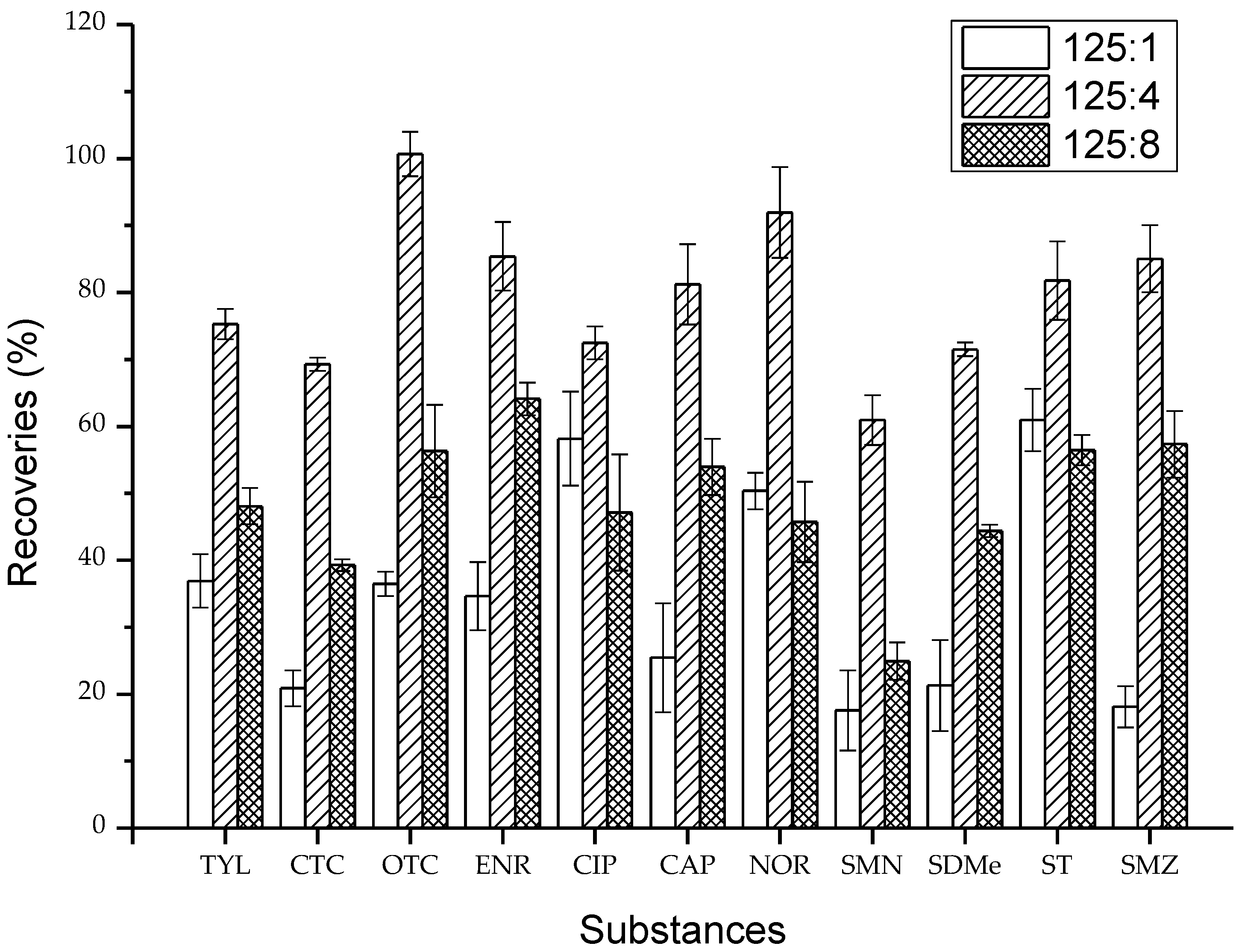
2.1. Validation of Extracting Agents
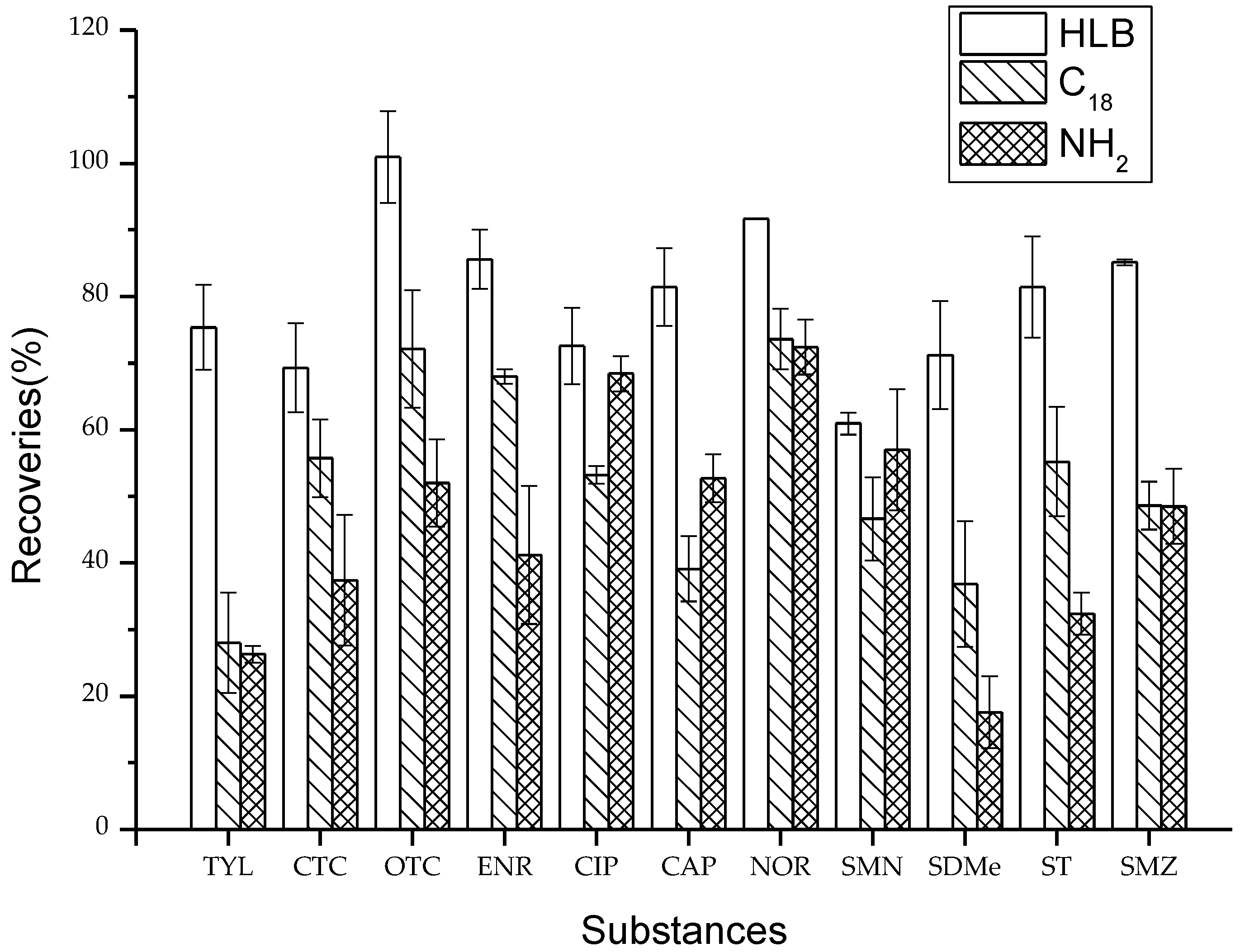
2.2. Optimization of SPE Cartridges
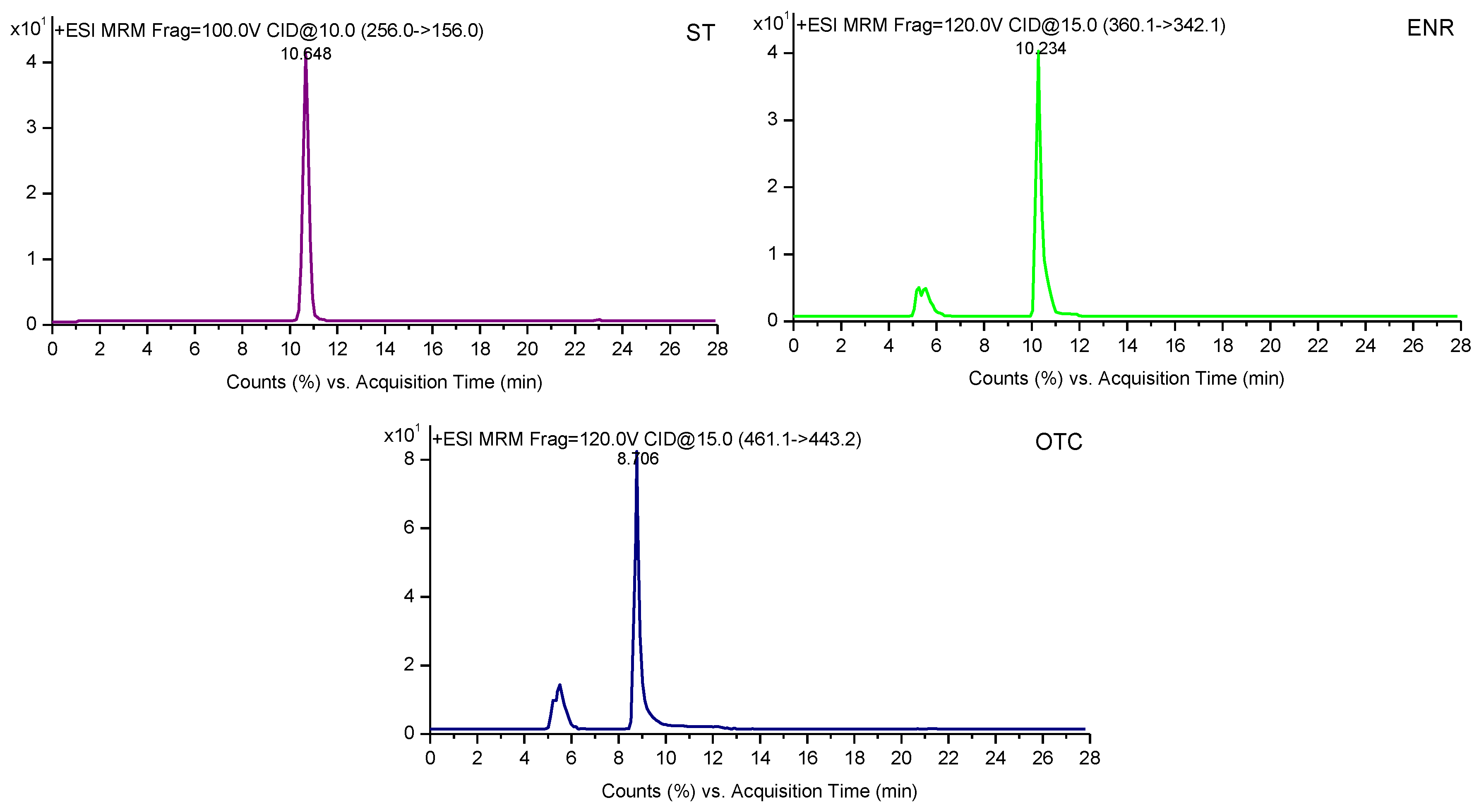
2.3. Method Efficiency
2.4. Method Precision and Accuracy
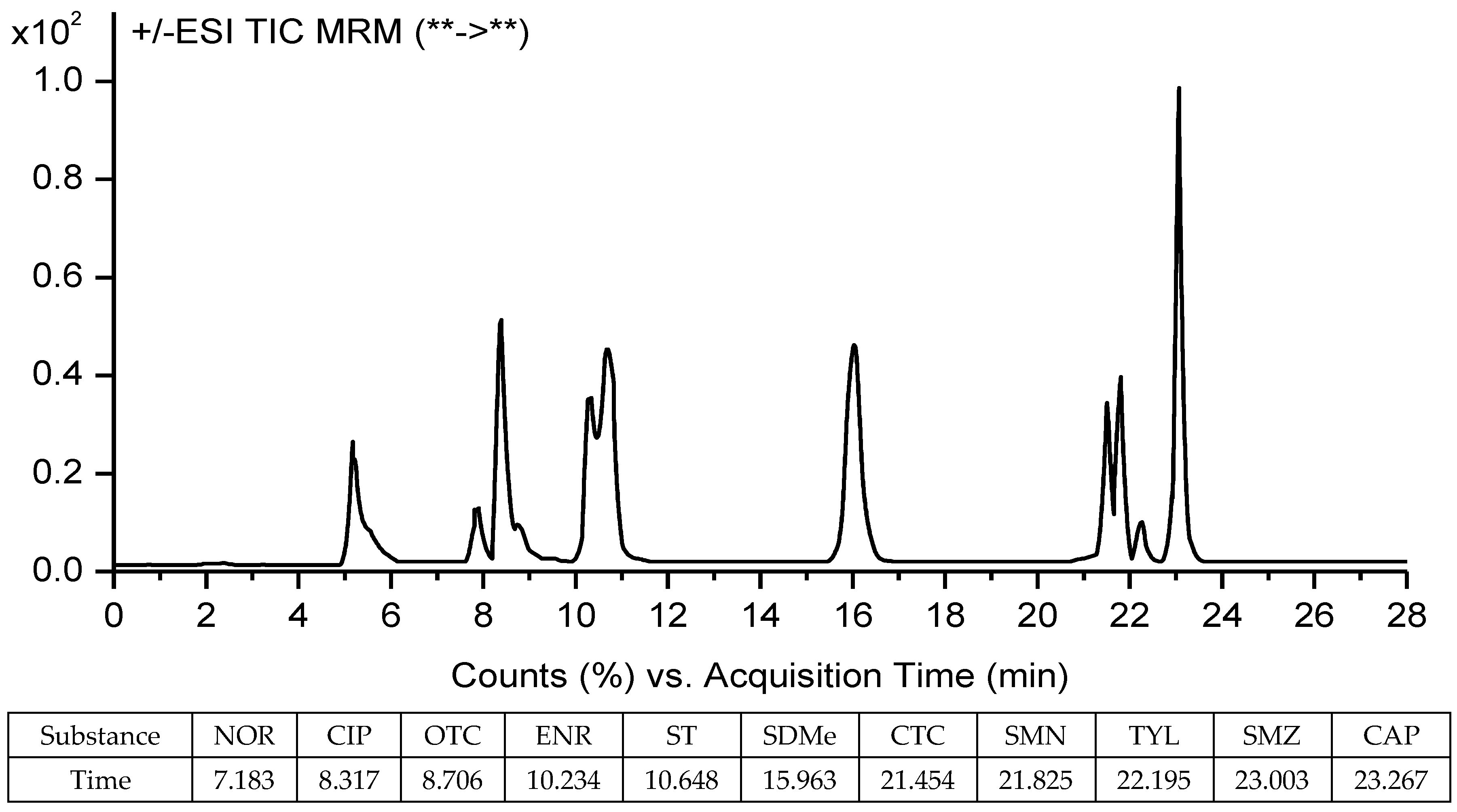
2.5. Samples Analyses
