
Role of Electron-Driven Proton-Transfer Processes in the Ultrafast Deactivation of Photoexcited Anionic 8-oxoGuanine-Adenine and 8-oxoGuanine-Cytosine Base Pairs


Abstract
:1. Introduction
2. Results
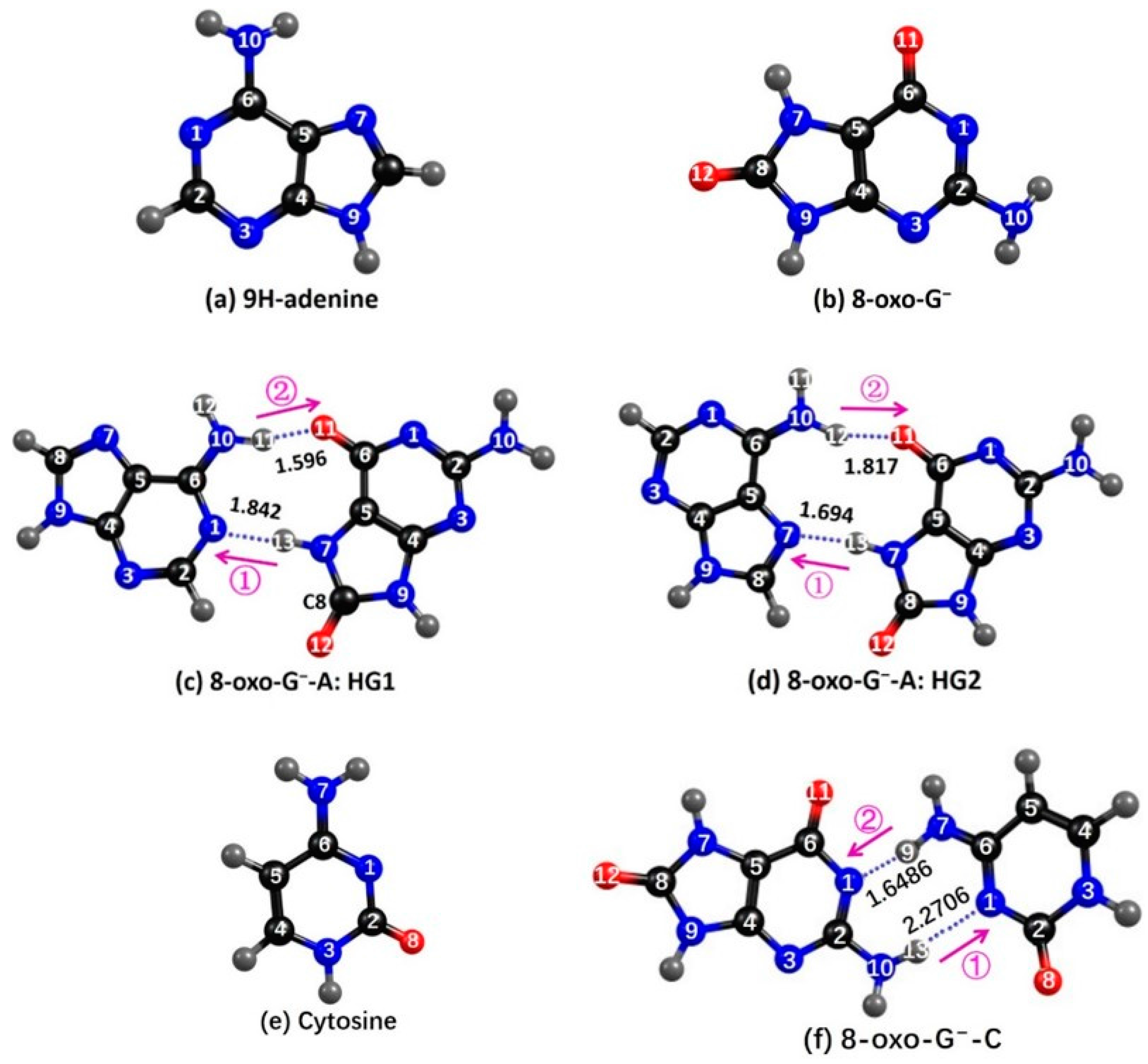
2.1. Ground State Geometries
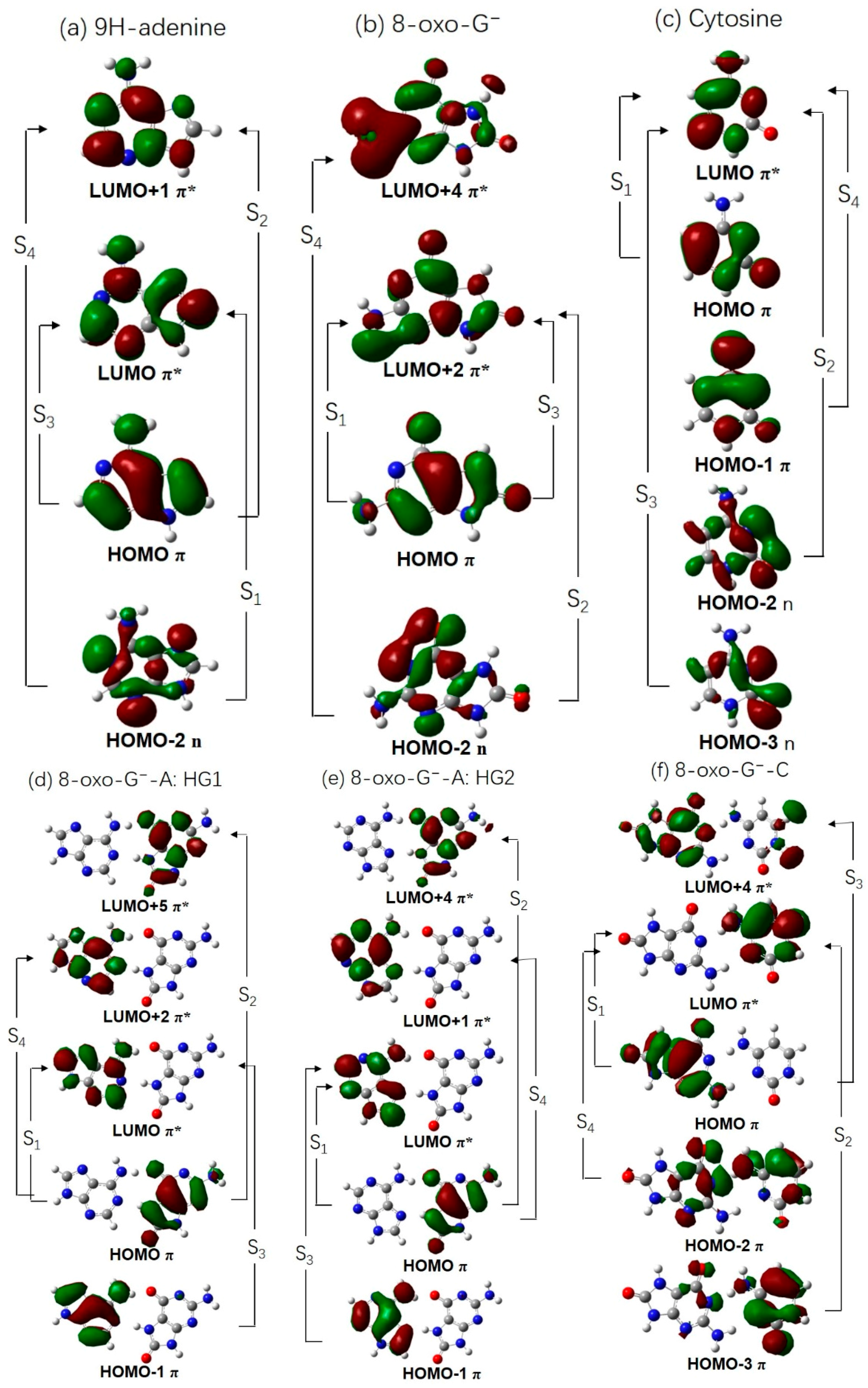
2.2. Vertical Excitation Energies
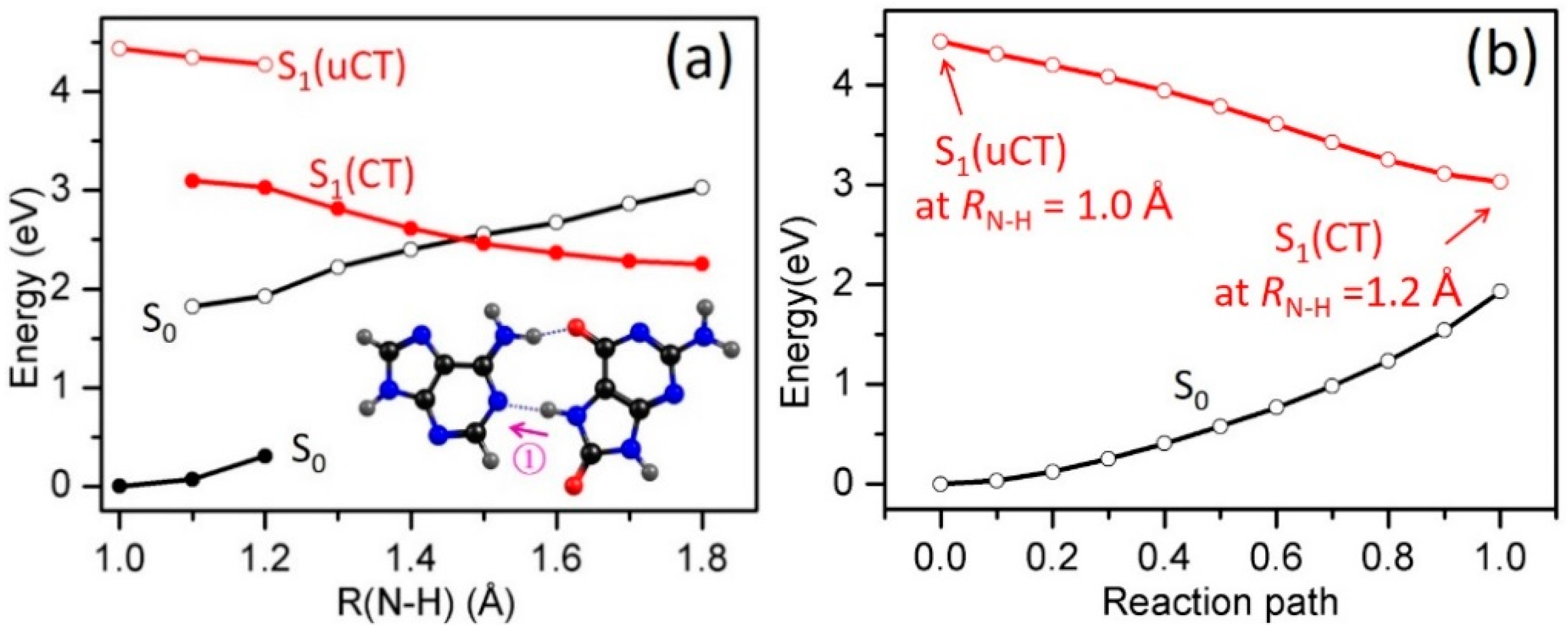
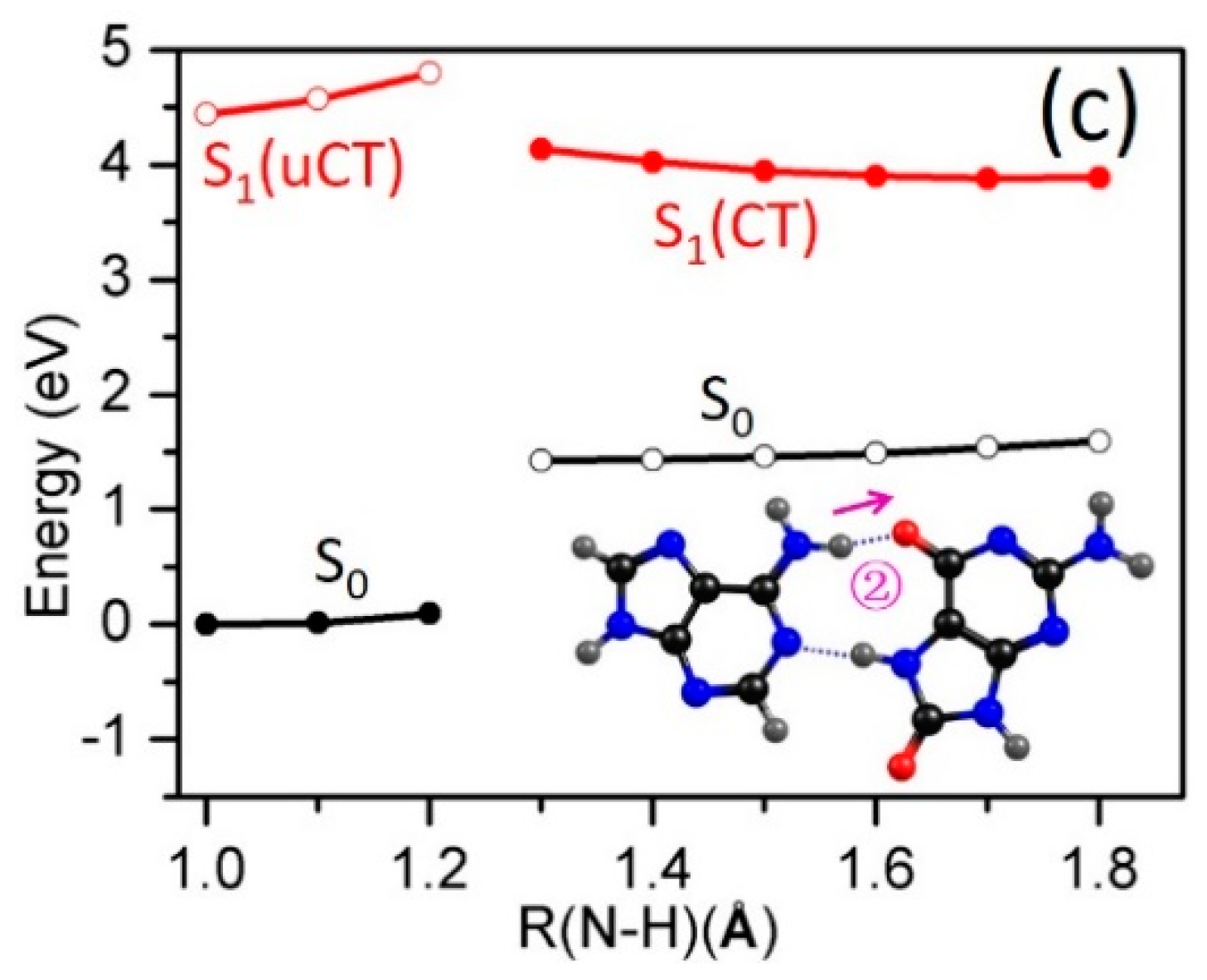
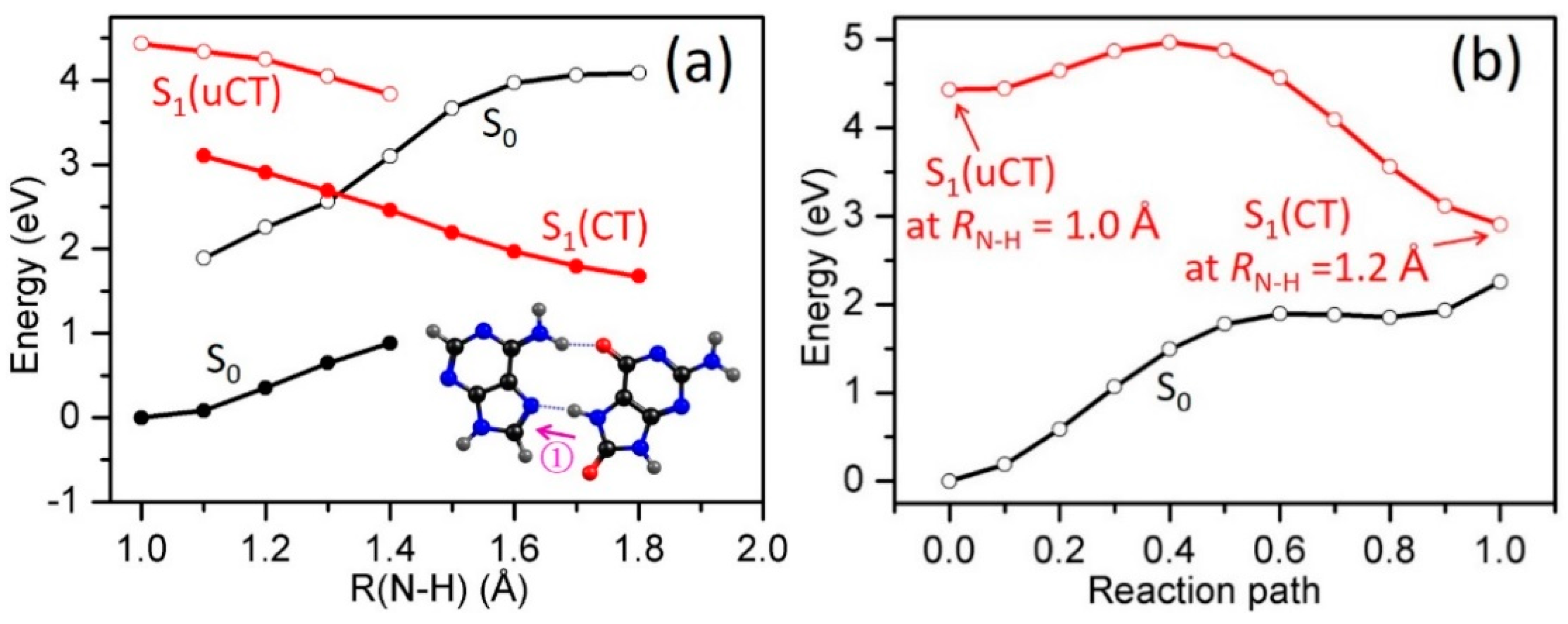
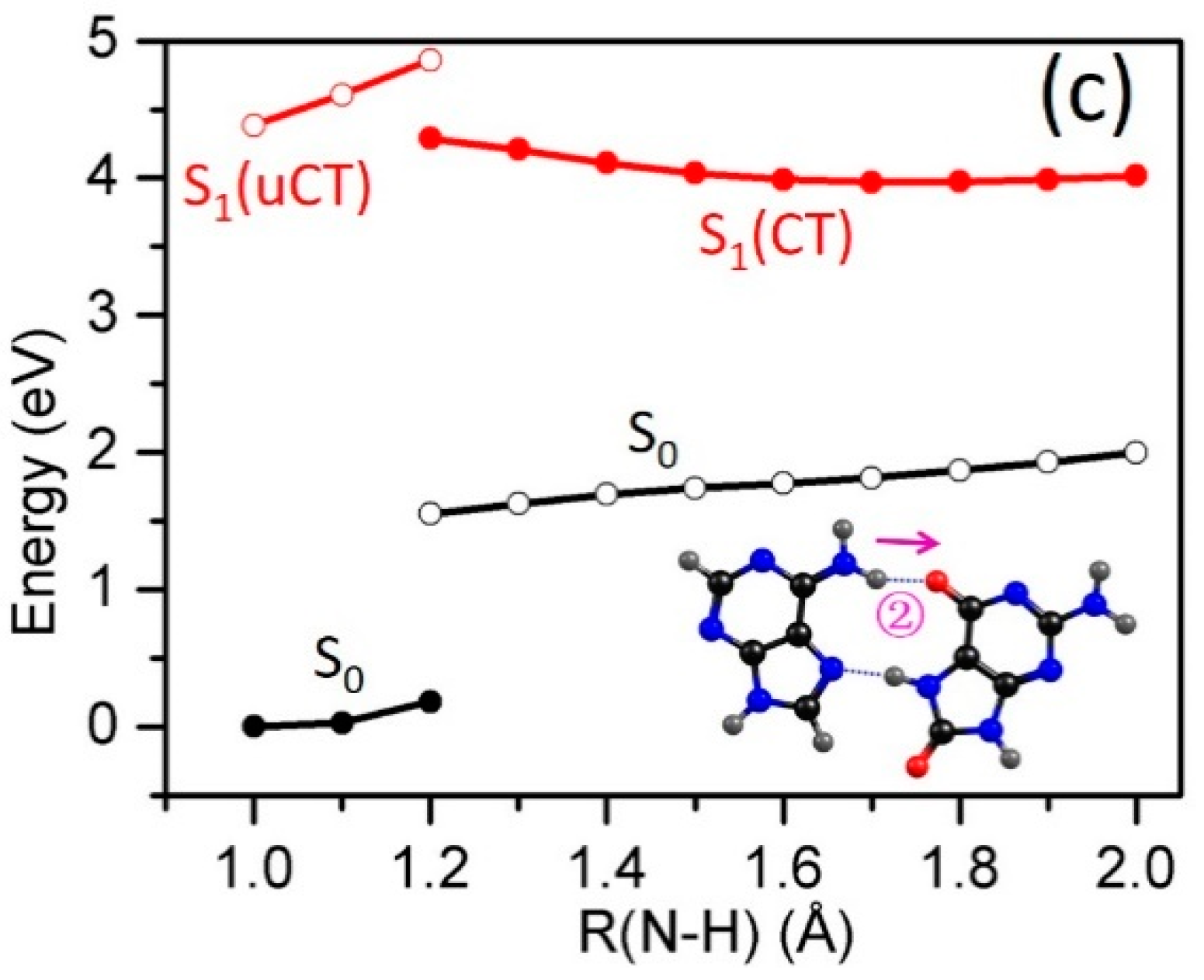
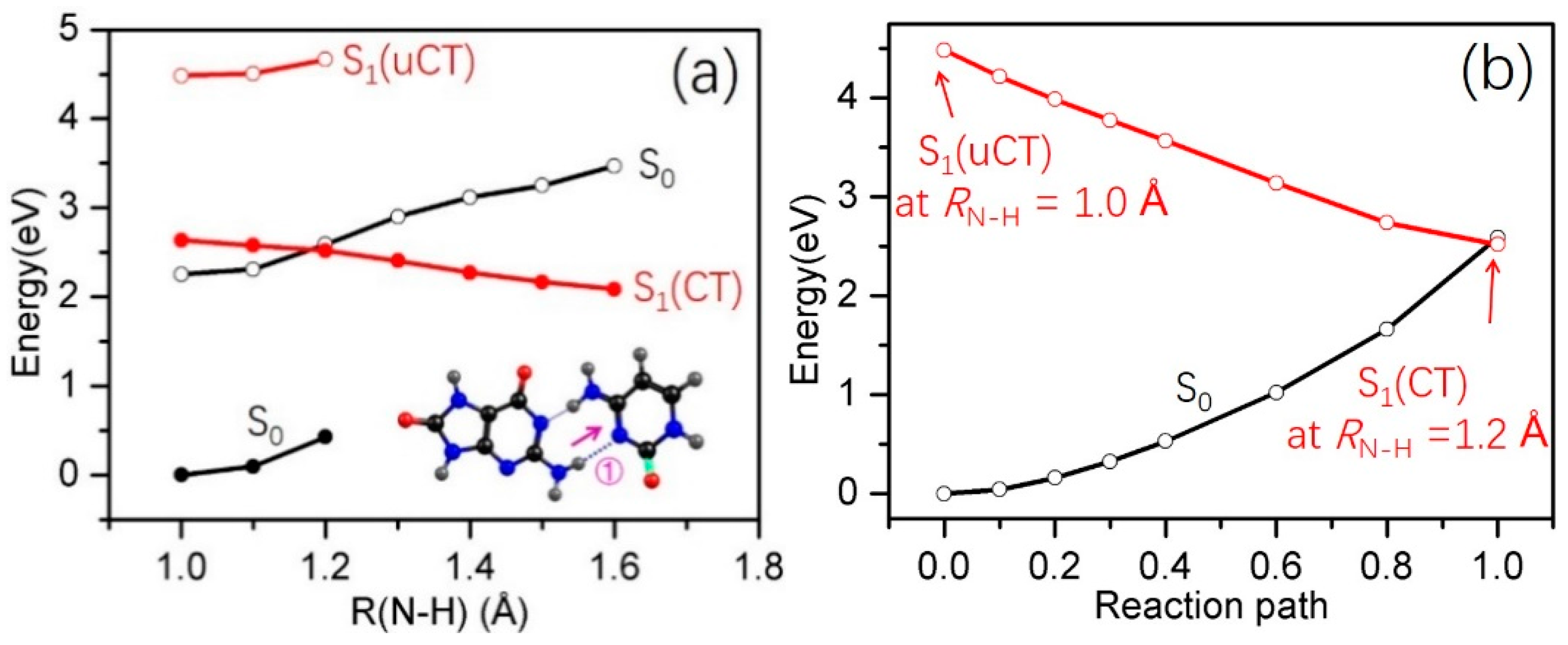
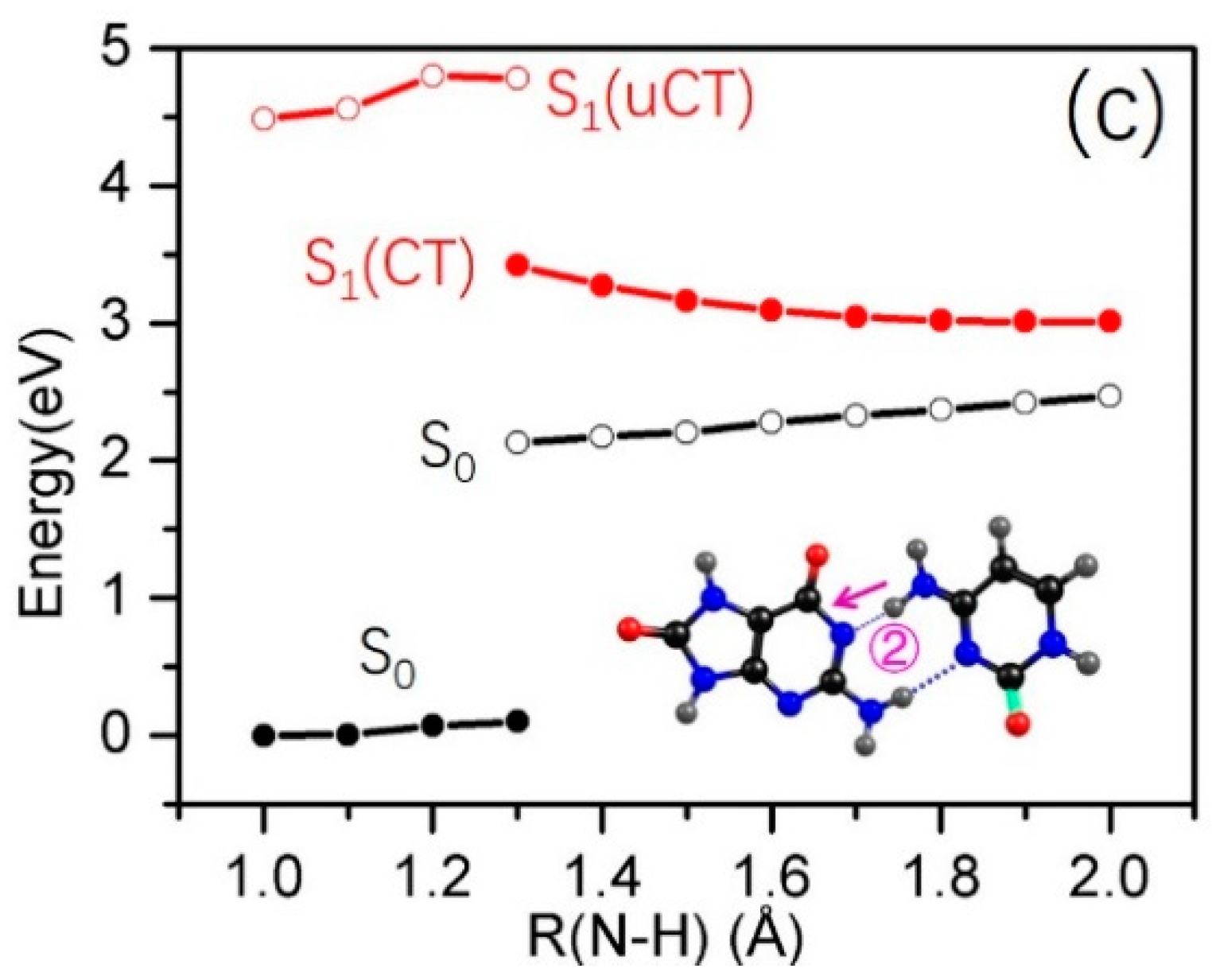
2.3. Electron-Driven Proton-Transfer Decay Paths
2.3.1. 8-oxoG−-A
2.3.2. 8-oxo-G−-C
3. General Discussions and Conclusions
4. Computational Methods
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Pecourt, J.-M.L.; Peon, J.; Kohler, B. DNA excited-state dynamics: Ultrafast internal conversion and vibrational cooling in a series of nucleosides. J. Am. Chem. Soc. 2001, 123, 10370–10378. [Google Scholar] [CrossRef]
- Kim, N.J.; Jeong, G.; Kim, Y.S.; Sung, J.; Keun Kim, S.; Park, Y.D. Resonant two-photon ionization and laser induced fluorescence spectroscopy of jet-cooled adenine. J. Chem. Phys. 2000, 113, 10051–10055. [Google Scholar] [CrossRef]
- Nir, E.; Kleinermanns, K.; Grace, L.; de Vries, M.S. On the photochemistry of purine nucleobases. J. Phys. Chem. A 2001, 105, 5106–5110. [Google Scholar] [CrossRef]
- Canuel, C.; Mons, M.; Piuzzi, F.; Tardivel, B.; Dimicoli, I.; Elhanine, M. Excited states dynamics of DNA and RNA bases: Characterization of a stepwise deactivation pathway in the gas phase. J. Chem. Phys. 2005, 122, 074316. [Google Scholar] [CrossRef] [PubMed]
- Ismail, N.; Blancafort, L.; Olivucci, M.; Kohler, B.; Robb, M.A. Ultrafast decay of electronically excited singlet cytosine via a π,π* to no,π* state switch. J. Am. Chem. Soc. 2002, 124, 6818–6819. [Google Scholar] [CrossRef] [PubMed]
- Sobolewski, A.; Domcke, W. On the mechanism of nonradiative decay of DNA bases: ab initio and tddft results for the excited states of 9H-adenine. Eur. Phys. J. D 2002, 20, 369–374. [Google Scholar] [CrossRef]
- Merchán, M.; Serrano-Andrés, L. Ultrafast internal conversion of excited cytosine via the lowest ππ* electronic singlet state. J. Am. Chem. Soc. 2003, 125, 8108–8109. [Google Scholar] [CrossRef] [PubMed]
