
AM-2201 Inhibits Multiple Cytochrome P450 and Uridine 5′-Diphospho-Glucuronosyltransferase Enzyme Activities in Human Liver Microsomes



Abstract
:1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. Materials and Reagents
3.2. Inhibitory Effect of AM-2201 on Eight Major CYP Activities in Human Liver Microsomes
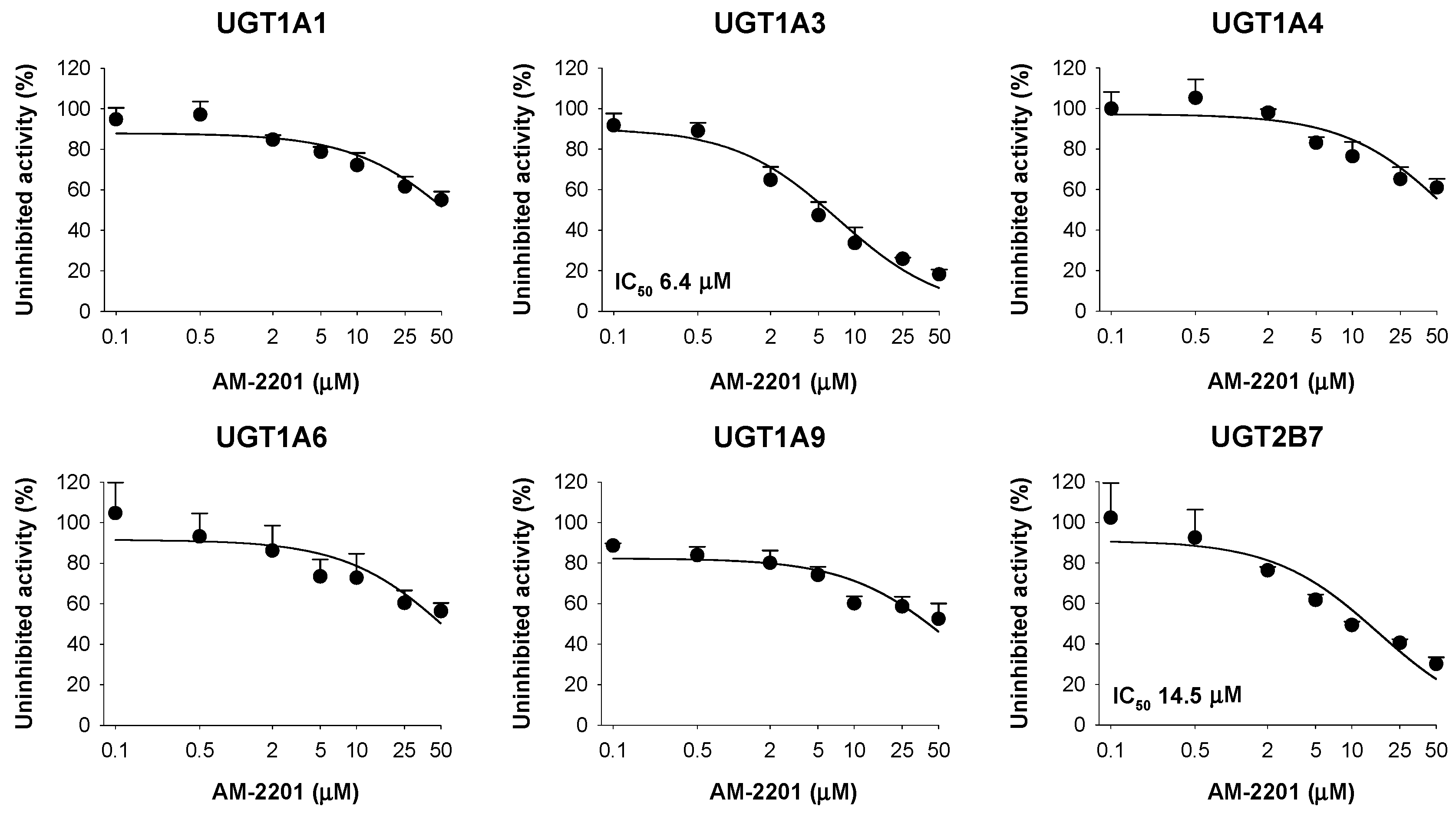
3.3. Inhibitory Effects of AM-2201 on Six Major UGT Activities in Human Liver Microsomes
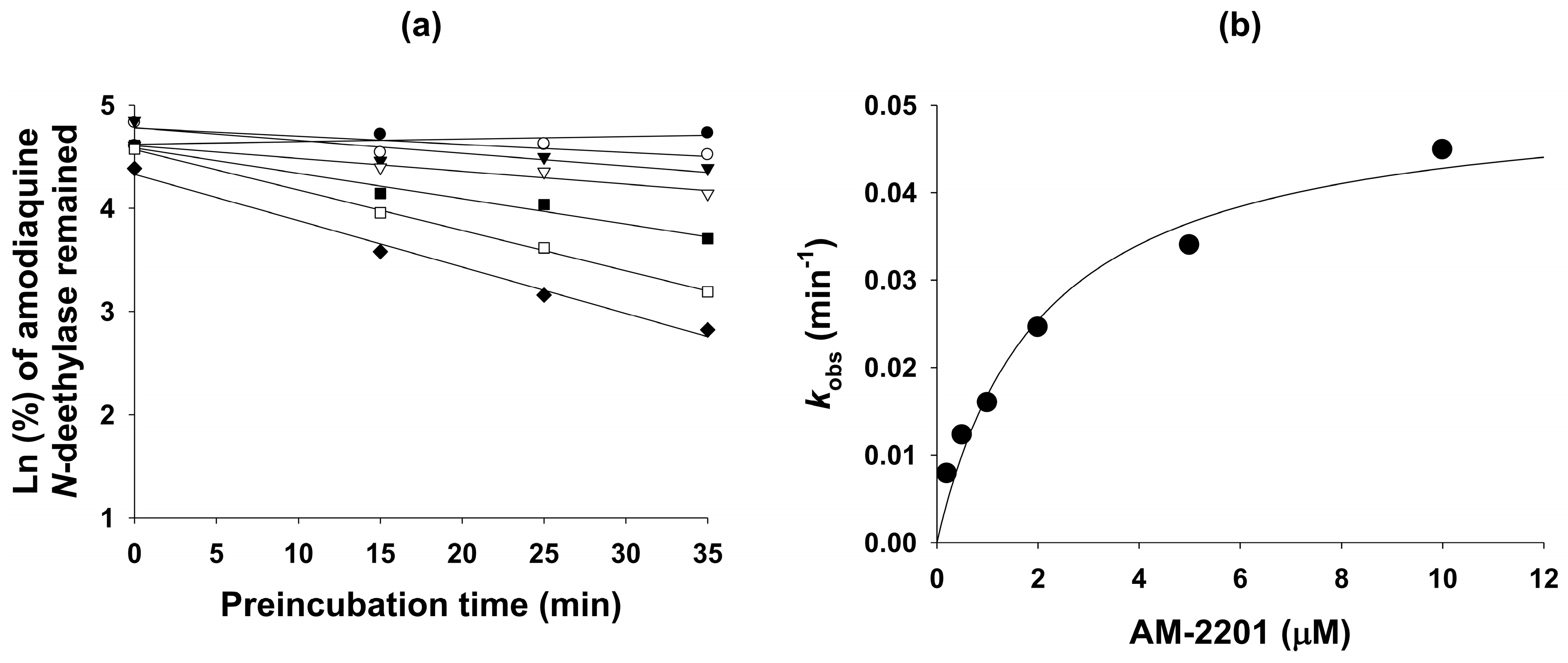
3.4. Mechanism-based Inhibition of CYP2C8 Activity by AM-2201
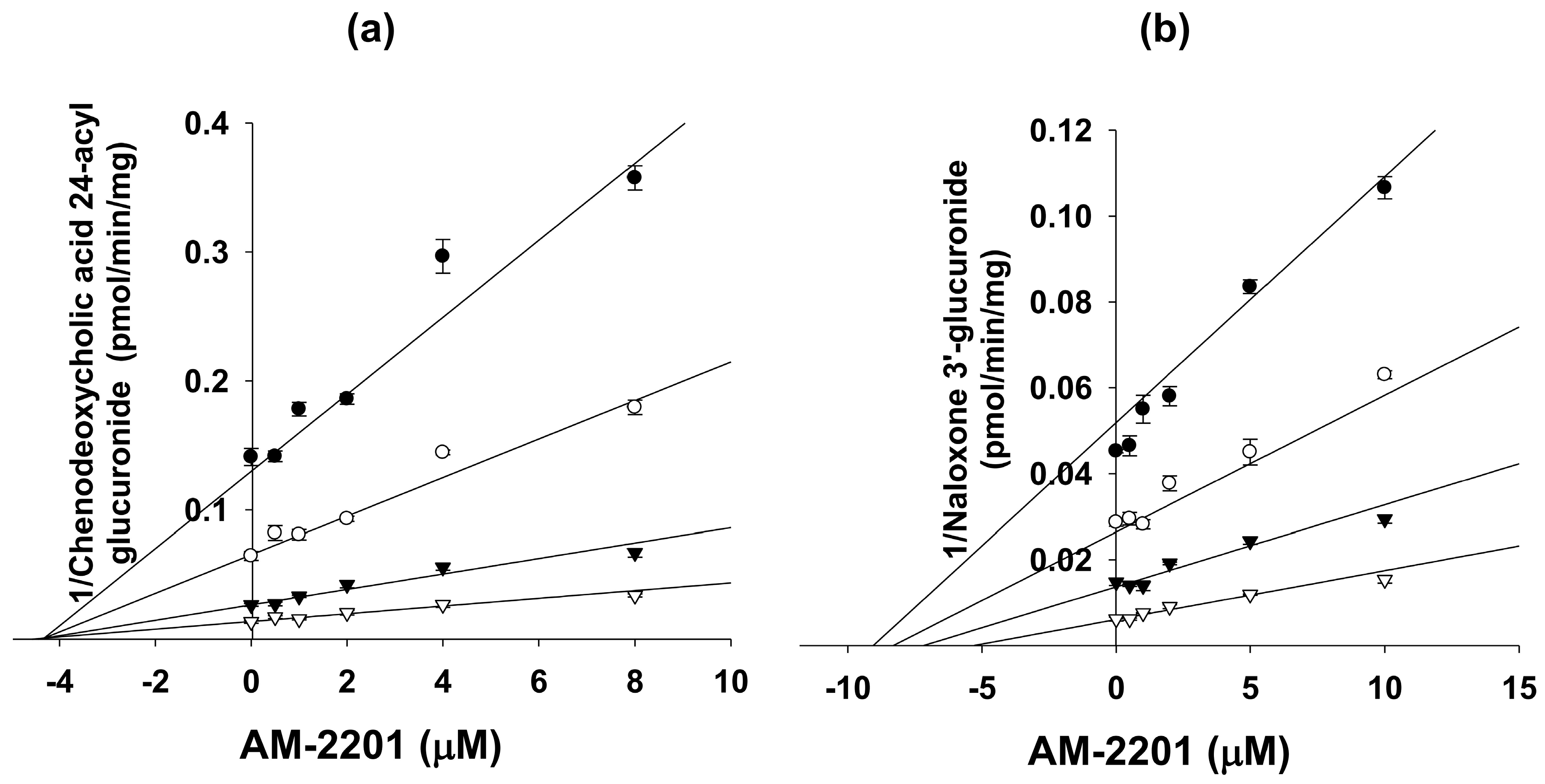
3.5. Kinetic Analysis
3.6. LC-MS/MS Analysis
3.7. Data Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fattore, L.; Fratta, W. Beyond THC: The new generation of cannabinoid designer drugs. Front. Behav. Neurosci. 2011, 5, 60. [Google Scholar] [CrossRef] [PubMed]
- Synthetic cannabinoids in Europe. Available online: http://www.emcdda.europa.eu/topics/pods/synthetic-cannabinoids (accessed on 22 February 2017).