3. Materials and Methods
3.1. Standards and Chemicals
3.2. Vegetable Samples
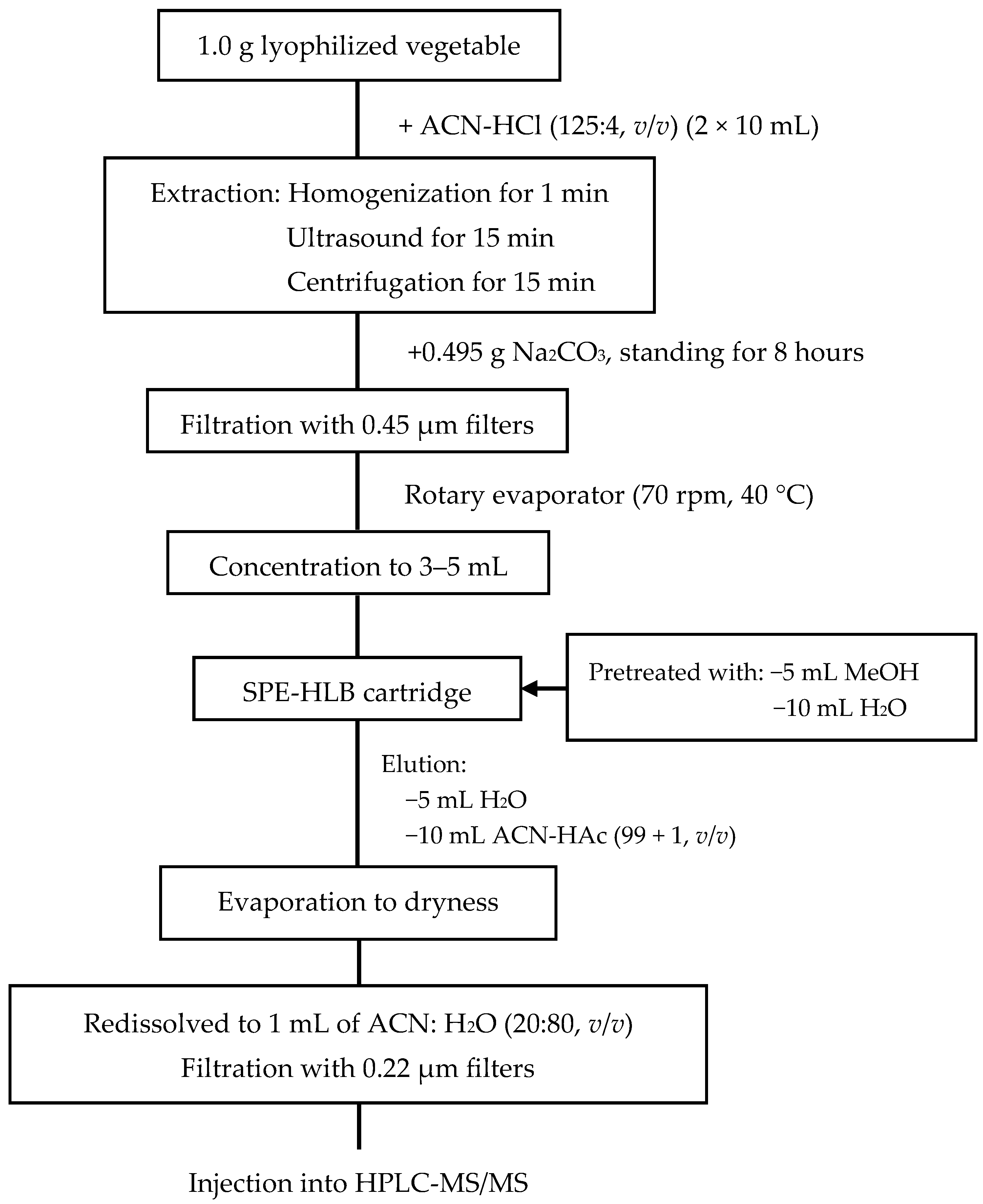
3.3. Sample Preparation
3.4. Sample Extraction and Clean-Up
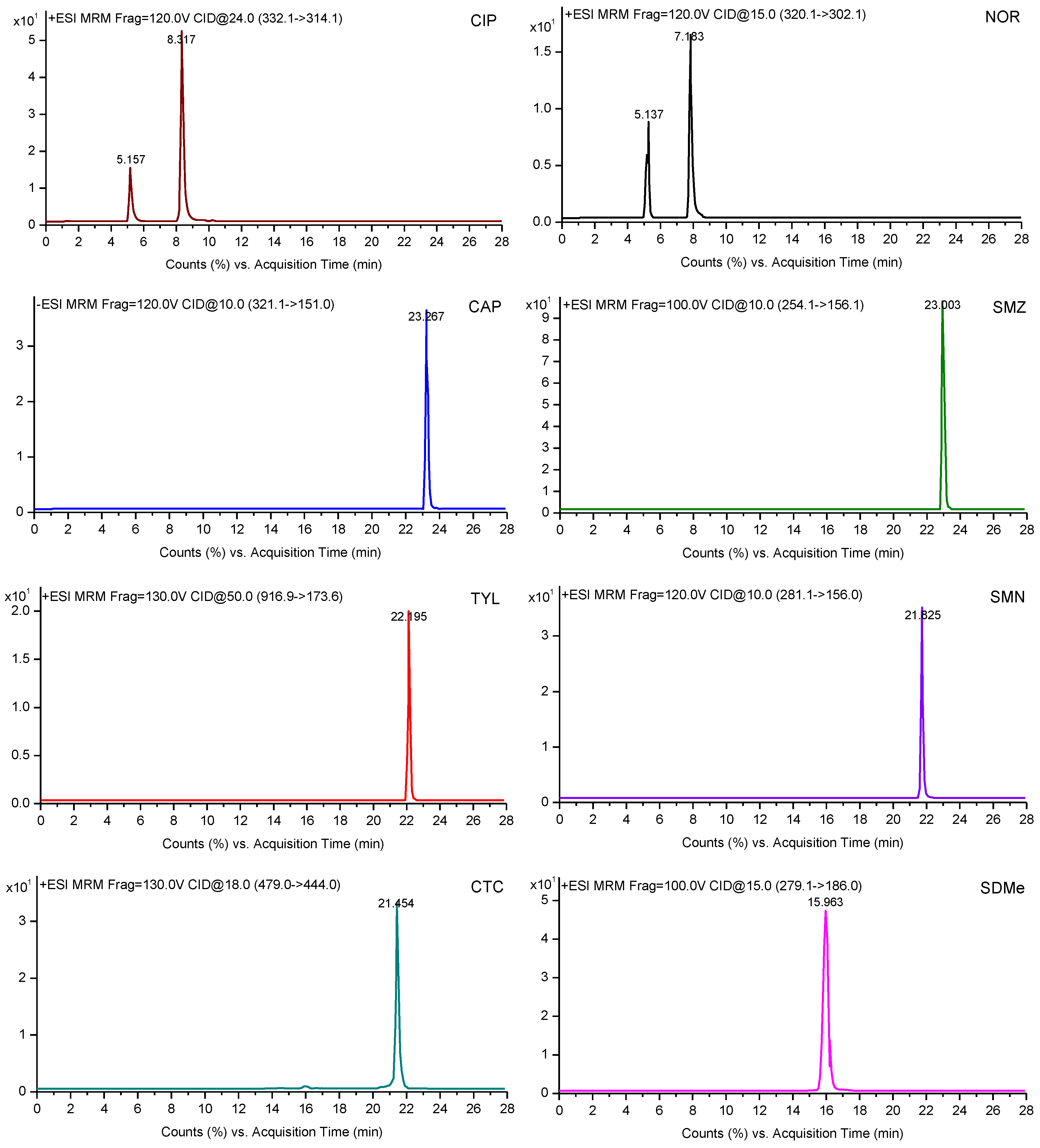
3.5. HPLC-MS/MS Analysis
3.6. Detection of Antibiotics in Vegetables
3.7. Statistical Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Gustafson, R.H.; Bowen, R.E. Antibiotic use in animal agriculture. J. Appl. Microbiol. 1997, 83, 531–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elena, M.C.; Carmen, G.B.; Sigrid, S.; Oliver, G. Environmental monitoring study of selected veterinary antibiotics in animal manure and soils in Austria. Environ. Pollut. 2007, 148, 570–579. [Google Scholar]
- FDA (Food and Drug Administration). Summary Report on Antimicrobials Sold or Distributed for Use in Food-Producing Animals; Food and Drug Administration, Department of Health and Human Services: Silver Spring, MD, USA, 2014.