- Matsika, S. Radiationless decay of excited states of uracil through conical intersections. J. Phys. Chem. A 2004, 108, 7584–7590. [Google Scholar] [CrossRef]
- Perun, S.; Sobolewski, A.L.; Domcke, W. Ab initio studies on the radiationless decay mechanisms of the lowest excited singlet states of 9H-adenine. J. Am. Chem. Soc. 2005, 127, 6257–6265. [Google Scholar] [CrossRef] [PubMed]
- Perun, S.; Sobolewski, A.; Domcke, W. Photostability of 9H-adenine: Mechanisms of the radiationless deactivation of the lowest excited singlet states. Chem. Phys. 2005, 313, 107–112. [Google Scholar] [CrossRef]
- Marian, C.M. A new pathway for the rapid decay of electronically excited adenine. J. Chem. Phys. 2005, 122, 104314. [Google Scholar] [CrossRef] [PubMed]
- Zgierski, M.Z.; Patchkovskii, S.; Fujiwara, T.; Lim, E.C. On the origin of the ultrafast internal conversion of electronically excited pyrimidine bases. J. Phys. Chem. A 2005, 109, 9384–9387. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Li, S. Theoretical study toward understanding ultrafast internal conversion of excited 9H-adenine. J. Phys. Chem. A 2005, 109, 8443–8446. [Google Scholar] [CrossRef] [PubMed]
- Blancafort, L. Excited-State potential energy surface for the photophysics of adenine. J. Am. Chem. Soc. 2006, 128, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Perun, S.; Sobolewski, A.L.; Domcke, W. Conical intersections in thymine. J. Phys. Chem. A 2006, 110, 13238–13244. [Google Scholar] [CrossRef] [PubMed]
- Buchner, F.; Ritze, H.-H.; Lahl, J.; Lübcke, A. Time-Resolved photoelectron spectroscopy of adenine and adenosine in aqueous solution. Phys. Chem. Chem. Phys. 2013, 15, 11402–11408. [Google Scholar] [CrossRef] [PubMed]
- Camillis, S.D.; Miles, J.; Alexander, G.; Ghafur, O.; Williams, I.D.; Townsend, D.; Greenwood, J.B. Ultrafast non-radiative decay of gas-phase nucleosides. Phys. Chem. Chem. Phys. 2015, 17, 23643–23650. [Google Scholar] [CrossRef] [PubMed]
- Gustavsson, T.; Sarkar, N.; Vaya, I.; Jimenez, M.C.; Markovitsi, D.; Improta, R. A joint experimental/theoretical study of the ultrafast excited state deactivation of deoxyadenosine and 9-methyladenine in water and acetonitrile. Photochem. Photobiol. Sci. 2013, 12, 1375–1386. [Google Scholar] [CrossRef] [PubMed]
- Pecourt, J.-M.L.; Peon, J.; Kohler, B. Ultrafast internal conversion of electronically excited RNA and DNA nucleosides in water. J. Am. Chem. Soc. 2000, 122, 9348–9349. [Google Scholar] [CrossRef]