- Seely, K.A.; Lapoint, J.; Moran, J.H.; Fattore, L. Spice drugs are more than harmless herbal blends: A review of the pharmacology and toxicology of synthetic cannabinoids. Prog. Neuropsychopharmacol. Biol. Psychiatry 2012, 39, 234–243. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.; Choi, H.; Heo, S.; Kim, E.; Lee, J. Synthetic cannabinoids abused in South Korea: Drug identifications by the national forensic service from 2009 to June 2013. Forensic Toxicol. 2014, 32, 82–88. [Google Scholar] [CrossRef]
- Helander, A.; Backberg, M.; Hulten, P.; Al-Saffar, Y.; Beck, O. Detection of new psychoactive substance use among emergency room patients: Results from the Swedish STRIDA project. Forensic Sci. Int. 2014, 243, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Langer, N.; Lindigkeit, R.; Schiebel, H.M.; Ernst, L.; Beuerle, T. Identification and quantification of synthetic cannabinoids in ‘Spice-like’ herbal mixtures: A snapshot of the german situation in the autumn of 2012. Drug Test. Anal. 2014, 6, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Musshoff, F.; Madea, B.; Kernbach–Wighton, G.; Bicker, W.; Kneisel, S.; Hutter, M.; Auwarter, V. Driving under the influence of synthetic cannabinoids (“Spice”): A case series. Int. J. Legal Med. 2014, 128, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Andreeva-Gateva, P.A.; Nankova, V.H.; Angelova, V.T.; Gatev, T.N. Synthetic cannabimimetics in Bulgaria 2010–2013. Drug Alcohol Depend. 2015, 157, 200–204. [Google Scholar] [CrossRef] [PubMed]
- Tournebize, J.; Gibaja, V.; Kahn, J.P. Acute effects of synthetic cannabinoids: Update 2015. Subst. Abus. 2016, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, J.; Takahashi, M.; Nonaka, R.; Seto, T.; Suzuki, J.; Yoshida, M.; Kanai, C.; Hamano, T. Identification and quantitation of a benzoylindole (2-methoxyphenyl)(1-pentyl-1H-indol-3-yl)methanone and a naphthoylindole 1-(5-fluoropentyl-1H-indol-3-yl)-(naphthalene-1-yl)methanone (AM-2201) found in illegal products obtained via the internet and their cannabimimetic effects evaluated by in vitro [35s]GTPγs binding assays. Forensic Toxicol. 2011, 29, 132–141. [Google Scholar]
- Schneir, A.B.; Baumbacher, T. Convulsions associated with the use of a synthetic cannabinoid product. J. Med. Toxicol. 2012, 8, 62–64. [Google Scholar] [CrossRef] [PubMed]
- Derungs, A.; Schwaninger, A.E.; Mansella, G.; Bingisser, R.; Kraemer, T.; Liechti, M.E. Symptoms, toxicities, and analytical results for a patient after smoking herbs containing the novel synthetic cannabinoid MAM-2201. Forensic Toxicol. 2013, 31, 164–171. [Google Scholar] [CrossRef]
- McQuade, D.; Hudson, S.; Dargan, P.I.; Wood, D.M. First European case of convulsions related to analytically confirmed use of the synthetic cannabinoid receptor agonist AM-2201. Eur. J. Clin. Pharmacol. 2013, 69, 373–376. [Google Scholar] [CrossRef] [PubMed]
- Patton, A.L.; Chimalakonda, K.C.; Moran, C.L.; McCain, K.R.; Radominska-Pandya, A.; James, L.P.; Kokes, C.; Moran, J.H. K2 toxicity: Fatal case of psychiatric complications following AM-2201 exposure. J. Forensic Sci. 2013, 58, 1676–1680. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, N.; Kawamura, M.; Kikura-Hanajiri, R.; Goda, Y. URB-754: A new class of designer drug and 12 synthetic cannabinoids detected in illegal products. Forensic Sci. Int. 2013, 227, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Lonati, D.; Buscaglia, E.; Papa, P.; Valli, A.; Coccini, T.; Giampreti, A.; Petrolini, V.M.; Vecchio, S.; Serpelloni, G.; Locatelli, C.A. MAM-2201 (analytically confirmed) intoxication after “synthacaine” consumption. Ann. Emerg. Med. 2014, 64, 629–632. [Google Scholar] [CrossRef] [PubMed]
