3. Experimental Section
3.1. General Considerations
Thin-layer chromatography was performed on 200 μm aluminum-foil-backed silica gel plates for the 2′-deoxynucleosides and on Merck 60F
254 (Merck, Billerica, MA, USA) for the ribose analogues. Column chromatographic purifications were performed on 100–200 mesh silica gel. CH
2Cl
2 for the chlorination reactions was distilled over CaH
2. Precursors
3a and
3b were prepared as described previously [
35]. The yield of
3a was 71% on a 2.52 mmol scale and the yield of
3b was 71% on a 2.1 mmol scale. TMSCl was redistilled prior to use and all other commercially available compounds were used without further purification.
1H-NMR spectra were recorded at 500 MHz or at 400 MHz in the solvents indicated under the individual compound headings and are referenced to residual protonated solvent resonances.
13C-NMR spectra were recorded at 125 MHz or at 100 MHz in the solvents indicated under the individual compound headings and are referenced to the solvent resonances (
Supplementary Materials). Chemical shifts (δ) are reported in parts per million (ppm), and coupling constants (
J) are in hertz (Hz). Standard abbreviations are used to designate resonance multiplicities (s = singlet, d = doublet, t = triplet, dd = doublet of doublet, ddd = doublet of doublet of doublet, quint = quintet, m = multiplet, br = broad, app = apparent). The saccharide carbons of the nucleoside are numbered 1′ through 5′ starting at the anomeric carbon atom and proceeding via the carbon chain to the primary carbinol carbon atom. The purinyl proton is designated as H-8 and the saccharide protons are designated on the basis of the carbon atom they are attached to.
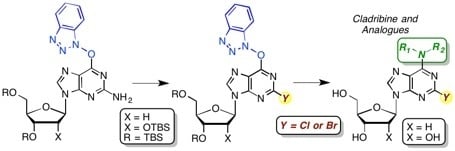
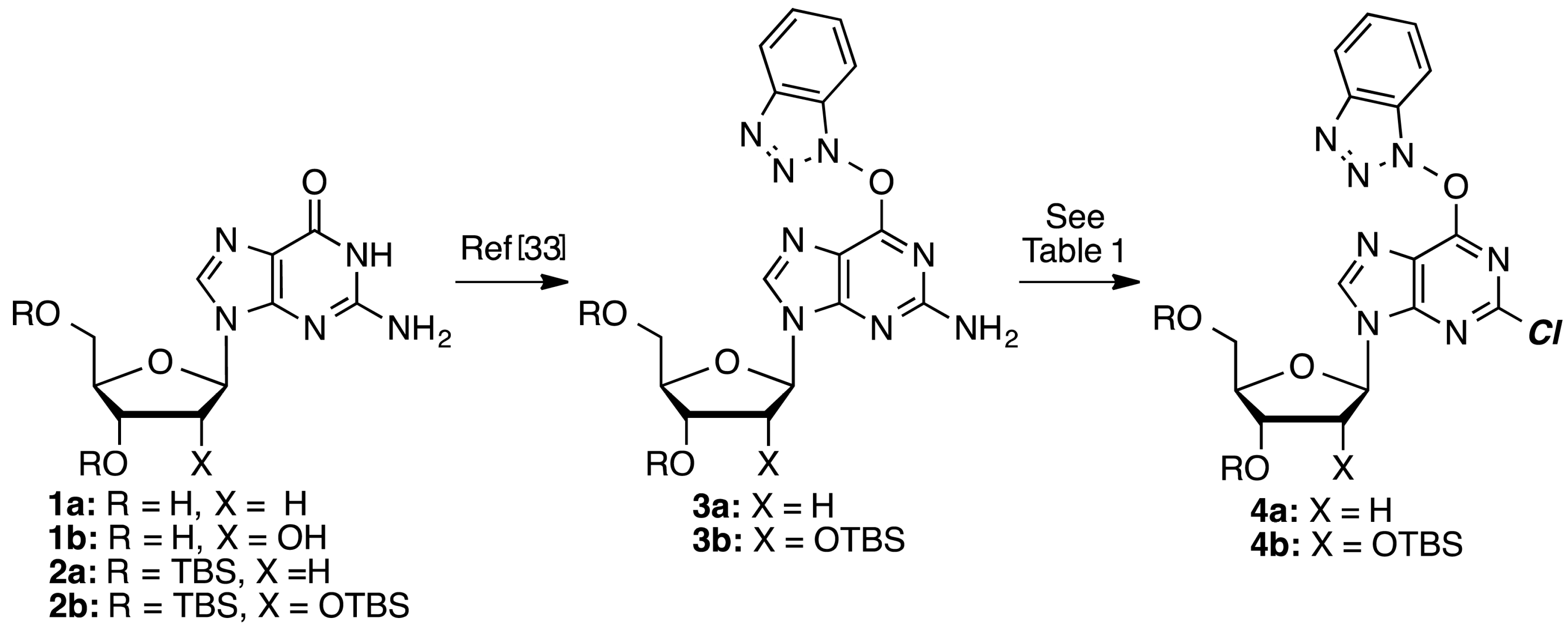
O6-(Benzotriazol-1-yl)-2-chloro-9-[2-deoxy-3,5-di-O-(t-butyldimethylsilyl)-β-d-ribofuranosyl]purine (4a): A mixture of compound 3a (1.0 g, 1.63 mmol) and SbCl3 (520.6 mg, 2.28 mmol) in dry CH2Cl2 (16.3 mL) was cooled to −15 °C using dry ice and acetone, in a nitrogen atmosphere. t-BuONO (0.678 mL, 5.70 mmol) was added dropwise and the mixture was stirred at −10 to −15 °C for 3.5 h, at which time TLC indicated the reaction to be complete. The reaction mixture was poured into ice-cold, saturated aqueous NaHCO3 (25 mL) with stirring. The mixture was filtered using vacuum (note: use of vacuum for this filtration is critical for maximizing product recovery) and the residue was washed with CH2Cl2 (25 mL). The organic layer was separated and the aqueous layer was back extracted with CH2Cl2 (2 × 15 mL). The combined organic layer was washed with water (15 mL) and brine (15 mL), dried over anhydrous Na2SO4, and evaporated under reduced pressure. Purification of the crude material on a silica gel column sequentially eluted with hexanes, 5% EtOAc in hexanes, followed by 20% EtOAc in hexanes gave 0.67 g (65% yield) of compound 4a as a white foam. Rf (SiO2 and 30% EtOAc in hexanes) = 0.55. 1H-NMR (500 MHz, CDCl3): δ 8.47 (s, 1H, H-8), 8.09 (d, J = 8.3 Hz, 1H, Ar-H), 7.52–7.41 (m, 3H, Ar-H), 6.45 (t, J = 5.8 Hz, 1H, H-1′), 4.62 (m, 1H, H-3′), 4.01 (m, 1H, H-4′), 3.89 (dd, J = 3.4, 11.2 Hz, 1H, H-5′), 3.76 (dd, J = 2.4, 11.2 Hz, 1H, H-5′), 2.61 (app quint, Japp ~ 6.2 Hz, 1H, H-2′), 2.49–2.48 (app quint, Japp ~ 5.8 Hz, 1H, H-2′), 0.88 (s, 18H, t-Bu), 0.08 and 0.07 (2s, 12H, SiCH3). 13C-NMR (125 MHz, CDCl3): δ 159.2, 154.9, 152.6, 144.4, 143.5, 129.0, 128.8, 125.0, 120.7, 119.1, 108.6, 88.4, 85.3, 71.6, 62.6, 41.6, 26.1, 25.9, 18.5, 18.1, −4.5, −4.7, −5.2, −5.3. HRMS (TOF) calcd for C28H43ClN7O4Si2 [M + H]+ 632.2598, found 632.2583.
2-Chloro-3′,5′-di-O-(t-butyldimethylsilyl)-2′-deoxyadenosine (5a): To a solution of compound 4a (126.0 mg, 0.2 mmol) in 1,2-DME (2 mL), 28%–30% aqueous ammonia (32 μL) was added, and the mixture was stirred at room temperature for 1.5 h. The mixture was diluted with EtOAc (5 mL) and washed with 5% aqueous NaCl (5 mL). The organic layer was separated and the aqueous layer was back extracted with EtOAc (5 mL). The combined organic layer was dried over anhydrous Na2SO4 and evaporated under reduced pressure. The crude material was chromatographed on a silica gel column by sequential elution with 20% EtOAc in hexanes followed by 5% MeOH in CH2Cl2 to afford 85.0 mg (82% yield) of compound 5a as a white solid. Rf (SiO2 and 10% MeOH in CH2Cl2) = 0.40. 1H-NMR (500 MHz, CDCl3): δ 8.12 (s, 1H, H-8), 6.54 (br s, 2H, NH2), 6.38 (t, J = 6.1 Hz, 1H, H-1′), 4.61 (m, 1H, H-3′), 3.99 (app q, Japp ~ 3.4 Hz, 1H, H-4′) 3.88 (dd, J = 3.9, 11.2 Hz, 1H, H-5′), 3.76 (dd, J = 3.0, 11.2 Hz, 1H, H-5′), 2.61 (app quint, Japp ~ 6.4 Hz, 1H, H-2′), 2.44–2.39 (ddd, J = 3.1, 6.3, 13.2 Hz, 1H, H-2′), 0.90 (s, 18H, t-Bu), 0.09 and 0.08 (2s, 12H, SiCH3). 13C-NMR (125 MHz, CDCl3): δ 156.2, 154.0, 150.4, 139.5, 118.7, 87.9, 84.5, 71.7, 62.6, 41.3, 25.9, 25.7, 18.4, 18.0, −4.7, −4.8, −5.4, −5.5. HRMS (TOF) calcd for C22H40ClN5O3Si2Na [M + Na]+ 536.2250, found 536.2252.