- Peon, J.; Zewail, A.H. DNA/RNA nucleotides and nucleosides: Direct measurement of excited-state lifetimes by femtosecond fluorescence up-conversion. Chem. Phys. Lett. 2001, 348, 255–262. [Google Scholar] [CrossRef]
- Schwalb, N.K.; Temps, F. Ultrafast electronic relaxation in guanosine is promoted by hydrogen bonding with cytidine. J. Am. Chem. Soc. 2007, 129, 9272–9273. [Google Scholar] [CrossRef] [PubMed]
- Stavros, V.G.; Verlet, J.R. Gas-Phase femtosecond particle spectroscopy: A bottom-up approach to nucleotide dynamics. Annu. Rev. Phys. Chem. 2016, 67, 211–232. [Google Scholar] [CrossRef] [PubMed]
- Tuna, D.; Domcke, W. Excited-State deactivation in 8-oxo-deoxyguanosine: Comparison between anionic and neutral forms. Phys. Chem. Chem. Phys. 2016, 18, 947–955. [Google Scholar] [CrossRef] [PubMed]
- Tuna, D.; Sobolewski, A.L.; Domcke, W. Mechanisms of ultrafast excited-state deactivation in adenosine. J. Phys. Chem. A 2013, 118, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Zgierski, M.Z.; Alavi, S. Quantum chemical study of biradical decay channels in cytidine nucleosides. Chem. Phys. Lett. 2006, 426, 398–404. [Google Scholar] [CrossRef]
- Zhang, Y.; Dood, J.; Beckstead, A.; Chen, J.; Li, X.-B.; Burrows, C.J.; Lu, Z.; Matsika, S.; Kohler, B. Ultrafast excited-state dynamics and vibrational cooling of 8-oxo-7,8-dihydro-2′-deoxyguanosine in D2O. J. Phys. Chem. A 2013, 117, 12851–12857. [Google Scholar] [CrossRef] [PubMed]
- Abo-Riziq, A.; Grace, L.; Nir, E.; Kabelac, M.; Hobza, P.; de Vries, M.S. Photochemical selectivity in guanine-cytosine base-pair structures. Proc. Natl. Acad. Sci. USA 2005, 102, 20–23. [Google Scholar] [CrossRef] [PubMed]
- Gobbo, J.P.; Saurí, V.; Roca-Sanjuán, D.; Serrano-Andrés, L.; Merchán, M.; Borin, A.C. On the deactivation mechanisms of adenine-thymine base pair. J. Phys. Chem. B 2012, 116, 4089–4097. [Google Scholar] [CrossRef] [PubMed]
- Groenhof, G.; Schäfer, L.V.; Boggio-Pasqua, M.; Goette, M.; Grubmüller, H.; Robb, M.A. Ultrafast deactivation of an excited cytosine−guanine base pair in DNA. J. Am. Chem. Soc. 2007, 129, 6812–6819. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, B.; Karsili, T.N.V.; Ashfold, M.N.R.; Domcke, W. A ‘bottom up’, ab initio computational approach to understanding fundamental photophysical processes in nitrogen containing heterocycles, DNA bases and base pairs. Phys. Chem. Chem. Phys. 2016, 18, 20007–20027. [Google Scholar] [CrossRef] [PubMed]