- Tomiyama, K.; Funada, M. Cytotoxicity of synthetic cannabinoids on primary neuronal cells of the forebrain: The involvement of cannabinoid CB1 receptors and apoptotic cell death. Toxicol. Appl. Pharmacol. 2014, 274, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Kong, T.Y.; Kim, J.H.; Kim, J.Y.; In, M.K.; Choi, K.H.; Kim, H.S.; Lee, H.S. Rapid analysis of drugs of abuse and their metabolites in human urine using dilute and shoot liquid chromatography-tandem mass spectrometry. Arch. Pharm. Res. 2016, 40, 180–196. [Google Scholar] [CrossRef] [PubMed]
- Chimalakonda, K.C.; Seely, K.A.; Bratton, S.M.; Brents, L.K.; Moran, C.L.; Endres, G.W.; James, L.P.; Hollenberg, P.F.; Prather, P.L.; Radominska-Pandya, A.; et al. Cytochrome P450-mediated oxidative metabolism of abused synthetic cannabinoids found in K2/Spice: Identification of novel cannabinoid receptor ligands. Drug Metab. Dispos. 2012, 40, 2174–2184. [Google Scholar] [CrossRef] [PubMed]
- Kiang, T.K.; Ensom, M.H.; Chang, T.K. UDP-glucuronosyltransferases and clinical drug-drug interactions. Pharmacol. Ther. 2005, 106, 97–132. [Google Scholar] [CrossRef] [PubMed]
- Zanger, U.M.; Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef] [PubMed]
- Roth, M.D.; Marques-Magallanes, J.A.; Yuan, M.; Sun, W.; Tashkin, D.P.; Hankinson, O. Induction and regulation of the carcinogen-metabolizing enzyme CYP1A1 by marijuana smoke and delta (9)-tetrahydrocannabinol. Am. J. Respir. Cell Mol. Biol. 2001, 24, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Yamaori, S.; Kushihara, M.; Yamamoto, I.; Watanabe, K. Characterization of major phytocannabinoids, cannabidiol and cannabinol, as isoform-selective and potent inhibitors of human CYP1 enzymes. Biochem. Pharmacol. 2010, 79, 1691–1698. [Google Scholar] [CrossRef] [PubMed]
- Yamaori, S.; Ebisawa, J.; Okushima, Y.; Yamamoto, I.; Watanabe, K. Potent inhibition of human cytochrome P450 3A isoforms by cannabidiol: Role of phenolic hydroxyl groups in the resorcinol moiety. Life Sci. 2011, 88, 730–736. [Google Scholar] [CrossRef] [PubMed]
- Yamaori, S.; Maeda, C.; Yamamoto, I.; Watanabe, K. Differential inhibition of human cytochrome P450 2A6 and 2B6 by major phytocannabinoids. Forensic Toxicol. 2011, 29, 117–124. [Google Scholar] [CrossRef]
- Yamaori, S.; Okamoto, Y.; Yamamoto, I.; Watanabe, K. Cannabidiol, a major phytocannabinoid, as a potent atypical inhibitor for CYP2D6. Drug Metab. Dispos. 2011, 39, 2049–2056. [Google Scholar] [CrossRef] [PubMed]
- Yamaori, S.; Koeda, K.; Kushihara, M.; Hada, Y.; Yamamoto, I.; Watanabe, K. Comparison in the in vitro inhibitory effects of major phytocannabinoids and polycyclic aromatic hydrocarbons contained in marijuana smoke on cytochrome P450 2C9 activity. Drug Metab. Pharmacokinet. 2012, 27, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Stout, S.M.; Cimino, N.M. Exogenous cannabinoids as substrates, inhibitors, and inducers of human drug metabolizing enzymes: A systematic review. Drug Metab. Rev. 2014, 46, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Zendulka, O.; Dovrtelova, G.; Noskova, K.; Turjap, M.; Sulcova, A.; Hanus, L.; Jurica, J. Cannabinoids and cytochrome P450 interactions. Curr. Drug Metab. 2016, 17, 206–226. [Google Scholar] [CrossRef] [PubMed]