3.2. General Procedure for the Synthesis of Cladribine Analogues
To a solution of compound 4a (126.0 mg, 0.2 mmol) in 1,2-DME (2 mL) the appropriate amine (1.5 equiv) was added, and the mixture was stirred at room temperature. The mixture was diluted with EtOAc (5 mL for compounds 6a, 9a, 11a, and 15 mL for compounds 7a, 8a, 10a) and washed 5% aqueous NaCl (5 mL for compounds 6a, 9a, 11a, and 15 mL for compounds 7a, 8a, 10a). The organic layer was separated and the aqueous layer was back extracted with EtOAc (5 mL for compounds 6a, 9a, 11a, and 15 mL for compounds 7a, 8a, 10a). The combined organic layer was dried over anhydrous Na2SO4 and evaporated under reduced pressure. The crude material was chromatographed on a silica gel column; see individual compound headings for details.
2-Chloro-N6-methyl-3′,5′-di-O-(t-butyldimethylsilyl)-2′-deoxyadenosine (6a): Compound 6a (85.0 mg, 80% yield) was obtained as a white, foamy solid after chromatography on a silica gel column by sequential elution with 5% EtOAc in hexanes and 15% EtOAc in hexanes. Rf (SiO2 and 30% EtOAc in hexanes) = 0.26. 1H-NMR (500 MHz, CDCl3): δ 8.02 (s, 1H, H-8), 6.37 (t, J = 6.4 Hz, 1H, H-1′), 6.17 (br s, 1H, NH), 4.60 (m, 1H, H-3′), 3.97 (app q, Japp ~ 3.4 Hz, 1H, H-4′), 3.87 (dd, J = 4.1, 11.2 Hz, 1H, H-5′), 3.75 (dd, J = 3.0, 11.2 Hz, 1H, H-5′), 2.61 (app quint, Japp ~ 6.4 Hz, 1H, H-2′), 2.42–2.37 (ddd, J = 2.8, 6.1, 12.7 Hz, 1H, H-2′), 0.90 (s, 18H, t-Bu), 0.09 and 0.08 (2s, 12H, SiCH3). 13C-NMR (125 MHz, CDCl3): δ 155.9, 154.5, 149.2; 138.7, 119.1, 87.9, 84.4, 71.8, 62.7, 41.2, 27.5, 25.9, 25.7, 18.4, 18.0, −4.7, −4.8, −5.4, −5.5. HRMS (TOF) calcd for C23H42ClN5O3Si2Na [M + Na]+ 550.2407, found 550.2419.
2-Chloro-N6,N6-dimethyl-3′,5′-di-O-(t-butyldimethylsilyl)-2′-deoxyadenosine (7a): Compound 7a (91.1 mg, 84% yield) was obtained as a colorless, thick gum after chromatography on a silica gel column by sequential elution with hexanes, 5% EtOAc in hexanes, and 20% EtOAc in hexanes. Rf (SiO2 and 30% EtOAc in hexanes) = 0.70. 1H-NMR (500 MHz, CDCl3): δ 7.93 (s, 1H, H-8), 6.36 (t, J = 6.3 Hz, 1H, H-1′), 4.58 (m, 1H, H-3′), 3.95 (m, 1H, H-4′), 3.83 (dd, J = 4.4, 11.2 Hz, 1H, H-5′), 3.73 (dd, J = 2.9, 11.2 Hz, 1H, H-5′), 3.50 (br s, 6H, N(CH3)2), 2.58 (app quint, Japp ~ 6.5 Hz, 1H, H-2′), 2.39–2.35 (m, 1H, H-2′), 0.88 (s, 18H, t-Bu), 0.08 and 0.06 (2s, 12H, SiCH3). 13C-NMR (125 MHz, CDCl3): δ 155.2, 153.8, 151.4, 137.2, 119.6, 87.9, 84.4, 72.1, 62.9, 41.1, 26.1, 26.0, 25.9, 25.8, 18.6, 18.2, −4.5, −4.6, −5.2, −5.3. HRMS (TOF) calcd for C24H45ClN5O3Si2 [M + H]+ 542.2744, found 542.2749.
2-Chloro-6-(pyrrolidin-1-yl)-9-[2-deoxy-3,5-di-O-(t-butyldimethylsilyl)-β-d-ribofuranosyl]purine (8a): Compound 8a (93.3 mg, 82% yield) was obtained as a colorless, thick gum after chromatography on a silica gel column by sequential elution with hexanes, 5% EtOAc in hexanes, and 20% EtOAc in hexanes. Rf (SiO2 and 30% EtOAc in hexanes) = 0.65. 1H-NMR (500 MHz, CDCl3): δ 7.92 (s, 1H, H-8), 6.37 (t, J = 6.6 Hz, 1H, H-1′), 4.59 (m, 1H, H-3′), 4.13 (br s, 2H, pyrrolidinyl NCH), 3.96 (m, 1H, H-4′), 3.83 (dd, J = 4.9, 11.2 Hz, 1H, H-5′), 3.74 (dd, J = 3.9, 11.2 Hz, 1H, H-5′), 3.79–3.73 (br m, 2H, pyrrolidinyl NCH), 2.61 (app quint, Japp ~ 6.5 Hz, 1H, H-2′), 2.39–2.35 (m, 1H, H-2′), 2.10–2.0 (br m, 2H, pyrrolidinyl CH), 1.96–1.85 (br m, 2H, pyrrolidinyl CH), 0.90 (s, 18H, t-Bu), 0.09 and 0.07 (2s, 12H, SiCH3). 13C-NMR (125 MHz, CDCl3): δ 154.3, 153.5, 151.0, 137.8, 119.8, 88.1, 84.4, 72.3, 63.1, 49.1, 41.08, 26.4, 26.2, 25.9, 24.3, 18.6, 18.2, −4.5, −4.6, −5.2, −5.3. HRMS (TOF) calcd. for C26H47ClN5O3Si2 [M + H]+ 568.2900, found 568.2906.
2-Chloro-6-(piperidin-1-yl)-9-[2-deoxy-3,5-di-O-(t-butyldimethylsilyl)-β-d-ribofuranosyl]purine (9a): Compound 9a (99.0 mg, 85% yield) was obtained as a white, foamy solid after chromatography on a silica gel column by sequential elution with 5% EtOAc in hexanes and 15% EtOAc in hexanes. Rf (SiO2 and 30% EtOAc in hexanes) = 0.65. 1H-NMR (500 MHz, CDCl3): δ 7.92 (s, 1H, H-8), 6.37 (t, J = 6.6 Hz, 1H, H-1′), 4.59 (m, 1H, H-3′), 4.45–3.73 (br m, 4H, piperidinyl N(CH2)2), 3.96 (app q, Japp ~ 3.6 Hz, 1H, H-4′), 3.83 (dd, J = 4.9, 11.2 Hz, 1H, H-5′), 3.74 (dd, J = 3.4, 11.2 Hz, 1H, H-5′), 2.59 (app quint, Japp ~ 6.3 Hz, 1H, H-2′), 2.40–2.35 (ddd, J = 3.3, 6.2, 13.3 Hz, 1H, H-2′), 1.68 (br m, 6H, piperidinyl CH), 0.90 (s, 18H, t-Bu), 0.09 and 0.07 (2s, 12H, SiCH3). 13C-NMR (125 MHz, CDCl3): δ 153.8, 151.4, 136.7, 119.0, 109.9, 87.8, 84.2, 72.0, 62.8, 46.3, 40.9, 26.1, 25.9, 25.7, 24.6, 18.4, 18.0, −4.7, −4.8, −5.4, −5.5. HRMS (TOF) calcd for C27H49ClN5O3Si2 [M + H] + 582.3057 found 582.3074.
2-Chloro-6-(morpholin-4-yl)-9-[2-deoxy-3,5-di-O-(t-butyldimethylsilyl)-β-d-ribofuranosyl]purine (10a): Compound 10a (97.4 mg, 83% yield) was obtained as a colorless, thick gum after chromatography on a silica gel column by sequential elution with hexanes, 5% EtOAc in hexanes, and 20% EtOAc in hexanes. Rf (SiO2 and 30% EtOAc in hexanes) = 0.68. 1H-NMR (500 MHz, CDCl3): δ 7.98 (s, 1H, H-8), 6.37 (t, J = 6.3 Hz, 1H, H-1′), 4.58 (m, 1H, H-3′), 4.55–3.98 (br m, 4H, morpholinyl N(CH2)2), 3.96 (m, 1H, H-4′), 3.84 (dd, J = 4.4, 11.2 Hz, 1H, H-5′), 3.78 (t, J = 4.6 Hz, 4H, morpholinyl CH2OCH2), 3.74 (dd, J = 2.9, 11.2 Hz, 1H, H-5′), 2.56 (app quint, Japp ~ 6.5 Hz, 1H, H-2′), 2.40–2.36 (m, 1H, H-2′), 0.89 (s, 18H, t-Bu), 0.08 and 0.07 (2s, 12H, SiCH3). 13C-NMR (125 MHz, CDCl3): δ 154.0, 153.9, 151.8, 137.6, 119.3, 88.0, 84.4, 72.0, 67.1, 62.9, 43.5 (br), 41.3, 26.1, 26.0, 25.9, 18.6, 18.2, −4.5, −4.6, −5.2, −5.3. HRMS (TOF) calcd for C26H47ClN5O4Si2 [M + H]+ 584.2850, found 584.2855.
