
Facilitated Visual Interpretation of Scores in Principal Component Analysis by Bioactivity-Labeling of 1H-NMR Spectra—Metabolomics Investigation and Identification of a New α-Glucosidase Inhibitor in Radix Astragali



Abstract
:
1. Introduction
2. Results and Discussion
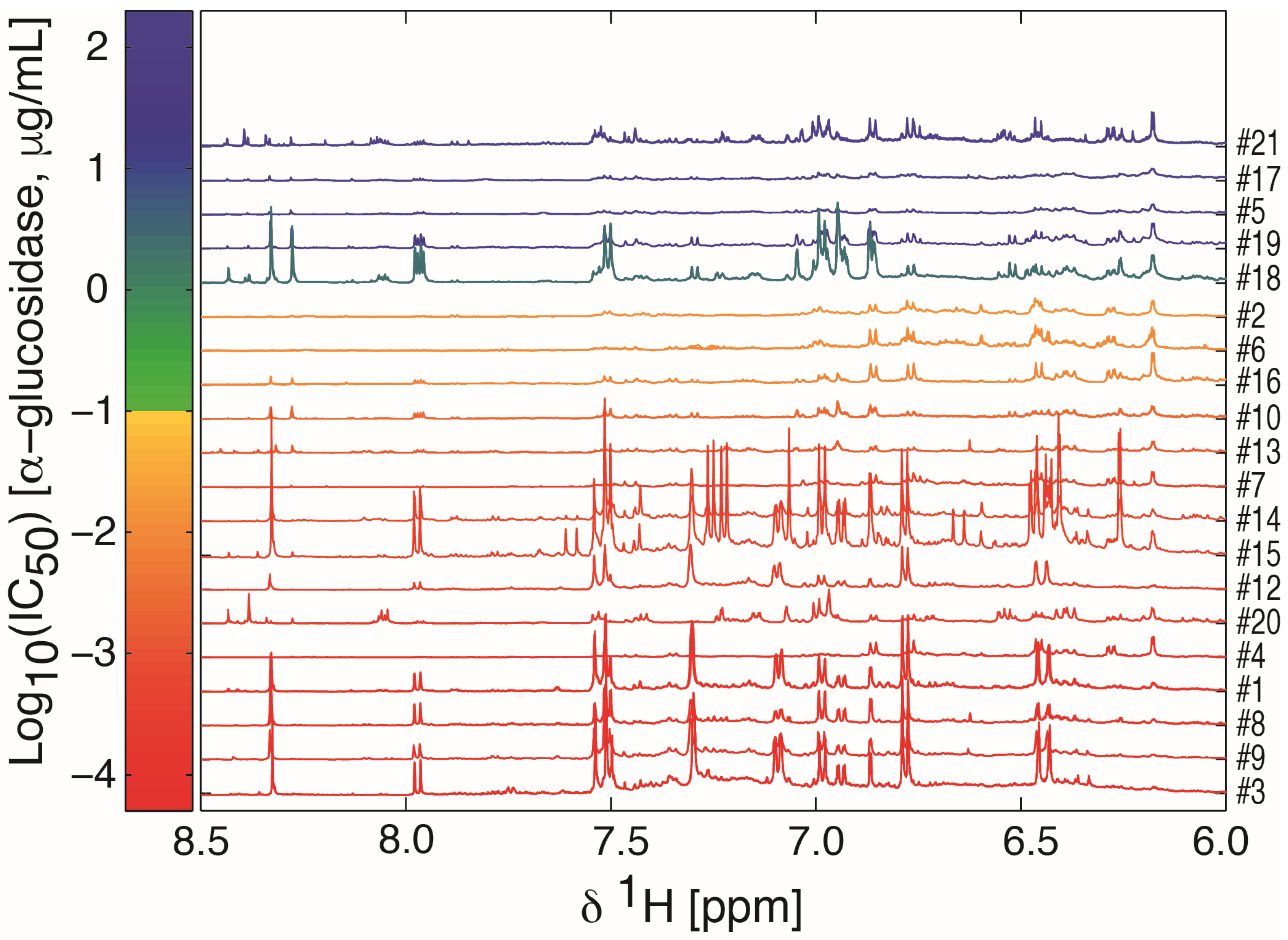
2.1. Sample Preparation, IC50 Assessment, and 1H-NMR Data Acquisition
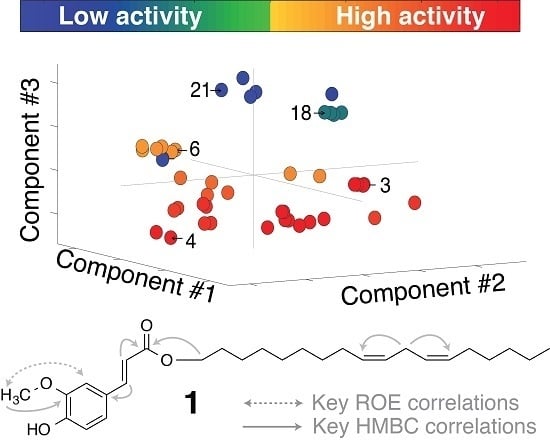
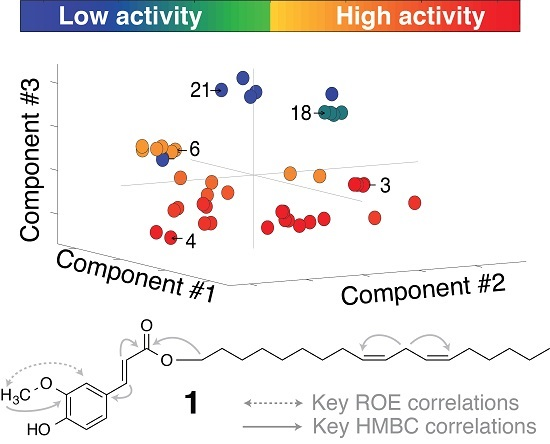
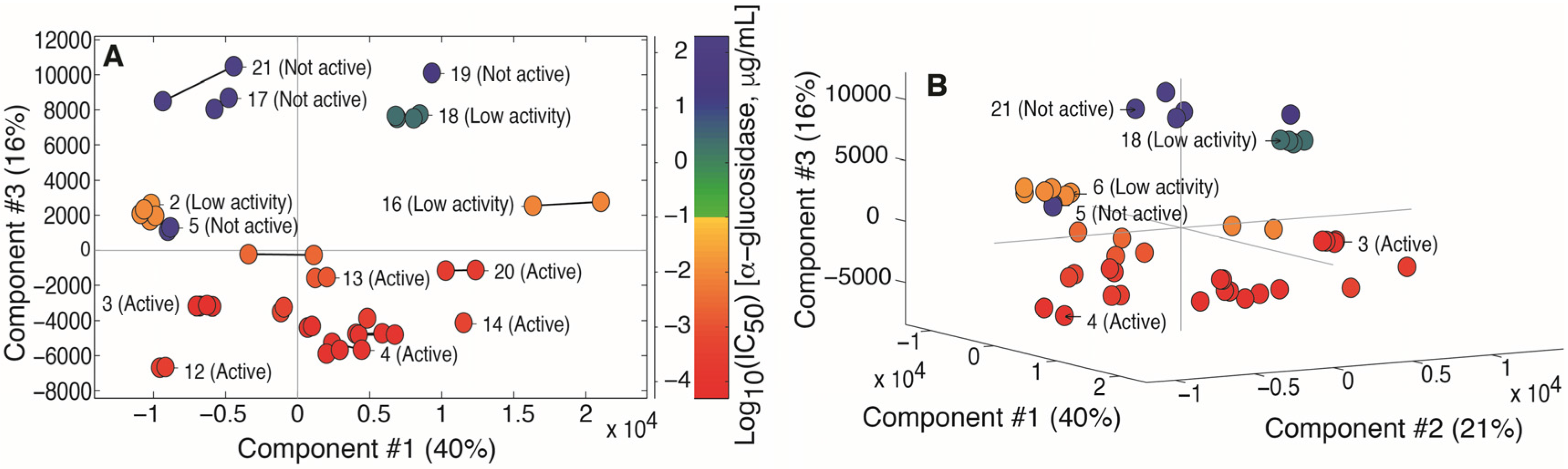
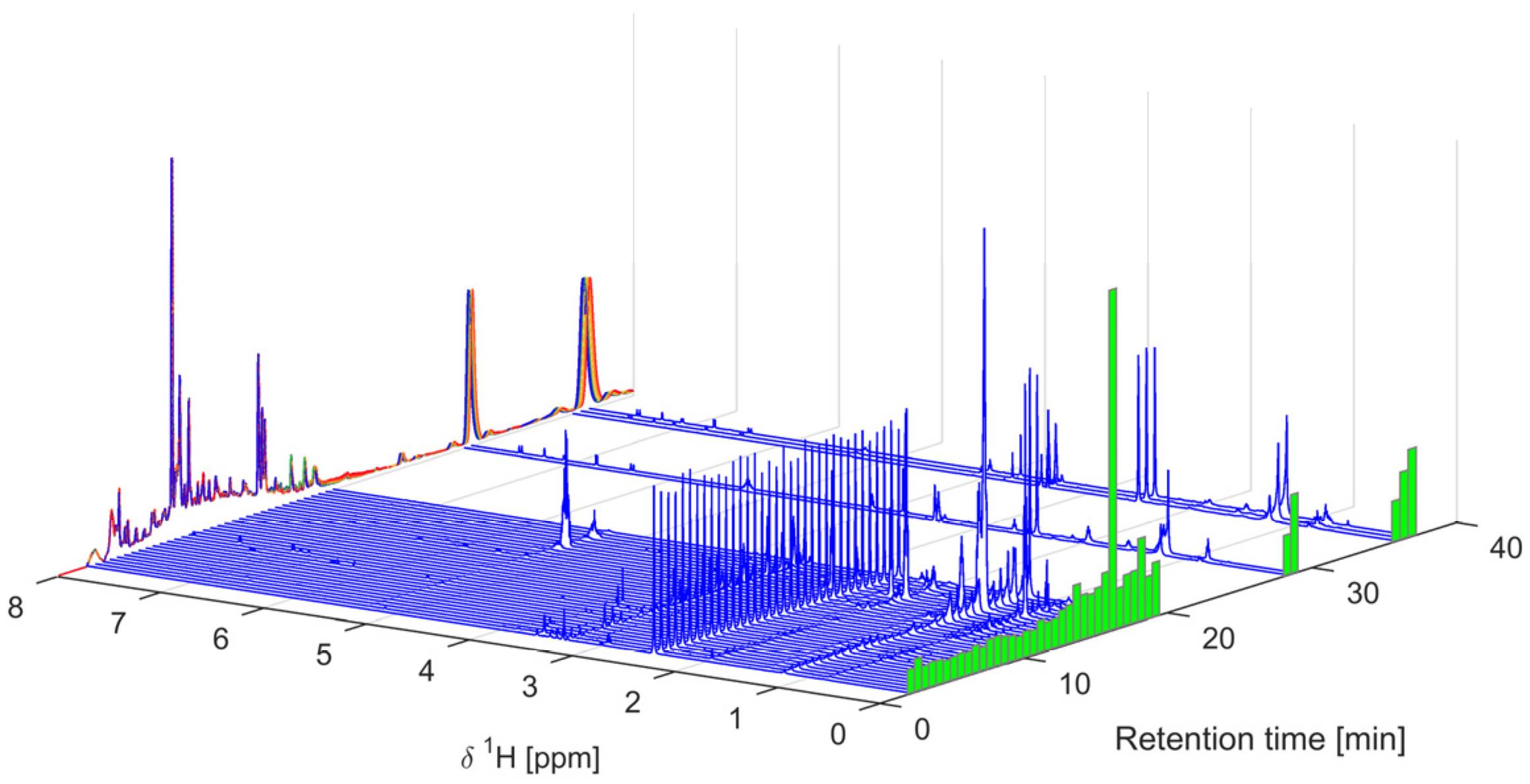
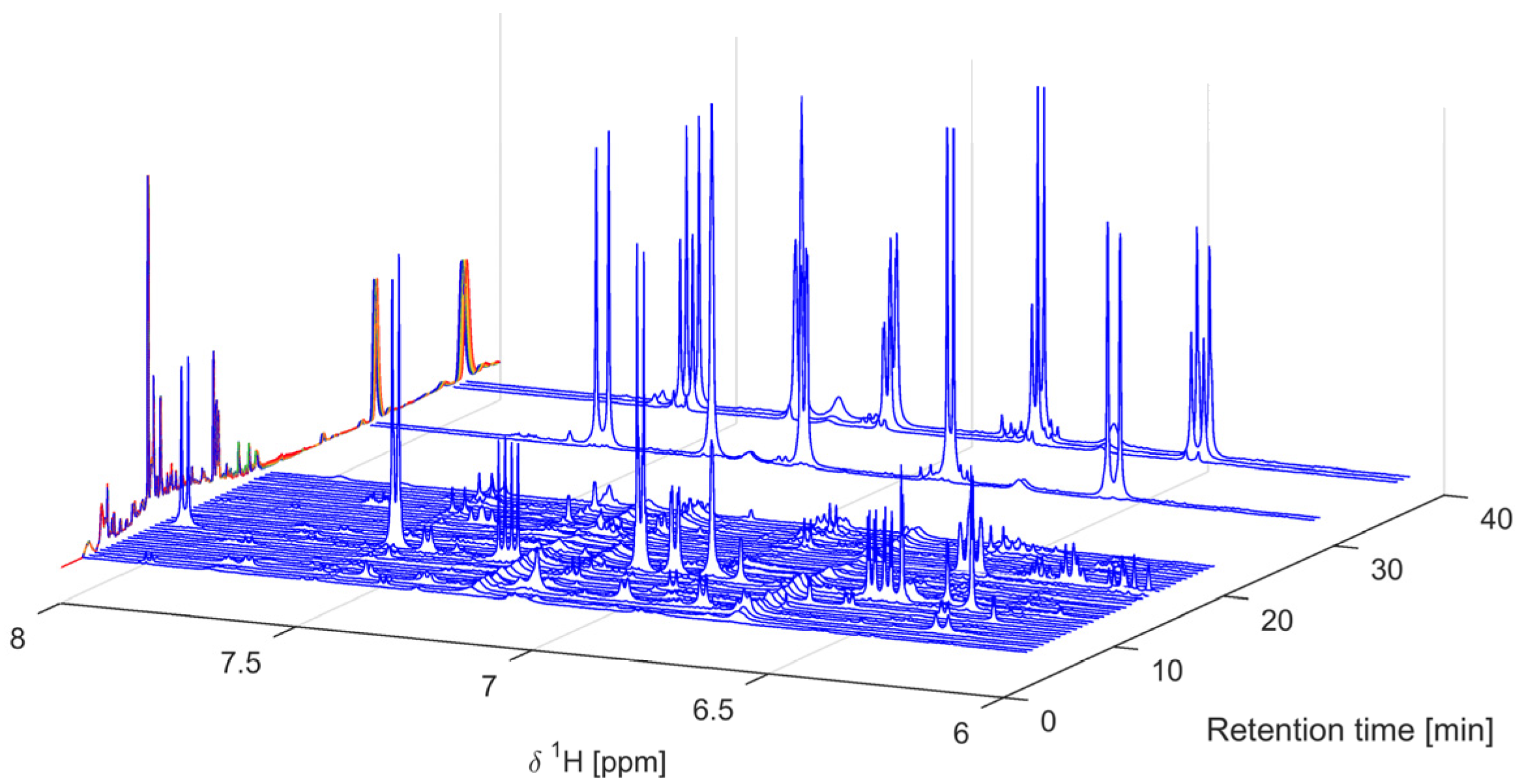
2.2. Multivariate Data Analysis
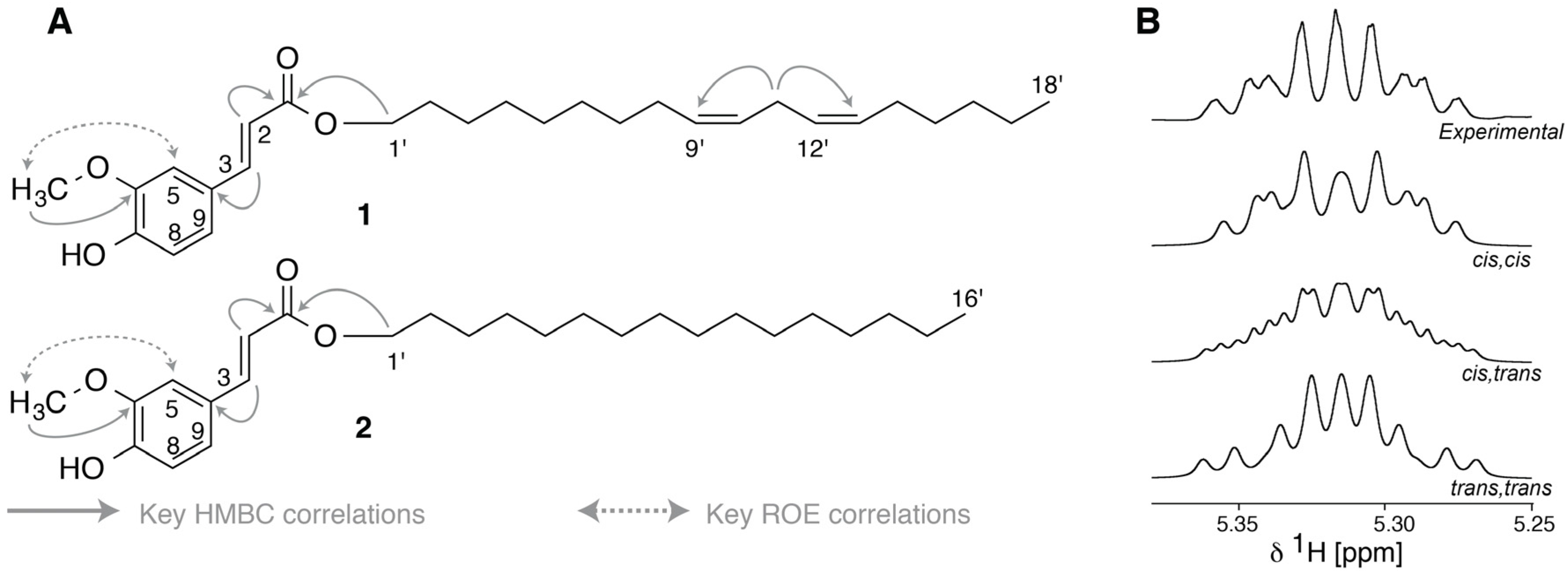
2.3. Identification of α-Glucosidase Inhibitors
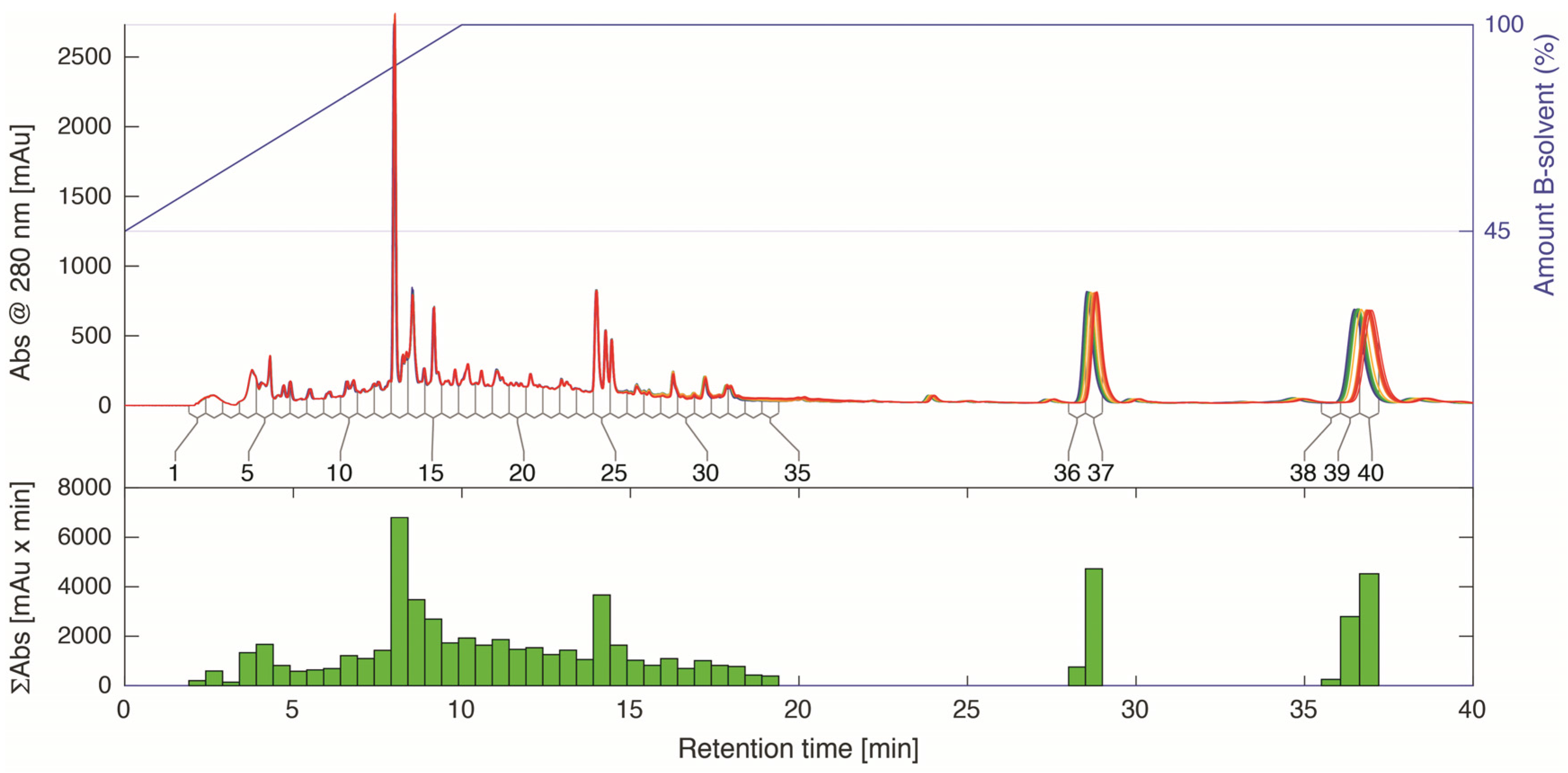
2.4. LC-HRMS and NMR Analysis of HPLC Fractions
2.5. α-Glucosidase Inhibitory Activity of Active Compounds
3. Materials and Methods
3.1. Plant Material and Extraction
3.2. 1H-NMR Analysis of Mixtures
3.3. Data Analysis and Principal Component Analysis
3.4. α-Glucosidase Inhibition Assay
3.5. Time-Sliced HPLC Separation
3.6. LC-HRMS and NMR Analysis
3.7. GC-MS Analysis of Alcohol Moieties
3.8. Preparative-Scale Isolation and Determination of IC50 Values
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chiba, S. Molecular mechanism in alpha-glucosidase and glucoamylase. Biosci. Biotechnol. Biochem. 1997, 61, 1233–1239. [Google Scholar] [CrossRef] [PubMed]