- Al Saabi, A.; Allorge, D.; Sauvage, F.L.; Tournel, G.; Gaulier, J.M.; Marquet, P.; Picard, N. Involvement of UDP-glucuronosyltransferases UGT1A9 and UGT2B7 in ethanol glucuronidation, and interactions with common drugs of abuse. Drug Metab. Dispos. 2013, 41, 568–574. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.F.; Zhou, Z.W.; Yang, L.P.; Cai, J.P. Substrates, inducers, inhibitors and structure-activity relationships of human cytochrome P450 2C9 and implications in drug development. Curr. Med. Chem. 2009, 16, 3480–3675. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.F. Drugs behave as substrates, inhibitors and inducers of human cytochrome P450 3A4. Curr. Drug Metab. 2008, 9, 310–322. [Google Scholar] [CrossRef] [PubMed]
- Pang, C.Y.; Mak, J.W.; Ismail, R.; Ong, C.E. In vitro modulatory effects of flavonoids on human cytochrome P450 2C8 (CYP2C8). Naunyn Schmiedebergs Arch. Pharmacol. 2012, 385, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Polasek, T.M.; Elliot, D.J.; Lewis, B.C.; Miners, J.O. Mechanism-based inactivation of human cytochrome P450 2C8 by drugs in vitro. J. Pharmacol. Exp. Ther. 2004, 311, 996–1007. [Google Scholar] [CrossRef] [PubMed]
- Ogilvie, B.W.; Zhang, D.; Li, W.; Rodrigues, A.D.; Gipson, A.E.; Holsapple, J.; Toren, P.; Parkinson, A. Glucuronidation converts gemfibrozil to a potent, metabolism-dependent inhibitor of CYP2C8: Implications for drug-drug interactions. Drug Metab. Dispos. 2006, 34, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Lai, X.S.; Yang, L.P.; Li, X.T.; Liu, J.P.; Zhou, Z.W.; Zhou, S.F. Human CYP2C8: Structure, substrate specificity, inhibitor selectivity, inducers and polymorphisms. Curr. Drug Metab. 2009, 10, 1009–1047. [Google Scholar] [CrossRef] [PubMed]
- Alonen, A.; Finel, M.; Kostiainen, R. The human UDP-glucuronosyltransferase UGT1A3 is highly selective towards N2 in the tetrazole ring of losartan, candesartan, and zolarsartan. Biochem. Pharmacol. 2008, 76, 763–772. [Google Scholar] [CrossRef] [PubMed]
- Erichsen, T.J.; Aehlen, A.; Ehmer, U.; Kalthoff, S.; Manns, M.P.; Strassburg, C.P. Regulation of the human bile acid UDP-glucuronosyltransferase 1A3 by the farnesoid X receptor and bile acids. J. Hepatol. 2010, 52, 570–578. [Google Scholar] [CrossRef] [PubMed]
- Jeong, E.S.; Kim, Y.W.; Kim, H.J.; Shin, H.J.; Shin, J.G.; Kim, K.H.; Chi, Y.H.; Paik, S.H.; Kim, D.H. Glucuronidation of fimasartan, a new angiotensin receptor antagonist, is mainly mediated by UGT1A3. Xenobiotica 2015, 45, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Su, M.K.; Seely, K.A.; Moran, J.H.; Hoffman, R.S. Metabolism of classical cannabinoids and the synthetic cannabinoid JWH-018. Clin. Pharmacol. Ther. 2015, 97, 562–564. [Google Scholar] [CrossRef] [PubMed]
- Mano, Y.; Usui, T.; Kamimura, H. Predominant contribution of UDP-glucuronosyltransferase 2B7 in the glucuronidation of racemic flurbiprofen in the human liver. Drug Metab. Dispos. 2007, 35, 1182–1187. [Google Scholar] [CrossRef] [PubMed]