2-Chloro-6-[(2-dimethylamino)ethyl)(methyl)amino)]-9-[2-deoxy-3,5-di-O-(t-butyldimethylsilyl)-β-d-ribofuranosyl]purine (11a): Compound 11a (77.0 mg, 64% yield) was obtained as a pale-yellow, thick gum after chromatography on a silica gel column by sequential elution with 10% EtOAc in hexanes, 20% EtOAc in hexanes, and 10% MeOH in CH2Cl2. Rf (SiO2 and 10% MeOH in CH2Cl2) = 0.10. 1H-NMR (500 MHz, CDCl3): δ 7.94 (s, 1H, H-8), 6.36 (t, J = 6.3 Hz, 1H, H-1′), 4.62 (m, 1H, H-3′), 4.28–3.84 (br m, 2H, NCH2), 3.99 (app q, Japp ~ 3.7 Hz, 1H, H-4′), 3.85 (dd, J = 4.4, 11.2 Hz, 1H, H-5′), 3.78 (dd, J = 3.4, 11.2 Hz, 1H, H-5′), 3.47 (br s, 3H, NCH3), 2.65–2.59 (m, 2H, NCH2 and 1H, H-2′), 2.41–2.37 (ddd, J = 4.2, 6.2, 13.1 Hz, 1H, H-2′), 2.34 (s, 6H, N(CH3)2), 0.92 and 0.91 (s, 18H, t-Bu), 0.11, 0.083, and 0.079 (3s, 12H, SiCH3). 13C-NMR (125 MHz, CDCl3): δ 154.9, 153.8, 151.4, 137.2, 119.4, 87.9, 84.4, 72.0, 62.9, 57.0, 48.6, 45.7, 41.0, 36.9, 26.0, 25.8, 18.4, 8.0, −4.6, −4.8, −5.4, −5.5. HRMS (TOF) calcd for C27H52ClN6O3Si2 [M + H]+ 599.3322 found 599.3313.
O6-(Benzotriazol-1-yl)-2-chloro-9-[2,3,5-tri-O-(t-butyldimethylsilyl)-β-d-ribofuranosyl]purine (4b): To a solution of t-BuONO (2.78 g, 26.91 mmol) in CH2Cl2 (197 mL) was added TMSCl (1.46 g, 13.45 mmol) at 0 °C. To this mixture a solution of 3b (2 g, 2.69 mmol) in CH2Cl2 (197 mL) was added dropwise, and then the reaction mixture was stirred at 0 °C for 1 h at which time the reaction was completed as indicated by TLC. The reaction mixture was diluted with CH2Cl2 (200 mL), washed with saturated NaHCO3 (100 mL), H2O (100 mL), and brine (100 mL). The organic layer was dried over anhydrous Na2SO4 and evaporated under reduced pressure. Purification of the crude material on a silica gel column sequentially eluted with hexanes, 5% EtOAc in hexanes, and 20% EtOAc in hexanes gave 1.4 g (68% yield) of compound 4b as a white foam. Rf (SiO2 and 30% EtOAc in hexanes) = 0.55. 1H-NMR (400 MHz, CDCl3): δ 8.56 (s, 1H, H-8), 8.14 (d, J = 8.0 Hz, 1H, Ar-H), 7.55–7.45 (m, 3H, Ar-H), 6.05 (d, J = 4.0 Hz, 1H, H-1′), 4.57 (t, J = 4.2 Hz, 1H, H-2′), 4.34 (t, J = 4.4 Hz, 1H, H-3′), 4.18–4.15 (m, 1H, H-4′), 4.08 (dd, J = 3.6, 11.6 Hz, 1H, H-5′), 3.83 (dd, J = 2.4, 11.6 Hz, 1H, H-5′), 0.96, 0.93, and 0.88 (3s, 27H, t-Bu), 0.15, 0.11, 0.02, and −0.11 (4s, 18H, SiCH3). 13C-NMR (100 MHz, CDCl3): δ 159.0, 154.9, 152.6, 144.4, 143.4, 128.9, 128.7, 124.9, 120.6, 119.0, 108.5, 89.4, 85.3, 71.2, 62.0, 26.1, 25.8, 25.6, 18.5, 18.0, 17.9, −4.3, −4.6, −4.7, −4.9, −5.3, −5.4. HRMS (TOF) calcd for C34H57ClN7O5Si3 [M + H]+ 762.3412, found 762.3427.
2-Chloro-2′,3′,5′-tri-O-(t-butyldimethylsilyl)adenosine (5b): To a solution of compound 4b (500 mg, 0.655 mmol) in 1,2-DME (8 mL), 28%–30% aqueous ammonia (64 μL) was added and the mixture was stirred at room temperature for 1.5 h. The reaction mixture was diluted with EtOAc (25 mL) and washed 5% aqueous NaCl (25 mL). The organic layer was separated and the aqueous layer was back extracted with EtOAc (25 mL). The combined organic layer was dried over anhydrous Na2SO4 and evaporated under reduced pressure. The crude material was chromatographed on a silica gel column by sequential elution with 20% EtOAc in hexanes and 5% MeOH in EtOAc to afford 310 mg (75% yield) of compound 5b as an off-white solid. Rf (SiO2 and 10% MeOH in EtOAc) = 0.21. 1H-NMR (400 MHz, CDCl3): δ 8.12 (s, 1H, H-8), 6.17 (br s, 2H, NH2), 5.93 (d, J = 4.8 Hz, 1H, H-1′), 4.69 (t, J = 4.6 Hz, 1H, H-2′), 4.32 (t, J = 4.6 Hz, H-3′), 4.14–4.06 (m, 1H, H-4′), 4.06 (dd, J = 4.8, 11.6 Hz, 1H, H-5′), 3.80 (dd, J = 2.8, 11.2 Hz, 1H, H-5′), 0.94, 0.91, and 0.84 (3s, 27H, t-Bu), 0.14, 0.09, −0.01, and −0.17 (4s, 18H, SiCH3). 13C-NMR (100 MHz, CDCl3): δ 156.1, 154.0, 150.7, 140.2, 118.9, 88.9, 85.4, 75.5, 71.7, 62.3, 26.0, 25.8, 25.7, 18.5, 18.0, 17.9, −4.3, −4.5, −4.7, −5.0, −5.3, −5.4. HRMS (TOF) calcd. for C28H55ClN5O4Si3 [M + H]+ 644.3245 found 644.3217.
3.3. General Procedure for the Synthesis of Ribose Analogues of Cladribine
To a solution of compound 4b (500 mg, 0.655 mmol) in 1,2-DME (8 mL) was added the appropriate amine (1.5 equiv) and the mixture was stirred at room temperature. The mixture was diluted with EtOAc (25 mL) and washed with 5% aqueous NaCl (15 mL). The organic layer was separated and the aqueous layer was back extracted with EtOAc (15 mL). The combined organic layers were dried over anhydrous Na2SO4 and evaporated under reduced pressure. The crude material was chromatographed on a silica gel column. See individual compound headings for details.
2-Chloro-N6-methyl-2′,3′,5′-tri-O-(t-butyldimethylsilyl)adenosine (6b): Compound 6b (340 mg, 80% yield) was obtained as a pale-yellow, thick gum after chromatography on a silica gel column by sequential elution with 10% EtOAc in hexanes, 20% EtOAc in hexanes, and 50% EtOAc in hexanes. Rf (SiO2 and EtOAc) = 0.20. 1H-NMR (400 MHz, CDCl3): δ 8.02 (s, 1H, H-8), 6.04 (br s, 1H, NH), 5.91 (d, J = 5.2 Hz, 1H, H-1′), 4.72 (t, J = 4.6 Hz, 1H, H-2′), 4.32 (t, J = 4.0 Hz, 1H, H-3′), 4.12–4.10 (m, 1H, H-4′), 4.06 (dd, J = 5.2, 11.6 Hz, 1H, H-5′), 3.79 (dd, J = 2.8, 11.2 Hz, 1H, H-5′), 3.17 (s, 3H, CH3), 0.94, 0.92, and 0.81 (3s, 27H, t-Bu), 0.13, 0.06, −0.02, and −0.19 (4s, 18H, SiCH3). 13C-NMR (100 MHz, CDCl3): δ 155.9, 154.5, 139.3, 119.3, 88.8, 85.4, 75.3, 71.5, 65.2, 62.4, 26.0, 25.8, 25.7, 18.4, 18.0, 17.8, 14.0, 11.0, −4.4, −4.7, −5.1, −5.4, −5.7. HRMS (TOF) calcd for C29H57ClN5O4Si3 [M + H]+ 658.3401, found 658.3416.