- Krentz, A.J.; Bailey, C.J. Oral antidiabetic agents: Current role in type 2 diabetes mellitus. Drugs 2005, 65, 385–411. [Google Scholar] [CrossRef]
- Van de Laar, F.A.; Lucassen, P.L.; Akkermans, R.P.; van de Lisdonk, E.H.; Rutten, G.E.; van Weel, C. Alpha-glucosidase inhibitors for patients with type 2 diabetes: Results from a Cochrane systematic review and meta-analysis. Diabetes Care 2005, 28, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Van de Laar, F.A.; Lucassen, P.L.B.J.; Akkermans, R.P.; van de Lisdonk, E.H.; de Grauw, W.J.C. Alpha-glucosidase inhibitors for people with impaired glucose tolerance or impaired fasting blood glucose. Cochrane Database Syst. Rev. 2006, 4, CD005061. [Google Scholar] [CrossRef]
- Whiting, D.R.; Guariguata, L.; Weil, C.; Shaw, J. IDF diabetes atlas: Global estimates of the prevalence of diabetes for 2011 and 2030. Diabetes Res. Clin. Pract. 2011, 94, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Wang, L.M.; He, J.; Bi, Y.F.; Li, M.; Wang, T.G.; Wang, L.H.; Jiang, Y.; Dai, M.; Lu, J.L.; et al. Prevalence and control of diabetes in Chinese adults. J. Am. Med. Assoc. 2013, 310, 948–958. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.W.; Liu, E.H.; Chu, C.; Peng, Y.B.; Cai, H.X.; Li, P. Anti-diabetic agents from natural products—An update from 2004 to 2009. Curr. Top. Med. Chem. 2010, 10, 434–457. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, J.S.; Nyberg, N.T.; Staerk, D. Assessment of constituents in Allium by multivariate data analysis, high-resolution α-glucosidase inhibition assay and HPLC-SPE-NMR. Food Chem. 2014, 161, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Kongstad, K.T.; Qinglei, S.; Nyberg, N.T.; Jäger, A.K.; Staerk, D. Dual high-resolution α-glucosidase and radical scavenging profiling combined with HPLC-HRMS-SPE-NMR for identification of minor and major constituents directly from the crude extract of Pueraria lobata. J. Nat. Prod. 2015, 78, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Tahtah, Y.; Kongstad, K.T.; Wubshet, S.G.; Nyberg, N.T.; Jønsson, L.H.; Jäger, A.K.; Qinglei, S.; Staerk, D. Triple aldose reductase/α-glucosidase/radical scavenging high-resolution profiling combined with high-performance liquid chromatography—High-resolution mass spectrometry—Solid-phase extraction—Nuclear magnetic resonance spectroscopy for identification of antidiabetic constituents in crude extract of Radix Scutellariae. J. Chromatogr. A 2015, 1408, 125–132. [Google Scholar] [PubMed]
- Pharmacopoeia of the Peoples Republic of China; People’s Medical Publishing House: Beijing, China, 2005.
- European Pharmacopoeia; Council of Europe: Strasbourg, France, 2010.
- Zhao, H.; Zhang, Y.; Guo, Y.; Shi, S. Identification of major α-glucosidase inhibitors in Radix Astragali and its human microsomal metabolites using ultrafiltration HPLC-DAD-MSn. J. Pharm. Biomed. Anal. 2015, 104, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Kan, H.; Li, P.; Liu, S.; Liu, Z. Screening and structural characterization of potential α-glucosidase inhibitors from Radix Astragali flavonoids extract by ultrafiltration LC-DAD-ESI-MSn. Anal. Methods 2015, 7, 123–128. [Google Scholar] [CrossRef]