- Markwick, P.R.L.; Doltsinis, N.L. Ultrafast repair of irradiated DNA: Nonadiabatic ab initio simulations of the guanine-cytosine photocycle. J. Chem. Phys. 2007, 126, 175102. [Google Scholar] [CrossRef] [PubMed]
- Nir, E.; Plützer, C.; Kleinermanns, K.; de Vries, M. Properties of isolated DNA bases, base pairs and nucleosides examined by laser spectroscopy. Eur. Phys. J. D 2002, 20, 317–329. [Google Scholar] [CrossRef]
- Perun, S.; Sobolewski, A.L.; Domcke, W. Role of electron-driven proton-transfer processes in the excited-state deactivation of the adenine-thymine base pair. J. Phys. Chem. A 2006, 110, 9031–9038. [Google Scholar] [CrossRef] [PubMed]
- Sobolewski, A.L.; Domcke, W. Ab initio studies on the photophysics of the guanine-cytosine base pair. Phys. Chem. Chem. Phys. 2004, 6, 2763–2771. [Google Scholar] [CrossRef]
- Sobolewski, A.L.; Domcke, W.; Hättig, C. Tautomeric selectivity of the excited-state lifetime of guanine/cytosine base pairs: The role of electron-driven proton-transfer processes. Proc. Natl. Acad. Sci. USA 2005, 102, 17903–17906. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, S.; Taketsugu, T. Photoreaction channels of the guanine-cytosine base pair explored by long-range corrected TDDFT calculations. Phys. Chem. Chem. Phys. 2012, 14, 8866–8877. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.S. Unraveling the molecular pathway from sunlight to skin cancer. Acc. Chem. Res. 1994, 27, 76–82. [Google Scholar] [CrossRef]
- Sinha, R.P.; Häder, D.-P. UV-Induced DNA damage and repair: A review. Photochem. Photobiol. Sci. 2002, 1, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Schreier, W.J.; Gilch, P.; Zinth, W. Early events of DNA photodamage. Annu. Rev. Phys. Chem. 2015, 66, 497–519. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Li, S. Ab initio study on deactivation pathways of excited 9H-guanine. J. Chem. Phys. 2006, 124, 154315. [Google Scholar] [CrossRef] [PubMed]