2-Chloro-N6,N6-dimethyl- 2′,3′,5′-tri-O-(t-butyldimethylsilyl)adenosine (7b): Compound 7b (360 mg, 81% yield) was obtained as a pale-yellow, thick gum after chromatography on a silica gel column by sequential elution with 10% EtOAc in hexanes and 20% EtOAc in hexanes. Rf (SiO2 and 20% EtOAc in hexanes) = 0.40. 1H-NMR (400 MHz, CDCl3): δ 7.93 (s, 1H, H-8), 5.90 (d, J = 5.2 Hz, 1H, H-1′), 4.77 (t, J = 4.8 Hz, 1H, H-2′), 4.31 (t, J = 3.8 Hz, 1H, H-3′), 4.04–4.09 (m, 1H, H-4′), 4.06 (dd, J = 5.6, 11.2 Hz, 1H, H-5′), 3.78 (dd, J = 2.8, 11.2 Hz, 1H, H-5′), 3.58 (br s, 6H, CH3), 0.94, 0.92, and 0.82 (3s, 27H, t-Bu), 0.13, 0.10, −0.02, and −0.18 (4s, 18H, SiCH3). 13C-NMR (100 MHz, CDCl3): δ 155.9, 153.7, 151.4, 137.9, 119.7, 88.8, 85.4, 74.8, 72.0, 62.5, 38.4, 26.0, 25.8, 25.7, 18.4, 18.0, 17.9, −4.3, −4.7, −5.0, −5.4. HRMS (TOF) calcd for C30H59ClN5O4Si3 [M + H]+ 672.3558, found 672.3561.
2-Chloro-6-(pyrrolidin-1-yl)-9-[2,3,5-tri-O-(t-butyldimethylsilyl)-β-d-ribofuranosyl]purine (8b): Compound 8b (390 mg, 85% yield) was obtained as a pale-yellow, thick gum after chromatography on a silica gel column by sequential elution with 10% EtOAc in hexanes and 20% EtOAc in hexanes. Rf (SiO2 and 20% EtOAc in hexanes) = 0.30. 1H-NMR (400 MHz, CDCl3): δ 7.90 (s, 1H, H-8), 5.90 (d, J = 5.2 Hz, 1H, H-1′), 4.81 (t, J = 5.0 Hz, 1H, H-2′), 4.32 (t, J = 4.0 Hz, 1H, H-3′), 4.14–4.08 (m, 1H, H-4′ and br m, 2H, pyrrolidinyl NCH), 4.06 (dd, J = 5.6, 11.2 Hz, 1H, H-5′), 3.77–3.73 (m, 3H, H-5′ and pyrrolidinyl NCH), 2.04 (br m, 2H, pyrrolidinyl CH), 1.98 (br m, 2H, pyrrolidinyl CH), 0.93, 0.89, and 0.81 (3s, 27H, t-Bu), 0.12, 0.11, −0.03, and −0.20 (4s, 18H, SiCH3). 13C-NMR (100 MHz, CDCl3): δ 154.0, 153.4, 151.0, 138.0, 119.9, 88.7, 85.5, 74.6, 72.2, 62.6, 48.9, 47.6, 26.0, 25.8, 25.7, 18.4, 18.1, 17.9, −4.4, −4.6, −4.7, −5.0, −5.3. HRMS (TOF) calcd for C32H61ClN5O4Si3 [M + H]+ 698.3714 found 698.3731.
2-Chloro-6-(piperidin-1-yl)-9-[2,3,5-tri-O-(t-butyldimethylsilyl)-β-d-ribofuranosyl]purine (9b): Compound 9b (375 mg, 80% yield) was obtained as a pale-yellow, thick gum after chromatography on a silica gel column by sequential elution with 10% EtOAc in hexanes and 20% EtOAc in hexanes. Rf (SiO2 and 20% EtOAc in hexanes) = 0.50. 1H-NMR (400 MHz, CDCl3): δ 7.91 (s, 1H, H-8), 5.89 (d, J = 5.6 Hz, 1H, H-1′), 4.79 (t, J = 4.8 Hz, 1H, H-2′), 4.32 (t, J = 4.0 Hz, 1H, H-3′), 4.22 (br s, 4H, piperidinyl NCH2), 4.11–4.09 (m, 1H, H-4′), 4.06 (dd, J = 5.2, 10.8 Hz, 1H, H-5′), 3.77 (dd, J = 2.8, 10.8 Hz, 1H, H-5′), 1.69 (br m, 6H, piperidinyl CH2), 0.94, 0.90, and 0.84 (3s, 27H, t-Bu), 0.12, 0.10, −0.02, and −0.17 (4s, 18H, SiCH3). 13C-NMR (100 MHz, CDCl3): δ 153.9, 153.8, 151.6, 137.6, 119.4, 88.8, 85.3, 74.7, 71.9, 62.5, 29.6, 25.8, 25.7, 24.6, 22.6, 18.4, 18.0, 17.9, 14.0, −4.3, −4.7, −5.0, −5.3. HRMS (TOF) calcd for C33H63ClN5O4Si3 [M + H]+ 712.3871, found 712.3875.
2-Chloro-6-(morpholin-4-yl)-9-[2,3,5-tri-O-(t-butyldimethylsilyl)-β-d-ribofuranosyl]purine (10b): Compound 10b (430 mg, 90% yield) was obtained as a pale-yellow, thick gum after chromatography on a silica gel column by sequential elution with 10% EtOAc in hexanes and 20% EtOAc in hexanes. Rf (SiO2 and 20% EtOAc in hexanes) = 0.47. 1H-NMR (400 MHz, CDCl3): δ 7.98 (s, 1H, H-8), 5.92 (d, J = 4.8 Hz, 1H, H-1′), 4.73 (t, J = 4.8 Hz, 1H, H-2′), 4.31–4.20 (t, J = 4.2 Hz, 1H, H-3′and br m, 4H, morpholinyl N(CH2)2), 4.12–4.09 (m, 1H, H-4′), 4.06 (dd, J = 5.2, 11.2 Hz, 1H, H-5′), 3.83 (t, J = 5.0 Hz, 4H, morpholinyl CH2OCH2), 3.77 (dd, J = 3.2, 11.2 Hz, 1H, H-5′), 0.94, 0.92, and 0.83 (3s, 27H, t-Bu), 0.13, 0.10, −0.01, and −0.16 (4s, 18H, SiCH3). 13C-NMR (100 MHz, CDCl3): δ 153.9, 153.7, 151.8, 138.1, 119.4, 88.8, 85.3,77.3,77.0,76.6,75.0, 71.8, 66.9, 62.3, 45.60, 29.6, 29.3, 26.0,25.8, 25.7, 22.6, 18.4, 18.0, 17.8,14.0, −4.3, −4.7, −5.0, −5.4. HRMS (TOF) calcd for C32H61ClN5O5Si3 [M + H]+ 714.3664 found 714.3668.
2-Chloro-6-[(2-dimethylamino)ethyl)(methyl)amino)]-9-[2,3,5-tri-O-(t-butyldimethylsilyl)-β-d-ribofuranosyl] purine (11b): Compound 11b (280 mg, 58%) was obtained as a pale-yellow, thick gum after chromatography on a silica gel column by sequential elution with 10% MeOH in CH2Cl2, 20% MeOH in CH2Cl2, and 30% MeOH in CH2Cl2. Rf (SiO2 and 10% MeOH in CH2Cl2) = 0.10. 1H-NMR (500 MHz, CDCl3): δ 7.97 (s, 1H, H-8), 5.90 (d, J = 4.8 Hz, 1H, H-1′), 4.72 (t, J = 4.8 Hz, 1H, H-2′), 4.32 (t, J = 3.8 Hz, 1H, H-3′), 4.15–4.10 (m, 1H, H-4′ and br s, 2H, NCH2), 4.07 (dd, J = 5.2, 11.2 Hz, 1H, H-5′), 3.78 (dd, J = 2.8, 11.2 Hz, 1H, H-5′ and br m, 2H, NCH2), 2.59 (br s, 2H, NCH2), 2.32 (s, 6H, N(CH3)2), 0.94, 0.92, and 0.82 (3s, 27H, t-Bu), 0.13, 0.10, −0.01, and −0.15 (4s, 18H, SiCH3). 13C-NMR (100 MHz, CDCl3): δ 154.8, 153.7, 151.4, 137.9, 119.5, 88.8, 85.2, 75.0, 71.9, 62.4, 45.7, 29.6, 26.0, 25.8, 25.7, 18.4, 18.0, 17.9, −4.3, −4.7, −5.0, −5.4. HRMS (TOF) calcd for C33H66ClN6O4Si3 [M + H]+ 729.4136, found 729.4157.