- Uchaipichat, V.; Galetin, A.; Houston, J.B.; Mackenzie, P.I.; Williams, J.A.; Miners, J.O. Kinetic modeling of the interactions between 4-methylumbelliferone, 1-naphthol, and zidovudine glucuronidation by UDP-glucuronosyltransferase 2B7 (UGT2B7) provides evidence for multiple substrate binding and effector sites. Mol. Pharmacol. 2008, 74, 1152–1162. [Google Scholar] [CrossRef] [PubMed]
- Precht, J.C.; Schroth, W.; Klein, K.; Brauch, H.; Krynetskiy, E.; Schwab, M.; Murdter, T.E. The letrozole phase 1 metabolite carbinol as a novel probe drug for UGT2B7. Drug Metab. Dispos. 2013, 41, 1906–1913. [Google Scholar] [CrossRef] [PubMed]
- Chau, N.; Elliot, D.J.; Lewis, B.C.; Burns, K.; Johnston, M.R.; Mackenzie, P.I.; Miners, J.O. Morphine glucuronidation and glucosidation represent complementary metabolic pathways that are both catalyzed by UDP-glucuronosyltransferase 2B7: Kinetic, inhibition, and molecular modeling studies. J. Pharmacol. Exp. Ther. 2014, 349, 126–137. [Google Scholar] [CrossRef] [PubMed]
- Kronstrand, R.; Roman, M.; Andersson, M.; Eklund, A. Toxicological findings of synthetic cannabinoids in recreational users. J. Anal. Toxicol. 2013, 37, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.U.; Kong, T.Y.; Kwon, S.S.; Hong, S.W.; Yeon, S.H.; Choi, J.H.; Lee, J.Y.; Cho, Y.Y.; Lee, H.S. Effect of honokiol on cytochrome P450 and UDP-glucuronosyltransferase enzyme activities in human liver microsomes. Molecules 2013, 18, 10681–10693. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.S.; Kim, J.H.; Jeong, H.U.; Cho, Y.Y.; Oh, S.R.; Lee, H.S. Inhibitory effects of aschantin on cytochrome P450 and uridine 5′-diphospho-glucuronosyltransferase enzyme activities in human liver microsomes. Molecules 2016, 21, 554. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not available.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Marker Enzymes | CYP | IC50 (μM) | Ki (μM) (Kinact, min−1 or Inhibition Mode) | |
|---|---|---|---|---|
| No Preincubation | With Preincubation * | |||
| Phenacetin O-deethylase | 1A2 | NI | NI | - |
| Coumarin 7-hydroxylase | 2A6 | NI | NI | - |
| Bupropion hydroxylase | 2B6 | NI | 21.9 | - |
| Amodiaquine N-deethylase | 2C8 | 53.8 | 6.9 | 2.1 (kinact: 0.0516) |
| Diclofenac 4′-hydroxylase | 2C9 | 11.9 | 11.9 | 3.9 (competitive) |
| [S]-Mephenytoin 4′-hydroxylase | 2C19 | NI | 31.3 | - |
| Bufuralol 1′-hydroxylase | 2D6 | NI | NI | - |
| Midazolam 1′-hydroxylase | 3A4 | 6.9 | 3.8 | 4.0 (competitive) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.-H.; Kwon, S.-S.; Kong, T.Y.; Cheong, J.C.; Kim, H.S.; In, M.K.; Lee, H.S. AM-2201 Inhibits Multiple Cytochrome P450 and Uridine 5′-Diphospho-Glucuronosyltransferase Enzyme Activities in Human Liver Microsomes. Molecules 2017, 22, 443. https://doi.org/10.3390/molecules22030443
Kim J-H, Kwon S-S, Kong TY, Cheong JC, Kim HS, In MK, Lee HS. AM-2201 Inhibits Multiple Cytochrome P450 and Uridine 5′-Diphospho-Glucuronosyltransferase Enzyme Activities in Human Liver Microsomes. Molecules. 2017; 22(3):443. https://doi.org/10.3390/molecules22030443
Chicago/Turabian StyleKim, Ju-Hyun, Soon-Sang Kwon, Tae Yeon Kong, Jae Chul Cheong, Hee Seung Kim, Moon Kyo In, and Hye Suk Lee. 2017. "AM-2201 Inhibits Multiple Cytochrome P450 and Uridine 5′-Diphospho-Glucuronosyltransferase Enzyme Activities in Human Liver Microsomes" Molecules 22, no. 3: 443. https://doi.org/10.3390/molecules22030443