- Marian, C.M. The guanine tautomer puzzle: Quantum chemical investigation of ground and excited states. J. Phys. Chem. A 2007, 111, 1545–1553. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Andres, L.; Merchan, M.; Borin, A.C. A three-state model for the photophysics of guanine. J. Am. Chem. Soc. 2008, 130, 2473–2484. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, S.; Domcke, W. Ab initio studies on the photophysics of guanine tautomers: Out-of-plane deformation and NH dissociation pathways to conical intersections. J. Phys. Chem. A 2008, 112, 7090–7097. [Google Scholar] [CrossRef] [PubMed]
- Hudock, H.R.; Levine, B.G.; Thompson, A.L.; Satzger, H.; Townsend, D.; Gador, N.; Ullrich, S.; Stolow, A.; Martínez, T.J. Ab initio molecular dynamics and time-resolved photoelectron spectroscopy of electronically excited uracil and thymine. J. Phys. Chem. A 2007, 111, 8500–8508. [Google Scholar] [CrossRef] [PubMed]
- Zechmann, G.; Barbatti, M. Photophysics and deactivation pathways of thymine. J. Phys. Chem. A 2008, 112, 8273–8279. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Mendoza, M.; Banyasz, A.; Douki, T.; Markovitsi, D.; Ravanat, J.-L. Direct oxidative damage of naked DNA generated upon absorption of UV radiation by nucleobases. J. Phys. Chem. Lett. 2016, 7, 3945–3948. [Google Scholar] [CrossRef] [PubMed]
- Banyasz, A.; Martinez-Fernandez, L.; Ketola, T.-M.; Muñoz-Losa, A.; Esposito, L.; Markovitsi, D.; Improta, R. Excited state pathways leading to formation of adenine dimers. J. Phys. Chem. Lett. 2016, 7, 2020–2023. [Google Scholar] [CrossRef] [PubMed]
- Cadet, J.; Douki, T.; Ravanat, J.-L. Oxidatively generated damage to the guanine moiety of DNA: Mechanistic aspects and formation in cells. Acc. Chem. Res. 2008, 41, 1075–1083. [Google Scholar] [CrossRef] [PubMed]
- Kanvah, S.; Joseph, J.; Schuster, G.B.; Barnett, R.N.; Cleveland, C.L.; Landman, U. Oxidation of DNA: Damage to nucleobases. Acc. Chem. Res. 2010, 43, 280–287. [Google Scholar] [CrossRef] [PubMed]
- Markus, T.Z.; Daube, S.S.; Naaman, R.; Fleming, A.M.; Muller, J.G.; Burrows, C.J. Electronic structure of DNA-unique properties of 8-oxoguanosine. J. Am. Chem. Soc. 2009, 131, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Shibutani, S.; Takeshita, M.; Grollman, A.P. Insertion of specific bases during DNA synthesis past the oxidation-damaged base 8-oxodG. Nature 1991, 349, 431–434. [Google Scholar] [CrossRef] [PubMed]