3.4. General Procedure for the Desilylation of Cladribine Analogues
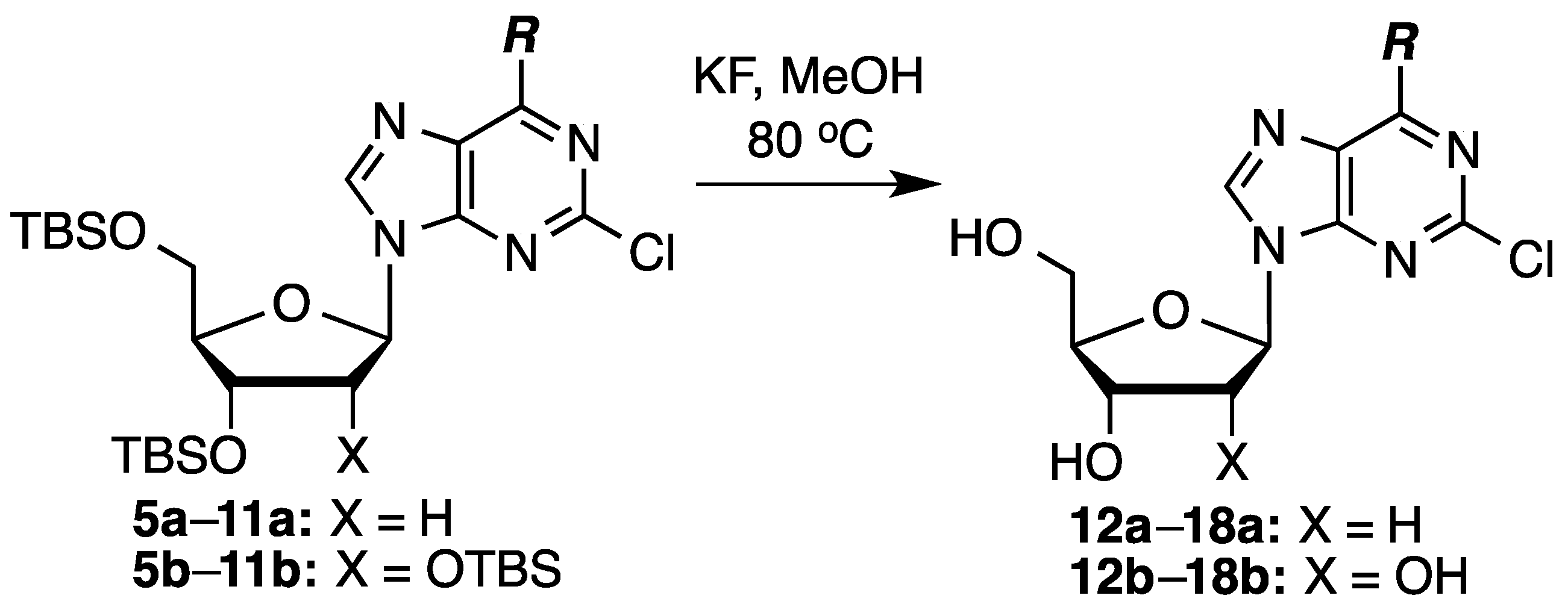
To a 0.1 M solution of the silylated compound in anhydrous MeOH, KF (2 equiv/silyl group) was added. The mixture was heated at 80 °C for 20–26 h, cooled, and silica gel was added. The mixture was evaporated to dryness and the compound-impregnated silica gel was loaded onto a wet-packed silica gel column. The products were obtained by elution with appropriate solvents (see the individual compound headings for details).
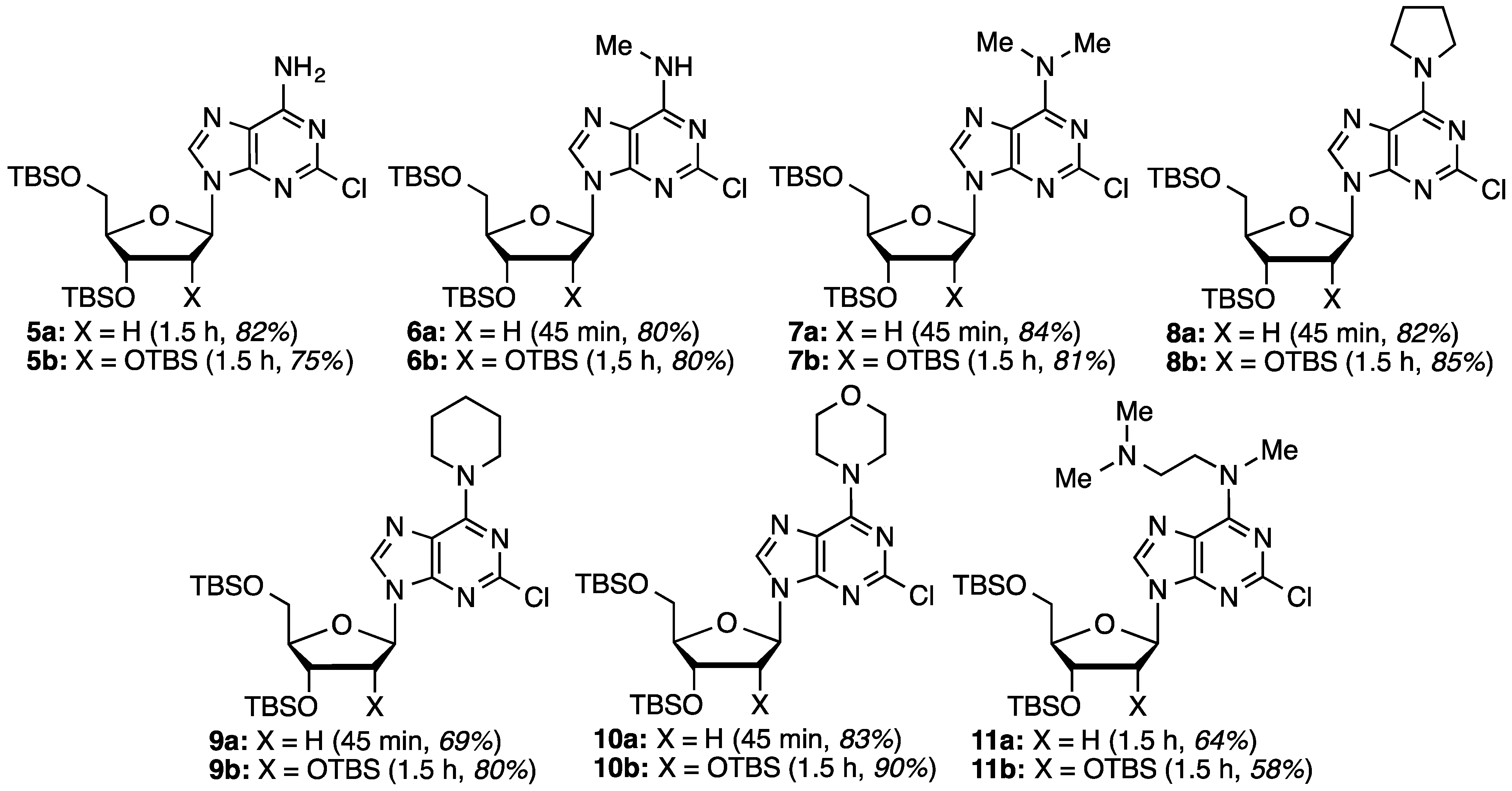
2-Chloro-2′-deoxyadenosine (12a): Prepared from compound 5a (60.0 mg, 0.117 mmol) and KF (27.0 mg, 0.467 mmol) in MeOH (1.17 mL). Chromatography on a silica gel column sequentially eluted with 5% MeOH in EtOAc and 10% MeOH in EtOAc gave 24.0 mg (72% yield) of compound 12a as an off-white solid. Rf (SiO2 and 10% MeOH in EtOAc) = 0.13. 1H-NMR (500 MHz, CD3OD): δ 8.28 (s, 1H, H-8), 6.36 (t, J = 6.8 Hz, 1H, H-1′), 4.57 (m, 1H, H-3′), 4.04 (m, 1H, H-4′), 3.84 (dd, J = 2.9, 12.2 Hz, 1H, H-5′), 3.74 (dd, J = 3.4, 12.2 Hz, 1H, H-5′), 2.76 (app quint, Japp ~ 6.7 Hz, 1H, H-2′), 2.43–2.39 (ddd, J = 2.9, 5.9, 13.2 Hz, 1H, H-2′). 13C-NMR (125 MHz, CD3OD): δ 158.3, 155.3, 151.4, 141.9, 119.8, 89.9, 87.0, 73.0, 63.6, 41.6. HRMS (TOF) calcd for C10H12ClN5O3Na [M + Na]+ 308.0521, found 308.0523.
2-Chloro-N6-methyl-2′-deoxyadenosine (
13a) [
50]: Prepared from compound
6a (80.0 mg, 0.154 mmol) and KF (36.0 mg, 0.618 mmol) in MeOH (1.54 mL). Chromatography on a silica gel column sequentially eluted with 2.5% MeOH in EtOAc and 5% MeOH in EtOAc gave 39.0 mg (85% yield) of compound
13a as a white, foamy solid.
Rf (SiO
2 and 10% MeOH in EtOAc) = 0.21.
1H-NMR (500 MHz, CD
3OD): δ 8.18 (s, 1H, H-8), 6.34 (t,
J = 6.8 Hz, 1H, H-1′), 4.56 (m, 1H, H-3′), 4.04 (m, 1H, H-4′), 3.84 (dd,
J = 2.4, 12.2 Hz, 1H, H-5′), 3.74 (dd,
J = 3.4, 12.2 Hz, 1H, H-5′), 3.05 (br s, 3H, NCH
3), 2.76 (app quint,
Japp ~ 6.8 Hz, 1H, H-2′), 2.42–2.38 (ddd,
J = 2.9, 5.8, 13.7 Hz, 1H, H-2′).