- Zhang, Q.Q.; Ying, G.G.; Pan, C.G.; Liu, Y.S.; Zhao, J.L. Comprehensive evaluation of antibiotics emission and fate in the river basins of China: Source analysis, multimedia modeling, and linkage to bacterial resistance. Environ. Sci. Technol. 2015, 49, 6772–6782. [Google Scholar] [CrossRef] [PubMed]
- Gbylik-Sikorska, M.; Posyniak, A.; Sniegocki, T.; Zmudzki, J. Liquid chromatography-tandem mass spectrometry multiclass method for the determination of antibiotics residues in water samples from water supply systems in food-producing animal farms. Chemosphere 2015, 119, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Pino, M.R.; Val, J.; Mainar, A.M.; Zuriaga, E.; Español, C.; Langa, E. Acute toxicological effects on the earthworm Eisenia fetida of 18 common pharmaceuticals in artificial soil. Sci. Total Environ. 2015, 518–519, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.G.; Zhou, Q.; Luo, Y. Occurrence and source analysis of typical veterinary antibiotics in manure, soil, vegetables and groundwater from organic vegetable bases, northern China. Environ. Pollut. 2010, 158, 2992–2998. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.H.; Lee, H.J.; Ryu, P.U. Public health risks: Chemical and antibiotic residues. Asian Australas. J. Anim. 2001, 14, 402–413. [Google Scholar] [CrossRef]
- Li, Z.J.; Xie, X.Y.; Zhang, S.Q.; Liang, Y.C. Negative effects of oxytetracycline on wheat (Triticum aestivum L.) growth, root activity, photosynthesis and chlorophyll contents. Agric. Sci. China 2011, 10, 545–553. [Google Scholar] [CrossRef]
- Li, Z.J.; Xie, X.Y.; Zhang, S.Q.; Liang, Y.C. Wheat growth and photosynthesis as affected by oxytetracycline as a soil contaminant. Pedosphere 2011, 21, 244–250. [Google Scholar] [CrossRef]
- Karmi, M. Detection and presumptive identification of antibiotic residues in poultry meat by using FPT. Glob. J. Pharmacol. 2014, 8, 160–165. [Google Scholar]
- Zhang, X.; Zhang, D.D.; Zhang, H.; Luo, Z.X.; Yan, C.Z. Occurrence, distribution, and seasonal variation of estrogenic compounds and antibiotic residues in Jiulongjiang River, South China. Environ. Sci. Pollut. Res. Int. 2012, 19, 1392–1404. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Qiang, Z.; Ben, W.; Zhu, B.; Liu, J. Rapid detection of multiple class pharmaceuticals in both municipal wastewater and sludge with ultra high performance liquid chromatography tandem mass spectrometry. J. Environ. Sci. 2014, 26, 1949–1959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.L.; Xiang, L.; Mo, C.H.; Li, Y.W.; Jiang, Y.N.; Yan, Q.Y.; Lv, X.; Huang, X.P. Determination of quinolones in vegetables using ultra performance liquid chromatography-electrospray ionization tandem mass spectrometry. Chin. J. Anal. Chem. 2013, 41, 876–881. (In Chinese) [Google Scholar] [CrossRef]
- Zhang, Y.; Li, Y.W.; Mo, C.H.; Tai, Y.P.; Bao, Y.P.; Wang, Q.J.; Yi, R.H. Determination of quinolones antibiotics in vegetable using high performance liquid chromatography-fluorescence. Guangdong Agric. Sci. 2009, 6, 176–180. (In Chinese) [Google Scholar]
- Yu, X.L.; Liu, H.; Pu, C.J.; Chen, J.H.; Sun, Y.; Hu, L. Determination of multiple antibiotics in leafy vegetables using QuEChERS–UHPLC–MS/MS. J. Sep. Sci. 2018, 41, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.P.; Li, Y.W.; Mo, C.H.; Yao, Y.; Tai, Y.P.; Wu, X.L.; Zhang, Y. Determination of six sulfonamide antibiotics in vegetables by solid phase extraction and high performance liquid chromatography. Environ. Chem. 2010, 29, 513–518. (In Chinese) [Google Scholar]
- He, Z.; Wang, Y.; Xu, Y.; Liu, X. Determination of antibiotics in vegetables using quechers-based method and liquid chromatography-quadrupole linear ion trap mass spectrometry. Food Anal. Methods 2018, 1–8. [Google Scholar] [CrossRef]