- Choi, C.W.; Choi, Y.H.; Cha, M.R.; Yoo, D.S.; Kim, Y.S.; Yon, G.H.; Hong, K.S.; Kim, Y.H.; Ryu, S.Y. Yeast alpha-glucosidase inhibition by isoflavones from plants of Leguminosae as an in vitro alternative to acarbose. J. Agric. Food Chem. 2010, 58, 9988–9993. [Google Scholar] [CrossRef] [PubMed]
- Van der Kooy, F.; Maltese, F.; Choi, Y.H.; Kim, H.K.; Verpoorte, R. Quality control of herbal material and phytopharmaceuticals with MS and NMR based metabolic fingerprinting. Planta Med. 2009, 75, 763–775. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.K.; Choi, Y.H.; Verpoorte, R. NMR-based plant metabolomics: Where do we stand, where do we go? Trends Biotechnol. 2011, 29, 267–275. [Google Scholar] [CrossRef]
- Agnolet, S.; Jaroszewski, J.W.; Verpoorte, R.; Staerk, D. 1H-NMR-based metabolomics combined with HPLC-PDA-MS-SPE-NMR for investigation of standardized Ginkgo biloba preparations. Metabolomics 2010, 6, 292–302. [Google Scholar] [CrossRef] [PubMed]
- Agnolet, S.; Wiese, S.; Verpoorte, R.; Staerk, D. Comprehensive analysis of commercial willow bark extracts by new technology platform: Combined use of metabolomics, high-performance liquid chromatography-solid-phase extraction-nuclear magnetic resonance spectroscopy and high-resolution radical scavenging assay. J. Chromatogr. A 2012, 1262, 130–137. [Google Scholar] [PubMed]