13C-NMR (125 MHz, CD
3OD): δ 157.3, 155.5, 150.1, 141.2, 120.4, 89.9, 87.0, 73.0, 63.7, 41.6, 27.8. HRMS (TOF) calcd for C
11H
14ClN
5O
3Na [M + Na]
+ 322.0677, found 322.0682.
2-Chloro-N6,N6-dimethyl-2′-deoxyadenosine (Cladribine, 14a): Prepared from compound 7a (80.0 mg, 0.147 mmol) and KF (34.3 mg, 0.590 mmol) in MeOH (1.4 mL). Chromatography on a silica gel column sequentially eluted with 50% EtOAc in hexanes, EtOAc, and 10% MeOH in EtOAc gave 38.0 mg (82% yield) of compound 14a as a white, foamy solid. Rf (SiO2 and 5% MeOH in EtOAc) = 0.29. 1H-NMR (500 MHz, CD3OD): δ 8.16 (s, 1H, H-8), 6.35 (t, J = 6.8 Hz, 1H, H-1′), 4.56 (m, 1H, H-3′), 4.04 (m, 1H, H-4′), 3.84 (dd, J = 2.9, 12.2 Hz, 1H, H-5′), 3.73 (dd, J = 3.4, 12.2 Hz, 1H, H-5′), 3.70–3.10 (br m, 6H, N(CH3)2), 2.74 (app quint, Japp ~ 6.8 Hz, 1H, H-2′), 2.41–2.36 (ddd, J = 2.9, 5.8, 13.2 Hz, 1H, H-2′). 13C-NMR (125 MHz, CD3OD): δ 156.4, 154.6, 152.2, 140.0, 120.72, 89.8, 86.9, 73.0, 63.7, 41.5, 39.0 (br). HRMS (TOF) calcd for C12H16ClN5O3Na [M + Na]+ 336.0834, found 336.0823.
2-Chloro-6-(pyrrolidin-1-yl)-9-(2-deoxy-β-d-ribofuranosyl)purine (15a): Prepared from compound 8a (60.0 mg, 0.105 mmol) and KF (24.5 mg, 0.422 mmol) in MeOH (1.0 mL). Chromatography on a silica gel column sequentially eluted with 50% EtOAc in hexanes, EtOAc, and 10% MeOH in EtOAc gave 29.1 mg (81% yield) of compound 15a as a white solid. Rf (SiO2 and 5% MeOH in EtOAc) = 0.19. 1H-NMR (500 MHz, CD3OD): δ 8.19 (s, 1H, H-8), 6.36 (t, J = 7.1 Hz, 1H, H-1′), 4.59 (m, 1H, H-3′), 4.14–4.07 (br m, 2H, pyrrolidinyl NCH), 4.04 (m, 1H, H-4′), 3.84 (dd, J = 3.4, 12.7 Hz, 1H, H-5′), 3.74 (dd, J = 3.4, 12.2 Hz, 1H, H-5′), 3.71–3.62 (br m, 2H, pyrrolidinyl NCH), 2.75 (app quint, Japp ~ 6.7 Hz, 1H, H-2′), 2.40–2.36 (ddd, J = 2.9, 5.8, 13.2 Hz, 1H, H-2′), 2.15–2.06 (br m, 2H, pyrrolidinyl CH), 2.04–1.92 (br m, 2H, pyrrolidinyl CH). 13C-NMR (125 MHz, CD3OD): δ 155.0, 154.7, 151.7, 140.6, 120.8, 89.9, 87.0, 73.0, 63.7, 50.2, 49.6, 41.6, 27.3, 25.2. HRMS (TOF) calcd for C14H19ClN5O3 [M + H]+ 340.1171, found 340.1149.
2-Chloro-6-(piperidin-1-yl)-9-(2-deoxy-β-d-ribofuranosyl)purine (16a): Prepared from compound 9a (70.0 mg, 0.120 mmol) and KF (28.0 mg, 0.481 mmol) in MeOH (1.20 mL). Chromatography on a silica gel column sequentially eluted with EtOAc and 2% MeOH in EtOAc gave 30.0 mg (71% yield) of compound 16a as a white, foamy solid. Rf (SiO2 and 10% MeOH in EtOAc) = 0.46. 1H-NMR (500 MHz, CD3OD): δ 8.15 (s, 1H, H-8), 6.34 (t, J = 6.6 Hz, 1H, H-1′), 4.55 (m, 1H, H-3′), 4.18 (br s, 4H, piperidinyl N(CH2)2), 4.03 (m, 1H, H-4′), 3.83 (dd, J = 2.4, 12.2 Hz, 1H, H-5′), 3.73 (dd, J = 3.4, 12.2 Hz, 1H, H-5′), 2.74 (app quint, Japp ~ 6.8 Hz, 1H, H-2′), 2.40–2.35 (ddd, J = 2.7, 5.9, 13.5 Hz, 1H, H-2′), 1.74 (br m, 2H, piperidinyl CH2), 1.65 (br m, 4H, piperidinyl CH2). 13C-NMR (125 MHz, CD3OD): δ 155.3, 154.9, 152.7, 139.6, 120.5, 89.8, 86.8, 73.0, 63.7, 47.8, 41.6, 27.3, 25.7. HRMS (TOF) calcd for C15H20ClN5O3Na [M + Na]+ 376.1147, found 376.1148.
2-Chloro-6-(morpholin-4-yl)-9-(2-deoxy-β-d-ribofuranosyl)purine (17a): Prepared from compound 10a (80.0 mg, 0.137 mmol) and KF (31.8 mg, 0.548 mmol) in MeOH (1.4 mL). Chromatography on a silica gel column sequentially eluted with 50% EtOAc in hexanes, EtOAc, and 10% MeOH in EtOAc gave 36.1 mg (74% yield) of compound 17a as a white, foamy solid. Rf (SiO2 and 5% MeOH in EtOAc) = 0.21. 1H-NMR (500 MHz, CD3OD): δ 8.22 (s, 1H, H-8), 6.37 (t, J = 7.1 Hz, 1H, H-1′), 4.56 (m, 1H, H-3′), 4.40–4.12 (br m, 4H, morpholinyl N(CH2)2), 4.04 (m, 1H, H-4′), 3.83 (dd, J = 2.9, 12.2 Hz, 1H, H-5′), 3.79 (t, J = 4.9 Hz, 4H, morpholinyl CH2OCH2), 3.74 (dd, J = 3.4, 12.2 Hz, 1H, H-5′), 2.74 (app quint, Japp ~ 6.7 Hz, 1H, H-2′), 2.41–2.37 (ddd, J = 2.9, 5.8, 13.2 Hz, 1H, H-2′). 13C-NMR (125 MHz, CD3OD): δ 155.2, 154.7, 152.7, 140.3, 120.6, 89.8, 86.8, 72.9, 68.0, 63.6, 47.0 (br), 41.5. HRMS (TOF) calcd for C14H19ClN5O4 [M + H]+ 356.1120, found 356.1107.
2-Chloro-6-[(2-dimethylamino)ethyl)(methyl)amino)]-9-(2-deoxy-β-d-ribofuranosyl)purine (18a): Prepared from compound 11a (70.0 mg, 0.117 mmol) and KF (27.0 mg, 0.467 mmol) in MeOH (1.17 mL). Chromatography on a silica gel column sequentially eluted with 10% MeOH in EtOAc and 20% MeOH in EtOAc gave 36.0 mg (84% yield) of compound 18a as a white, foamy solid. Rf (SiO2 and 10% MeOH in EtOAc) = 0.10. 1H-NMR (500 MHz, CD3OD): δ 8.14 (s, 1H, H-8), 6.35 (t, J = 6.9 Hz, 1H, H-1′), 4.56 (m, 1H, H-3′), 4.12 (br s, 2H, NCH2), 4.03 (m, 1H, H-4′), 3.83 (dd, J = 3.4, 12.2 Hz, 1H, H-5′), 3.74 (dd, J = 3.9, 12.2 Hz, 1H, H-5′), 3.48 (br s, 3H, NCH3), 2.76–2.71 (br m, 3H, NCH2 and 1H, H-2′), 2.42–2.41 (m, 1H, H-2′), 2.39 (s, 6H, N(CH3)2). 13C-NMR (125 MHz, CD3OD): δ 156.4, 154.7, 152.5, 140.1, 120.8, 89.8, 86.8, 73.0, 63.7, 57.5, 45.8, 41.6, 37.6, (one broadened resonance could not be identified). HRMS (TOF) calcd for C15H24ClN6O3 [M + H]+ 371.1598, found 371.1584.
3.5. General Procedure for the Desilylation of Ribose Cladribine Analogues
To a 0.1 M solution of the silylated compound in anhydrous MeOH, KF (2 equiv/silyl group) was added. The mixture was heated at 80 °C for 24 h, cooled, and silica gel was added. The mixture was evaporated to dryness and the compound-impregnated silica gel was loaded onto a wet-packed silica gel column. The products were obtained by elution with appropriate solvents (see the individual compound headings for details).