- Jacobsen, A.M.; Halling-Sørensen, B.; Ingerslev, F.; Hansen, S.H. Simultaneous extraction of tetracycline, macrolide and sulfonamide antibiotics from agricultural soils using pressurised liquid extraction, followed by solid-phase extraction and liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2004, 1038, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Schlusener, M.P.; Spiteller, M.; Kai, B. Determination of antibiotics from soil by pressurized liquid extraction and liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2003, 1003, 21–28. [Google Scholar] [CrossRef]
- Jacobsen, A.M.; Halling-Sørensen, B. Multi-component analysis of tetracyclines, sulfonamides and tylosin in swine manure by liquid chromatography-tandem mass spectrometry. Anal. Bioanal. Chem. 2006, 384, 1164–1174. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhong, D.L.; Chen, G.C.; Tang, F.B.; Song, Q.H.; Zhang, J.F. Determination of antibiotic residues in manure by liquid chromatography-tandem mass spectrometry coupled with solid phase extraction. Chin. J. Chromatogr. 2013, 31, 1010–1015. (In Chinese) [Google Scholar] [CrossRef]
- Feng, Y.; Wei, C.J.; Zhang, W.J.; Liu, Y.W.; Li, Z.J.; Hu, H.Y.; Xue, J.M.; Davis, M. A simple and economic method for simultaneous determination of 11 antibiotics in manure by solid-phase extraction and high-performance liquid chromatography. J. Soils Sediments 2016, 16, 2242–2251. [Google Scholar] [CrossRef]
- Tang, C.M.; Huang, Q.X.; Yu, Y.Y.; Peng, X.Z. Multiresidue determination of sulfonamides, macrolides, trimethoprim, and chloramphenicol in sewage sludge and sediment using ultrasonic extraction coupled with solid phase extraction and liquid chromatography-tandem mass spectrometry. Chin. J. Anal. Chem. 2009, 37, 1119–1124. [Google Scholar] [CrossRef]
- Lindberg, R.; Jarnheimer, P.; Olsen, B.; Johansson, M.; Tysklind, M. Determination of antibiotic substances in hospital sewage water using solid phase extraction and liquid chromatography/mass spectrometry and group analogue internal standards. Chemosphere 2004, 57, 1479–1488. [Google Scholar] [CrossRef] [PubMed]
- Han, R.W.; Zheng, N.; Yu, Z.N.; Wang, J.; Xu, X.M.; Qu, X.Y.; Li, S.L.; Zhang, Y.D.; Wang, J.Q. Simultaneous determination of 38 veterinary antibiotic residues in raw milk by UPLC-MS/MS. Food Chem. 2015, 181, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Leung, D. The challenges of developing a generic extraction procedure to analyze multiclass veterinary drug residues in milk and honey using ultra-high pressure liquid chromatography quadrupole time-of-flight mass spectrometry. Drug Test. Anal. 2012, 4, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Juan, C.; Moltó, J.C.; Mañes, J.; Font, G. Determination of macrolide and lincosamide antibiotics by pressurized liquid extraction and liquid chromatography-tandem mass spectrometry in meat and milk. Food Control 2010, 21, 1703–1709. [Google Scholar] [CrossRef]
- And, S.S.A.; Lee, L.S. Sorption of three tetracyclines by several soils: Assessing the role of pH and cation exchange. Environ. Sci. Technol. 2005, 39, 7452–7459. [Google Scholar]
- Willach, S.; Lutze, H.V.; Eckey, K.; Löppenberg, K.; Lüling, M.; Terhalle, J.; Wolbert, J.; Jochmann, M.A.; Karst, U.; Schmidt, T.C. Degradation of sulfamethoxazole using ozone and chlorine dioxide-Compound-specific stable isotope analysis, transformation product analysis and mechanistic aspects. Water Res. 2017, 122, 280–289. [Google Scholar] [CrossRef] [PubMed]
- Commission of the European Communities. Commission Decision 2002/657/EC Implementing Council Directive 96/23/EC Concerning the Performance of Analytical Methods and the Interpretation of Results; Commission of the European: Brussels, Belgium, 2002. [Google Scholar]