- Xiao, W.-L.; Motley, T.J.; Unachukwu, U.J.; Lau, C.B.S.; Jiang, B.; Hong, F.; Leung, P.-C.; Wang, Q.-F.; Livingston, P.O.; Cassileth, B.R.; et al. Chemical and genetic assessment of variability in commercial Radix Astragali (Astragalus spp.) by ion trap LC−MS and nuclear ribosomal DNA barcoding sequence analyses. J. Agric. Food Chem. 2011, 59, 1548–1556. [Google Scholar] [CrossRef] [PubMed]
- Wold, S.; Esbensen, K.; Geladi, P. Principal component analysis. Chemom. Intell. Lab. Syst. 1987, 2, 37–52. [Google Scholar] [CrossRef]
- Bernards, M.A.; Lewis, N.G. Alkyl ferulates in wound healing potato tubers. Phytochemistry 1992, 31, 3409–3412. [Google Scholar] [CrossRef]
- Molina, I.; Li-Beisson, Y.; Beisson, F.; Ohlrogge, J.B.; Pollard, M. Identification of an Arabidopsis feruloyl-coenzyme A transferase required for suberin synthesis. Plant Physiol. 2009, 151, 1317–1328. [Google Scholar] [CrossRef] [PubMed]
- Boher, P.; Serra, O.; Soler, M.; Molinas, M.; Figueras, M. The potato suberin feruloyl transferase FHT which accumulates in the phellogen is induced by wounding and regulated by abscisic and salicylic acids. J. Exp. Bot. 2013, 64, 3225–3236. [Google Scholar] [CrossRef] [PubMed]
- Lai, W.C.; Wu, Y.C.; Danko, B.; Cheng, Y.B.; Hsieh, T.J.; Hsieh, T.C.; Tsai, Y.C.; El-Shazly, M.; Martins, A.; Hohmann, J.; et al. Bioactive constituents of Cirsium japonicum var australe. J. Nat. Prod. 2014, 77, 1624–1631. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, J.S.; Lauridsen, M.B.; Dragsted, L.O.; Nielsen, J.; Staerk, D. Development of a bioassay-coupled HPLC-SPE-ttNMR platform for identification of alpha-glucosidase inhibitors in apple peel (Malus × domestica Borkh.). Food Chem. 2012, 135, 1692–1699. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Kongstad, K.T.; Wiese, S.; Jäger, A.K.; Staerk, D. Edible seaweed as future functional food: Identification of α-glucosidase inhibitors by combined use of high-resolution α-glucosidase inhibition profiling and HPLC-HRMS-SPE-NMR. Food Chem. 2016, 203, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not available.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Origin a | Voucher b | IC50 (µg/mL) c |
|---|---|---|---|
| 1 | Xihe, Gansu (cultivated) | HQ106 | 19 ± 0 |
| 2 | Kangding, Sichuan | HQ101 | 157 ± 13 |
| 3 | Dingxi, Gansu (cultivated) | HQ107 | 14 ± 1 |
| 4 | Songpan, Sichuan (cultivated) | HQ102 | 19 ± 1 |
| 5 | Longxi, Gansu (cultivated, 3 years) | HQ111 | >10,000 d |
| 6 | Litang, Sichuan (wild, >3 years) | HQ103 | 136 ± 7 |
| 7 | Litang, Sichuan (wild, 1–2 years) | HQ104 | 35 ± 1 |
| 8 | Weiyuan, Gansu (cultivated, 4–5 years) | HQ108 | 18 ± 4 |
| 9 | Weiyuan, Gansu (cultivated, 2–3 years) | HQ109 | 17 ± 1 |
| 10 | Zhangxian, Gansu (cultivated) | HQ119 | 71 ± 2 |
| 11 | Xiaojin, Sichuan (wild) | HQ105 | 107 ± 12 |
| 12 | Dangchang, Gansu (cultivated) | HQ110 | 27 ± 7 |
| 13 | Longxi, Gansu (cultivated) | HQ112 | 51 ± 2 |