2-Chloroadenosine (12b): Prepared from compound 5b (100 mg, 0.155 mmol) and KF (54.0 mg, 0.93 mmol) in MeOH (1.55 mL). Chromatography on a silica gel column sequentially eluted with 5% MeOH in EtOAc and 10% MeOH in EtOAc gave 35.0 mg (74% yield) of compound 12b as an off-white solid. Rf (SiO2 and 10% MeOH in EtOAc) = 0.13. 1H-NMR (400 MHz, CD3OD): δ 8.27 (s, 1H, H-8), 5.92 (d, J = 6.0 Hz, 1H, H-1′), 4.72 (t, J = 5.6, 1H, H-2′), 4.32 (dd, J = 2.8, 5.2 Hz, 1H, H-3′), 4.14 (m, 1H, H-4′), 3.90 (dd, J = 2.8, 12.4 Hz, 1H, H-5′), 3.76 (dd, J = 2.8, 12.4 Hz, 1H, H-5′). 13C-NMR (100 MHz, DMSO-d6): δ 174.5, 156.7, 152.9, 150.3, 140.0, 118.1, 87.4, 85.7, 73.8, 70.3, 61.3, 25.3. HRMS (TOF) calcd for C10H13ClN5O4 [M + H]+ 302.0651, found 302.0627.
2-Chloro-N6-methyladenosine (13b): Prepared from compound 6b (100 mg, 0.152 mmol) and KF (52.9 mg, 0.912 mmol) in MeOH (1.52 mL). Chromatography on a silica gel column sequentially eluted with 2.5% MeOH in EtOAc and 5% MeOH in EtOAc gave 36.0 mg (75% yield) of compound 13b as a white, foamy solid. Rf (SiO2 and 10% MeOH in EtOAc) = 0.21. 1H-NMR (400 MHz, CD3OD): δ 8.20 (s, 1H, H-8), 5.90 (d, J = 6.0 Hz, 1H, H-1′), 4.68 (t, J = 5.6 Hz, 1H, H-2′), 4.31 (dd, J = 2,8, 4.8 Hz, 1H, H-3′), 4.14 (m, 1H, H-4′), 3.96 (dd, J = 2.4, 12.4 Hz, 1H, H-5′), 3.76 (dd, J = 2.8, 12.4 Hz, 1H, H-5′), 3.07 (br s, 3H, NCH3). 13C-NMR (100 MHz, DMSO-d6): δ 155.5, 153.2, 149.2, 139.7, 118.6, 87.3, 85.6, 73.6, 70.6, 70.3, 61.6, 61.3, 53.9, 27.1. HRMS (TOF) calcd for C11H15ClN5O4 [M + H]+ 316.0807, found 316.0808.
2-Chloro-N6,N6-dimethyladenosine (14b): Prepared from compound 7b (100 mg, 0.148 mmol) and KF (51.8 mg, 0.892 mmol) in MeOH (1.48 mL). Chromatography on a silica gel column sequentially eluted with 50% EtOAc in hexanes, EtOAc, and 10% MeOH in EtOAc gave 36.8 mg (75% yield) of compound 14b as an off-white, foamy solid. Rf (SiO2 and 5% MeOH in EtOAc) = 0.32. 1H-NMR (400 MHz, CD3OD): δ 8.17 (s, 1H, H-8), 5.91 (d, J = 6.4 Hz, 1H, H-1′), 4.67 (t, J = 5.4 Hz, 1H, H-2′), 4.31 (dd, J = 2.8, 4.8 Hz, 1H, H-3′), 4.14 (m, 1H, H-4′), 3.90 (dd, J = 2.8, 12.4 Hz, 1H, H-5′), 3.76 (dd, J = 2.4, 12.4 Hz, 1H, H-5′), 3.60–3.49 (br m, H, N(CH3)2). 13C-NMR (100 MHz, DMSO-d6): δ 154.4, 152.5, 151.1, 138.6, 118.5, 87.2, 85.6, 73.6, 70.2, 61.2, 37.5 (br). HRMS (TOF) calcd for C12H17ClN5O4 [M + H]+ 330.0964, found 330.0964.
2-Chloro-6-(pyrrolidin-1-yl)-9-(β-d-ribofuranosyl)purine (15b): Prepared from compound 8b (100 mg, 0.143 mmol) and KF (49.8 mg, 0.858 mmol) in MeOH (1.43 mL). Chromatography on a silica gel column sequentially eluted with 50% EtOAc in hexanes, EtOAc, and 10% MeOH in EtOAc gave 43.3 mg (85% yield) of compound 15b as a white solid. Rf (SiO2 and 5% MeOH in EtOAc) = 0.38. 1H-NMR (400 MHz, CD3OD): δ 8.18 (s, 1H, H-8), 5.91 (d, J = 6.4 Hz, 1H, H-1′), 4.67 (t, J = 5.4 Hz, 1H, H-2′), 4.32 (dd, J = 2.8, 4.8 Hz, 1H, H-3′), 4.14 (m, 1H, H-4′), 4.13–4.10 (br m, 2H, pyrrolidinyl NCH), 3.90 (dd, J = 2.8, 12.8 Hz, 1H, H-5′), 3.74 (dd, J = 2.4, 12.4 Hz, 1H, H-5′), 3.72–3.65 (br m, 2H, pyrrolidinyl NCH), 2.13–2.07 (br m, 2H, pyrrolidinyl CH), 2.05–1.95 (br m, 2H, pyrrolidinyl CH). 13C-NMR (100 MHz, DMSO-d6): δ 152.7, 152.7, 150.7, 139.1, 118.7, 87.2, 85.6, 73.7, 70.2, 61.2, 48.6, 47.3, 25.6, 23.6. HRMS (TOF) calcd for C14H19ClN5O4 [M + H]+ 356.1120, found 356.1127.
2-Chloro-6-(piperidin-1-yl)-9-(β-d-ribofuranosyl)purine (16b): Prepared from compound 9b (100 mg, 0.140 mmol) and KF (48.9 mg, 0.842 mmol) in MeOH (1.40 mL). Chromatography on a silica gel column sequentially eluted with EtOAc and 2% MeOH in EtOAc gave 43.6 mg (84% yield) of compound 16b as a white, foamy solid. Rf (SiO2 and 10% MeOH in EtOAc) = 0.41. 1H-NMR (400 MHz, CD3OD): δ 8.13 (s, 1H, H-8), 5.87 (d, J = 6.0 Hz, 1H, H-1′), 4.64 (t, J = 5.4 Hz, 1H, H-2′), 4.27 (dd, J = 2.8, 5.2 Hz, 1H, H-3′), 4.19 (br s, 4H, piperidinyl N(CH2)2), 4.11 (m, 1H, H-4′), 3.89 (dd, J = 2.8, 12.2 Hz, 1H, H-5′), 3.73 (dd, J = 2.4, 12.2 Hz, 1H, H-5′), 1.74 (br m, 2H, piperidinyl CH2), 1.64 (br m, 4H, piperidinyl CH2). 13C-NMR (100 MHz, DMSO-d6): δ 153.1, 152.6, 151.4, 138.5, 118.2, 87.2, 85.6, 73.6, 70.4, 70.2, 70.1, 61.2, 44.8 (br), 25.6, 25.2, 23.9, 14.0. HRMS (TOF) calcd for C15H21ClN5O4 [M + H]+ 370.1277, found 370.1302.
2-Chloro-6-(morpholin-4-yl)-9-(β-d-ribofuranosyl)purine (17b): Prepared from compound 10b (100 mg, 0.139 mmol) and KF (48.7 mg, 0.839 mmol) in MeOH (1.40 mL). Chromatography on a silica gel column sequentially eluted with 50% EtOAc in hexanes, EtOAc, and 10% MeOH in EtOAc gave 40.6 mg (78% yield) of compound 17b as a white, foamy solid. Rf (SiO2 and 5% MeOH in EtOAc) = 0.21. 1H-NMR (400 MHz, CD3OD): δ 8.21 (s, 1H, H-8), 5.92 (d, J = 6.0 Hz, 1H, H-1′), 4.66 (t, J = 5.6 Hz, 1H, H-2′), 4.32 (dd, J = 2.8, 4.8 Hz, 1H, H-3′), 4.30–4.24 (br m, 4H, morpholinyl N(CH2)2), 4.14 (m, 1H, H-4′), 3.90 (dd, J = 2.8, 12.4 Hz, 1H, H-5′), 3.80 (t, J = 4.8 Hz, 4H, morpholinyl CH2OCH2), 3.76 (dd, J = 2.8, 12.4 Hz, 1H, H-5′). 13C-NMR (100 MHz, DMSO-d6): δ 153.3, 152.5, 151.6, 139.0, 118.3, 87.3, 85.6, 85.4, 73.7, 70.4, 70.2, 66.0, 61.4, 61.1, 48.5, 45.0 (br). HRMS (TOF) calcd for C14H19ClN5O5 [M + H]+ 372.1069, found 372.1056.