- Li, Z.J.; Xu, J.M.; Muhammad, A.; Ma, G.R. Effect of bound residues of metsulfuron-methyl in soil on rice growth. Chemosphere 2005, 58, 1177–1183. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Gao, J.Y.; Zhai, Z.G.; Liang, Q.L.; Wang, Y.M.; Bai, Y.Q.; Luo, G.A. An HPLC-ESI-MS method for simultaneous determination of fourteen metabolites of promethazine and caffeine and its application to pharmacokinetic study of the combination therapy against motion sickness. J. Pharm. Biomed. 2012, 62, 119–128. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 11 antibiotics are available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Matrix Media | Methods | Number and Types of Antibiotics | Detection Limits | References |
|---|---|---|---|---|
| Vegetables | UPLC-ESI-MS/MS | 4 (quinolones) | 0.021–0.092 μg/kg | [14] |
| HPLC-FLD | 4 (quinolones) | 0.575–1.538 μg/kg | [15] | |
| SPE-HPLC | 6 (sulfonamides) | 21.9–72.8 μg/kg | [16] | |
| UHPLC-MS/MS | 20 (fluoroquinolones, sulfonamides and tetracyclines) | 0.33–2.92 μg/kg | [17] | |
| LC-QqLIT-MS/MS | 49 (sulfonamides, quinolones, macrolides, β-lactams and tetracyclines) | 2–5 μg/kg | [18] | |
| Soils | PLE-SPE-LC-MS/MS | 5 (tetracyclines, macrolides and sulfonamides) | 0.6–5.6 μg/kg | [19] |
| 8 (macrolides, ionophores and tiamulin) | 0.2–1.6 μg/kg | [20] | ||
| Manure | LLE-SPE-LC-MS/MS | 11 (tetracyclines, sulfonamides and tylosin) | 2.7–32.1 μg/kg | [21] |
| SPE-HPLC-MS/MS | 3 (tetracyclines, quinolones and sulfadimidine) | 0.04–0.25 mg/kg | [22] | |
| SPE-HPLC | 11 (tetracyclines, quinolones, sulfonamides, tylosin and chloramphenicol) | 0.1–1.9 μg/kg | [23] | |
| Sewage sludge | USE-LC-MS/MS | 10 (sulfonamides, macrolides, trimethoprim and chloramphenicol) | 2.2–66.9 μg/kg | [24] |
| SPE-LC-MS/MS | 16 (fluoroquinolones, sulfonamides, trimethoprim, beta-lactams, nitroimidazoles and tetracyclines) | 0.1–3.6 μg/L | [25] | |
| Milk, honey and meat | SPE-UPLC-MS/MS | 38 (beta-lactams, sulfonamides, quinolones, tetracyclines, macrolides and lincosamide) | 0.1–5.0 μg/kg | [26] |
| SPE-UHPLC QqTOF MS | 104 (aminoglycosides, endectocides, fluoroquinolones, ionophores, β-lactams, macrolides, NSAIDs, phenicols, sulfonamides and tetracyclines) | — | [27] | |
| PLE-LC-MS/MS | 7 (macrolides and lincosamides) | 3–10 μg/kg (milk) 5–10 μg/kg (meat) | [28] |
| Substance | LOD (μg/kg) | LOQ (μg/kg) | Calibration Curve | Correlation Coefficient (r2) |
|---|---|---|---|---|
| TYL | 0.005 | 0.017 | y = 0.130x − 0.028 | 0.997 |
| CTC | 0.014 | 0.046 | y = 0.049x + 0.100 | 0.998 |
| OTC | 0.227 | 0.760 | y = 0.015x + 0.146 | 0.991 |
| ENR | 0.011 | 0.036 | y = 0.025x + 0.064 | 0.991 |
| CIP | 0.026 | 0.088 | y = 0.029x + 0.069 | 0.994 |
| CAP | 0.024 | 0.081 | y = 0.454x − 0.112 | 0.999 |
| NOR | 0.138 | 0.459 | y = 0.100x + 0.054 | 0.995 |
| SMN | 0.005 | 0.017 | y = 0.051x − 0.079 | 0.994 |
| SDMe | 0.007 | 0.024 | y = 0.015x − 0.076 | 0.993 |
| ST | 0.014 | 0.048 | y = 0.022x − 0.030 | 0.999 |
| SMZ | 0.005 | 0.015 | y = 0.022x − 0.033 | 0.999 |
| Substance | Spiked (μg/kg) | Leek | Celery | Lentil | Carob | Cauliflower | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Recovery (%) | RSD (%) | Recovery (%) | RSD (%) | Recovery (%) | RSD (%) | Recovery (%) | RSD (%) | Recovery (%) | RSD (%) | ||
| TYL | 5 | 89.2 | 4.0 | 81.6 | 3.1 | 81.4 | 6.7 | 81.1 | 7.6 | 79.2 | 3.5 |
| 10 | 86.8 | 3.3 | 79.3 | 4.9 | 82.8 | 5.5 | 82.0 | 5.5 | 73.1 | 5.4 | |
| 50 | 93.2 | 3.5 | 78.4 | 6.8 | 87.4 | 5.4 | 89.7 | 4.6 | 71.4 | 7.3 | |
| CTC | 5 | 91.4 | 3.9 | 82.2 | 4.6 | 92.7 | 4.3 | 92.8 | 4.5 | 97.1 | 3.5 |
| 10 | 89.9 | 6.1 | 83.6 | 5.5 | 91.6 | 2.6 | 93.7 | 3.9 | 92.2 | 5.6 | |
| 50 | 91.3 | 4.7 | 86.9 | 7.3 | 94.3 | 6.5 | 94.0 | 5.2 | 89.4 | 7.2 | |
| OTC | 5 | 76.2 | 4.7 | 91.8 | 4.2 | 91.1 | 4.7 | 93.2 | 7.7 | 91.9 | 4.3 |
| 10 | 93.0 | 7.5 | 89.2 | 3.6 | 93.0 | 2.1 | 96.5 | 5.1 | 93.0 | 7.2 | |
| 50 | 96.4 | 3.7 | 94.1 | 7.1 | 94.6 | 1.9 | 93.3 | 3.2 | 89.9 | 4.3 | |
| ENR | 5 | 100 | 4.6 | 90.3 | 7.7 | 98.3 | 4.2 | 92.9 | 2.5 | 93.2 | 3.2 |
| 10 | 90.1 | 3.1 | 97.8 | 5.1 | 93.7 | 3.3 | 99.2 | 1.3 | 91.5 | 4.2 | |
| 50 | 95.0 | 6.8 | 100.5 | 9.1 | 97.5 | 4.5 | 95.3 | 4.4 | 97.9 | 3.8 | |