- Greenman, C.; Stephens, P.; Smith, R.; Dalgliesh, G.L.; Hunter, C.; Bignell, G.; Davies, H.; Teague, J.; Butler, A.; Stevens, C.; et al. Patterns of somatic mutation in human cancer genomes. Nature 2007, 446, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Steenken, S.; Jovanovic, S.V.; Bietti, M.; Bernhard, K. The trap depth (in DNA) of 8-oxo-7,8-dihydro-2′deoxyguanosine as derived from electron-transfer equilibria in aqueous solution. J. Am. Chem. Soc. 2000, 122, 2373–2374. [Google Scholar] [CrossRef]
- Nguyen, K.V.; Burrows, C.J. A prebiotic role for 8-oxoguanosine as a flavin mimic in pyrimidine dimer photorepair. J. Am. Chem. Soc. 2011, 133, 14586–14589. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, K.V.; Burrows, C.J. Photorepair of cyclobutane pyrimidine dimers by 8-oxopurine nucleosides. J. Phys. Org. Chem. 2012, 25, 574–577. [Google Scholar] [CrossRef]
- Jayanth, N.; Ramachandran, S.; Puranik, M. Solution structure of the DNA damage lesion 8-oxoguanosine from ultraviolet resonance Raman spectroscopy. J. Phys. Chem. A 2009, 113, 1459–1471. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Beckstead, A.A.; Kohler, B.; Matsika, S. Excited state relaxation of neutral and basic 8-oxoguanine. J. Phys. Chem. B 2015, 119, 8293–8301. [Google Scholar] [CrossRef] [PubMed]
- Changenet-Barret, P.; Gustavsson, T.; Improta, R.; Markovitsi, D. Ultrafast excited-state deactivation of 8-hydroxy-2′-deoxyguanosine studied by femtosecond fluorescence spectroscopy and quantum-chemical calculations. J. Phys. Chem. A 2015, 119, 6131–6139. [Google Scholar] [CrossRef] [PubMed]
- Crespo-Hernandez, C.E.; Cohen, B.; Kohler, B. Base stacking controls excited-state dynamics in A-T DNA. Nature 2005, 436, 1141–1144. [Google Scholar] [CrossRef] [PubMed]
- Vayá, I.; Gustavsson, T.; Douki, T.; Berlin, Y.; Markovitsi, D. Electronic excitation energy transfer between nucleobases of natural DNA. J. Am. Chem. Soc. 2012, 134, 11366–11368. [Google Scholar] [CrossRef] [PubMed]
- Markovitsi, D. UV-induced DNA damage: The role of electronic excited states. Photochem. Photobiol. 2016, 92, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Dood, J.; Beckstead, A.A.; Li, X.-B.; Nguyen, K.V.; Burrows, C.J.; Improta, R.; Kohler, B. Photoinduced electron transfer in DNA: Charge shift dynamics between 8-oxo-guanine anion and adenine. J. Phys. Chem. B 2015, 119, 7491–7502. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Dood, J.; Beckstead, A.A.; Li, X.-B.; Nguyen, K.V.; Burrows, C.J.; Improta, R.; Kohler, B. Efficient UV-induced charge separation and recombination in an 8-oxoguanine-containing dinucleotide. Proc. Natl. Acad. Sci. USA 2014, 111, 11612–11617. [Google Scholar] [CrossRef] [PubMed]