| 14 | Lixian, Gansu (cultivated, 3 years) | HQ113 | 34 ± 1 |
| 15 | Minxian, Gansu (cultivated, 3 years) | HQ114 | 29 ± 1 |
| 16 | Jingyuan, Gansu (cultivated, 2 years) | HQ115 | 129 ± 38 |
| 17 | Jingyuan, Gansu (cultivated, 3–4 years) | HQ116 | >10,000 d |
| 18 | Zhangxian, Gansu (cultivated, 3 years) | HQ117 | 1457 ± 170 |
| 19 | Zhangxian, Gansu (cultivated, 2 years) | HQ118 | >5000 e |
| 20 | Wutaishan, Shanxi (cultivated) | HQ120 | 26 ± 1 |
| 21 | Neimenggu (cultivated) | HQ121 | >10,000 d |
| Pos. | 1 | 2 | ||
|---|---|---|---|---|
| δC a | δH (J in Hz) a,b | δC a | δH (J in Hz) a,b | |
| 1 | 166.7 | 166.9 | ||
| 2 | 114.4 | 6.43 d (15.9) | 114.3 | 6.44 d (16.0) |
| 3 | 144.8 | 7.52 d (15.9) | 144.8 | 7.52 d (16.0) |
| 4 | 124.3 | 123.1 | ||
| 5 | 111.1 | 7.28 d (1.7) | 110.8 | 7.29 d (1.7) |
| 6 | 148.2 | 148.3 | ||
| 7 | 149.2 | 149.1 | ||
| 8 | 115.4 | 6.78 d (8.1) | 115.3 | 6.78 d (8.1) |
| 9 | 123.0 | 7.09 dd (8.1, 1.7) | 123.1 | 7.09 dd (8.1, 1.7) |
| 1′ | 63.7 | 4.09 t (6.7) | 63.6 | 4.10 t (6.6) |
| 2′ | 28.1 | 1.61 m | 27.7 | 1.61 tt (7.3, 6.3) |
| 3′ | 25.3 | 1.32 m | 25.2 | 1.33 m |
| 4′ | 28.6 | 1.26 overlap | 28.6 | 1.22 overlap |
| 5′ | 28.6 | 1.26 overlap | 28.6 | 1.22 overlap |
| 6′ | 28.6 | 1.26 overlap | 28.6 | 1.22 overlap |
| 7′ | 28.8 | 1.26 overlap | 28.6 | 1.22 overlap |
| 8′ | 26.5 | 1.99 overlap | 28.6 | 1.22 overlap |
| 9′ | 129.6 | 5.31 m | 28.6 | 1.22 overlap |
| 10′ | 127.7 | 5.28 m | 28.6 | 1.22 overlap |