| CIP | 5 | 87.2 | 7.9 | 82.8 | 6.0 | 81.6 | 1.9 | 71.9 | 3.1 | 73.5 | 2.4 |
| 10 | 83.5 | 5.6 | 87.3 | 12.3 | 82.0 | 5.7 | 79.1 | 2.8 | 72.9 | 3.1 | |
| 50 | 85.9 | 5.2 | 104 | 13.4 | 89.2 | 2.8 | 88.0 | 3.3 | 78.3 | 4.0 | |
| NOR | 5 | 77.3 | 7.6 | 96.1 | 6.1 | 86.5 | 5.9 | 88.0 | 2.6 | 91.4 | 4.4 |
| 10 | 83.1 | 5.8 | 94.8 | 7.9 | 89.6 | 2.3 | 85.2 | 5.4 | 93.1 | 2.5 | |
| 50 | 85.6 | 4.6 | 96.7 | 8.9 | 91.3 | 1.7 | 87.5 | 1.7 | 90.4 | 3.3 | |
| SMN | 5 | 86.6 | 3.7 | 86.1 | 7.3 | 87.4 | 5.6 | 95.7 | 3.6 | 86.2 | 2.5 |
| 10 | 88.9 | 8.9 | 88.4 | 9.0 | 91.2 | 8.0 | 94.9 | 7.5 | 81.1 | 8.2 | |
| 50 | 89.4 | 5.6 | 89.8 | 2.5 | 95.1 | 4.5 | 97.2 | 4.1 | 89.4 | 5.3 | |
| SDMe | 5 | 96.5 | 7.2 | 94.5 | 8.3 | 93.7 | 5.7 | 93.9 | 5.0 | 97.9 | 1.9 |
| 10 | 91.4 | 5.0 | 96.2 | 5.9 | 91.8 | 1.2 | 96.2 | 2.4 | 95.7 | 3.2 | |
| 50 | 95.2 | 4.5 | 94.6 | 10.0 | 94.0 | 3.1 | 97.4 | 3.3 | 99.1 | 4.2 | |
| ST | 5 | 81.5 | 6.2 | 80.2 | 9.6 | 89.2 | 4.7 | 94.4 | 4.0 | 91.8 | 4.9 |
| 10 | 82.7 | 4.1 | 82.6 | 7.4 | 86.3 | 2.5 | 95.3 | 6.4 | 96.2 | 3.3 | |
| 50 | 83.1 | 3.0 | 87.9 | 3.9 | 88.6 | 6.4 | 97.1 | 4.3 | 93.8 | 3.2 | |
| SMZ | 5 | 86.6 | 3.8 | 84.1 | 5.1 | 92.1 | 7.5 | 87.5 | 2.7 | 85.9 | 2.6 |
| 10 | 88.9 | 4.9 | 87.4 | 6.8 | 97.5 | 8.0 | 84.9 | 1.9 | 88.7 | 1.5 | |
| 50 | 91.3 | 6.8 | 92.0 | 7.4 | 94.2 | 4.1 | 89.5 | 5.6 | 91.3 | 4.8 | |
| CAP | 5 | 93.0 | 3.5 | 73.1 | 6.7 | 86.9 | 4.3 | 74.7 | 3.5 | 86.2 | 3.7 |
| 10 | 92.4 | 6.8 | 77.7 | 5.5 | 92.3 | 2.9 | 80.6 | 5.4 | 72.8 | 5.2 | |
| 50 | 94.3 | 8.7 | 72.6 | 5.4 | 93.5 | 5.1 | 83.1 | 7.3 | 91.5 | 2.9 | |
| Substance | Freq 1 (%) | Residual Concentration (μg/kg) | |||
|---|---|---|---|---|---|
| Mean | Med. | Max | Min | ||
| OTC | 71 | 2.578 | 3.463 | 4.706 | ND 2 |
| CTC | 100 | 1.448 | 1.153 | 4.966 | 1.043 |
| ∑TCs 3 | 100 | 4.026 | 4.606 | 6.838 | 1.089 |
| ENR | 54 | 0.785 | 1.414 | 1.659 | ND |
| CIP | 71 | 0.935 | 1.302 | 1.414 | ND |
| NOR | 86 | 1.743 | 1.954 | 3.029 | ND |
| ∑QNs | 97 | 3.463 | 3.336 | 5.251 | ND |
| SMN | 66 | 0.023 | 0.008 | 0.328 | ND |
| SDMe | 51 | 0.002 | 0.001 | 0.010 | ND |
| ST | 63 | 0.083 | 0.003 | 1.940 | ND |
| SMZ | 71 | 0.015 | 0.004 | 0.261 | ND |
| ∑SAs | 97 | 0.123 | 0.023 | 1.956 | ND |
| CAP | 28 | 0.050 | ND | 0.698 | ND |
| TYL | 1 | 0.037 | ND | 0.425 | ND |
| Substance | Parent Ion (m/z) | Quantitative Ion (m/z) | Collision Energy (eV) | Qualitative Ion (m/z) | Collision Energy (eV) | Fragment Voltage (V) |
|---|---|---|---|---|---|---|
| TYL | 961.9 | 173.8 | 50 | 145.1 | 50 | 130 |
| CTC | 479.0 | 444.0 | 18 | 462.0 | 13 | 130 |
| OTC | 461.1 | 443.2 | 15 | 426.0 | 5 | 120 |
| CAP | −321.1 | 151.0 | 10 | 257.0 | 5 | 120 |
| SDMe | 279.1 | 186.0 | 15 | 156.0 | 15 | 100 |
| SMN | 281.1 | 156.0 | 10 | 188.0 | 10 | 120 |
| ST | 256.0 | 156..0 | 10 | 108.0 | 10 | 100 |
| SMZ | 254.1 | 156.1 | 10 | 160.1 | 15 | 100 |
| NOR | 320.1 | 302.1 | 15 | 276.6 | 10 | 120 |
| CIP | 332.1 | 314.1 | 24 | 231.0 | 34 | 120 |
| ENR | 360.1 | 342.1 | 15 | 316.1 | 15 | 120 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feng, Y.; Zhang, W.-J.; Liu, Y.-W.; Xue, J.-M.; Zhang, S.-Q.; Li, Z.-J. A Simple, Sensitive, and Reliable Method for the Simultaneous Determination of Multiple Antibiotics in Vegetables through SPE-HPLC-MS/MS. Molecules 2018, 23, 1953. https://doi.org/10.3390/molecules23081953
Feng Y, Zhang W-J, Liu Y-W, Xue J-M, Zhang S-Q, Li Z-J. A Simple, Sensitive, and Reliable Method for the Simultaneous Determination of Multiple Antibiotics in Vegetables through SPE-HPLC-MS/MS. Molecules. 2018; 23(8):1953. https://doi.org/10.3390/molecules23081953
Chicago/Turabian StyleFeng, Yao, Wen-Juan Zhang, Yuan-Wang Liu, Jian-Ming Xue, Shu-Qing Zhang, and Zhao-Jun Li. 2018. "A Simple, Sensitive, and Reliable Method for the Simultaneous Determination of Multiple Antibiotics in Vegetables through SPE-HPLC-MS/MS" Molecules 23, no. 8: 1953. https://doi.org/10.3390/molecules23081953