- Bucher, D.B.; Schlueter, A.; Carell, T.; Zinth, W. Watson-Crick base pairing controls excited-state decay in natural DNA. Angew. Chem. Int. Ed. 2014, 53, 11366–11369. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; de La Harpe, K.; Beckstead, A.A.; Improta, R.; Kohler, B. UV-induced proton transfer between DNA strands. J. Am. Chem. Soc. 2015, 137, 7059–7062. [Google Scholar] [CrossRef] [PubMed]
- Schultz, T.; Samoylova, E.; Radloff, W.; Hertel, I.V.; Sobolewski, A.L.; Domcke, W. Efficient deactivation of a model base pair via excited-state hydrogen transfer. Science 2004, 306, 1765–1768. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Sevilla, M.D. Excited state proton-coupled electron transfer in 8-oxoG-C and 8-oxoG-A base pairs: A time dependent density functional theory (TD-DFT) study. Photochem. Photobiol. Sci. 2013, 12, 1328–1340. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Schlick, T. Distinct energetics and closing pathways for DNA polymerase β with 8-oxog template and different incoming nucleotides. BMC Struct. Biol. 2007, 7, 7. [Google Scholar] [CrossRef] [PubMed]
- Hsu, G.W.; Ober, M.; Carell, T.; Beese, L.S. Error-Prone replication of oxidatively damaged DNA by a high-fidelity DNA polymerase. Nature 2004, 431, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.C.; Cahill, D.S.; Kasai, H.; Nishimura, S.; Loeb, L.A. 8-hydroxyguanine, an abundant form of oxidative DNA damage, causes G-T and A-C substitutions. J. Biol. Chem. 1992, 267, 166–172. [Google Scholar] [PubMed]
- Sobolewski, A.L.; Domcke, W. Computational studies of the photophysics of hydrogen-bonded molecular systems. J. Phys. Chem. A 2007, 111, 11725–11735. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, K.V.; Burrows, C.J. Whence flavins? Redox-active ribonucleotides link metabolism and genome repair to the RNA world. Acc. Chem. Res. 2012, 45, 2151–2159. [Google Scholar] [CrossRef] [PubMed]
- Møller, C.; Plesset, M.S. Note on an approximation treatment for many-electron systems. Phys. Rev. 1934, 46, 618–622. [Google Scholar] [CrossRef]
- Dunning, T.H., Jr. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Schirmer, J. Beyond the random-phase approximation: A new approximation scheme for the polarization propagator. Phys. Rev. A 1982, 26, 2395–2416. [Google Scholar] [CrossRef]