| 11′ | 25.1 | 2.71 t (6.6) | 28.6 | 1.22 overlap |
| 12′ | 127.7 | 5.28 m | 28.6 | 1.22 overlap |
| 13′ | 129.6 | 5.31 m | 28.6 | 1.22 overlap |
| 14′ | 26.5 | 1.99 overlap | 30.9 | 1.22 overlap |
| 15′ | 28.8 | 1.26 overlap | 22.0 | 1.24 overlap |
| 16′ | 28.8 | 1.26 overlap | 13.8 | 0.84 t (6.8) |
| 17′ | 21.9 | 1.24 overlap | - | - |
| 18′ | 13.9 | 0.85 t (6.8) | - | - |
| 6-OCH3 | 55.7 | 3.80 s | 55.5 | 3.80 s |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Nyberg, N.T.; Jäger, A.K.; Staerk, D. Facilitated Visual Interpretation of Scores in Principal Component Analysis by Bioactivity-Labeling of 1H-NMR Spectra—Metabolomics Investigation and Identification of a New α-Glucosidase Inhibitor in Radix Astragali. Molecules 2017, 22, 411. https://doi.org/10.3390/molecules22030411
Liu Y, Nyberg NT, Jäger AK, Staerk D. Facilitated Visual Interpretation of Scores in Principal Component Analysis by Bioactivity-Labeling of 1H-NMR Spectra—Metabolomics Investigation and Identification of a New α-Glucosidase Inhibitor in Radix Astragali. Molecules. 2017; 22(3):411. https://doi.org/10.3390/molecules22030411
Chicago/Turabian StyleLiu, Yueqiu, Nils T. Nyberg, Anna K. Jäger, and Dan Staerk. 2017. "Facilitated Visual Interpretation of Scores in Principal Component Analysis by Bioactivity-Labeling of 1H-NMR Spectra—Metabolomics Investigation and Identification of a New α-Glucosidase Inhibitor in Radix Astragali" Molecules 22, no. 3: 411. https://doi.org/10.3390/molecules22030411