- Hättig, C.; Weigend, F. CC2 excitation energy calculations on large molecules using the resolution of the identity approximation. J. Chem. Phys. 2000, 113, 5154–5161. [Google Scholar] [CrossRef]
- Turbomole, V. 4. A Development of the University of Karlsruhe and Forschungszentrum Karlsruhe GmBH: Karlsruhe, Germany, 2012.
- Sample Availability: Samples of the compounds are not available from the authors.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| State | ∆E/eV (f) | State | ∆E/eV (f) | State | ∆E/eV (f) |
|---|---|---|---|---|---|
| 8-oxo-G−-A HG1 | 8-oxo-G−-A HG2 | 8-oxo-G−-C | |||
| S1 1ππ*(O−→A) | 4.44 (0.0139) | S1 1ππ*(O−→A) | 4.39 (0.0091) | S1 1ππ*(O−→C) | 4.48 (0.0023) |
| S2 1ππ*(O−→O−) | 4.85 (0.2321) | S2 1ππ*(O−→O−) | 4.89 (0.1025) | S2 1ππ*(C→C) | 4.76 (0.0176) |
| S3 1ππ*(A→A) | 4.91 (0.1050) | S3 1ππ*(A→A) | 4.95 (0.0922) | S3 1ππ*(O−→O−) | 4.88 (0.0695) |
| S4 1ππ*(O−→A) | 5.09 (0.0109) | S4 1ππ*(O−→A) | 5.18 (0.0281) | S4 1ππ*(O−→C) | 4.94 (0.0782) |
| State | ∆E/eV (f) | State | ∆E/eV (f) | State | ∆E/eV (f) |
|---|---|---|---|---|---|
| 9H-adenine | 8-oxo-G− | Cytosine | |||
| S1 1nπ* | 5.13 (0.0051) | S1 1ππ* | 4.92 (0.0629) | S1 1ππ* | 4.65 (0.0545) |
| S2 1ππ* | 5.27 (0.0152) | S2 1nπ* | 5.16 (0.0000) | S2 1nπ* | 4.81 (0.0019) |
| S3 1ππ* | 5.40 (0.2856) | S3 1ππ* | 5.47 (0.2964) | S3 1nπ* | 5.29 (0.0016) |
| S4 1nπ* | 5.82 (0.0018) | S4 1nπ* | 5.54 (0.0003) | S4 1ππ* | 5.76 (0.1261) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, X.; Karsili, T.N.V.; Domcke, W. Role of Electron-Driven Proton-Transfer Processes in the Ultrafast Deactivation of Photoexcited Anionic 8-oxoGuanine-Adenine and 8-oxoGuanine-Cytosine Base Pairs. Molecules 2017, 22, 135. https://doi.org/10.3390/molecules22010135
Wu X, Karsili TNV, Domcke W. Role of Electron-Driven Proton-Transfer Processes in the Ultrafast Deactivation of Photoexcited Anionic 8-oxoGuanine-Adenine and 8-oxoGuanine-Cytosine Base Pairs. Molecules. 2017; 22(1):135. https://doi.org/10.3390/molecules22010135
Chicago/Turabian StyleWu, Xiuxiu, Tolga N. V. Karsili, and Wolfgang Domcke. 2017. "Role of Electron-Driven Proton-Transfer Processes in the Ultrafast Deactivation of Photoexcited Anionic 8-oxoGuanine-Adenine and 8-oxoGuanine-Cytosine Base Pairs" Molecules 22, no. 1: 135. https://doi.org/10.3390/molecules22010135