2-Chloro-6-[(2-dimethylamino)ethyl)(methyl)amino)]-9-(β-d-ribofuranosyl)purine (18b): Prepared from compound 11b (100 mg, 0.137 mmol) and KF (47.7 mg, 0.822 mmol) in MeOH (1.37 mL). Chromatography on a silica gel column sequentially eluted with 10% MeOH in EtOAc and 20% MeOH in EtOAc gave 34.5 mg (65% yield) of compound 18b as a white, foamy solid. Rf (SiO2 and 15% MeOH in EtOAc) = 0.10. 1H-NMR (400 MHz, CD3OD): δ 8.19 (s, 1H, H-8), 5.92 (d, J = 6 Hz, 1H, H-1′), 4.67 (t, 1H, J = 5.6 Hz, 1H, H-2′), 4.34 (dd, J = 3.2, 5.2 Hz 1H, H-3′), 4.14 (d, J = 2.4 Hz, 3H, H-4′), 3.90 (dd, J = 2.4, 12.4 Hz, 1H, H-5′), 3.76 (dd, J = 2.8, 12.4 Hz, 1H, H-5′), 3.49 (br s, 3H, NCH3), 2.83 (t, 2H, NCH2, J = 6.8 Hz ), 2.47 (s, 6H, N(CH3)2). 13C-NMR (100 MHz, CD3OD): δ 156.1, 154.7, 152.3, 140.5, 120.7, 90.8, 87.7, 75.3, 72.3, 63.2, 57.0, 49.6, 49.4, 49.2, 49.0, 48.7, 48.5, 48.3, 45.5, 37.4. HRMS (TOF) calcd for C15H24ClN6O4 [M + H]+ 387.1542, found 387.1513.
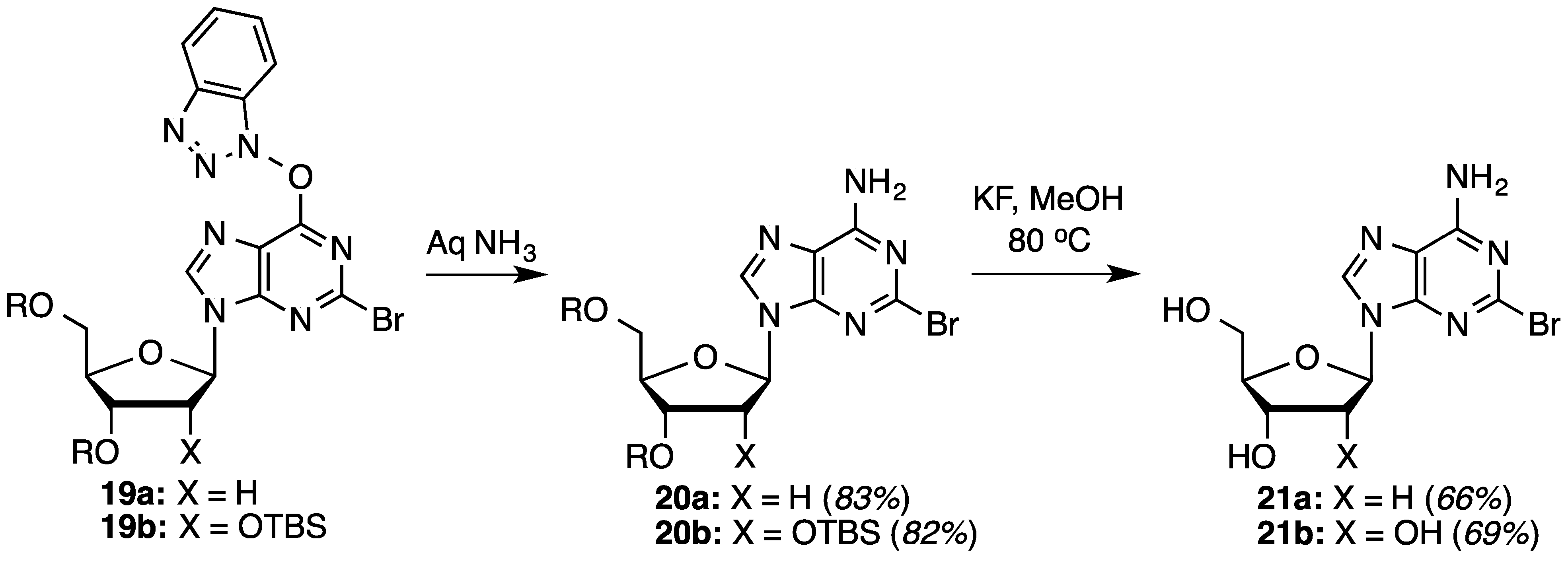
O6-(Benzotriazol-1-yl)-2-bromo-9-[2-deoxy-3,5-di-O-(t-butyldimethylsilyl)-β-d-ribofuranosyl]purine (19a): A mixture of compound 3a (300.0 mg, 0.489 mmol) and SbBr3 (247.7 mg, 0.685 mmol) in dry CH2Br2 (4.9 mL) was cooled to −15 °C using dry ice and acetone, in a nitrogen atmosphere. t-BuONO (203.8 μL, 1.713 mmol) was added dropwise and the mixture was stirred at −10 °C to −15 °C for 2 h. Because TLC indicated the presence of starting material, another aliquot of t-BuONO (203.8 μL, 1.713 mmol) was added and the reaction proceeded for 1 h at −15 °C, at which time TLC indicated the reaction to be complete. The reaction mixture was poured into ice-cold, saturated aqueous NaHCO3 (5 mL) with stirring. The mixture was filtered using vacuum (note: use of vacuum for this filtration is critical for maximizing product recovery) and the residue was washed with CH2Cl2 (5 mL). The organic layer was separated and the aqueous layer was back extracted with CH2Cl2 (2 × 5 mL). The combined organic layer was washed with water (5 mL) and brine (5 mL), dried over anhydrous Na2SO4, and evaporated under reduced pressure. Purification of the crude material on a silica gel column sequentially eluted with hexanes, 5% EtOAc in hexanes, and 30% EtOAc in hexanes gave 208.5 mg (63% yield) of compound 19a as a white foam. Rf (SiO2 and 30% EtOAc in hexanes) = 0.60. 1H-NMR (500 MHz, CDCl3): δ 8.45 (s, 1H, H-8), 8.13 (d, J = 8.3 Hz, 1H, Ar-H), 7.56–7.45 (m, 3H, Ar-H), 6.47 (t, J = 5.9 Hz, 1H, H-1′), 4.63 (m, 1H, H-3′), 4.03 (m, 1H, H-4′), 3.90 (dd, J = 3.4, 11.2 Hz, 1H, H-5′), 3.78 (dd, J = 1.5, 11.2 Hz, 1H, H-5′), 2.61 (app quint, Japp ~ 6.2 Hz, 1H, H-2′), 2.49 (app quint, Japp ~ 5.8 Hz, 1H, H-2′), 0.91 (s, 18H, t-Bu), 0.11 and 0.09 (2s, 12H, SiCH3). 13C-NMR (125 MHz, CDCl3): δ 158.7, 154.9, 144.3, 143.6, 142.6, 129.1, 128.9, 125.1, 120.8, 119.5, 108.7, 88.5, 85.4, 71.7, 62.7, 41.8, 26.2, 25.9, 18.6, 18.2, −4.4, −4.6, −5.2, −5.3. HRMS (TOF) calcd for C28H43BrN7O4Si2 [M + H]+ 676.2093, found 676.2078.
2-Bromo-3′,5′-di-O-(t-butyldimethylsilyl)-2′-deoxyadenosine (20a): To a solution of compound 19a (135.3 mg, 0.20 mmol) in 1,2-DME (2 mL), 28%–30% aqueous ammonia (48.6 μL) was added, and the mixture was stirred at room temperature for 45 min. The mixture was diluted with EtOAc (15 mL) and washed with 5% aqueous NaCl (10 mL). The organic layer was separated and the aqueous layer was back extracted with EtOAc (15 mL). The combined organic layer was dried over anhydrous Na2SO4 and evaporated under reduced pressure. The crude material was chromatographed on a silica gel column by sequential elution with hexanes, 20% EtOAc in hexanes and 40% EtOAc in hexanes to afford 93.1 mg (83% yield) of compound 20a as a white foam. Rf (SiO2 and EtOAc) = 0.50. 1H-NMR (500 MHz, CDCl3): δ 8.08 (s, 1H, H-8), 6.63 (br s, 2H, Ar-NH2), 6.37 (t, J = 6.1 Hz, 1H, H-1′), 4.61 (m, 1H, H-3′), 3.98 (m, 1H, H-4′), 3.88 (dd, J = 3.9, 11.2 Hz, 1H, H-5′), 3.75 (dd, J = 2.4, 11.2 Hz, 1H, H-5′), 2.61 (app quint, Japp ~ 6.3 Hz, 1H, H-2′), 2.43–2.38 (m, 1H, H-2′), 0.90 (s, 18H, t-Bu), 0.09 and 0.08 (2s, 12H, SiCH3). 13C-NMR (125 MHz, CDCl3): δ 156.3, 150.4, 144.9, 139.6, 119.3, 88.2, 84.7, 71.9, 62.9, 41.4, 26.1, 25.9, 18.6, 18.2, −4.4, −4.6, −5.2, −5.3. HRMS (TOF) calcd for C22H41BrN5O3Si2 [M + H]+ 558.1926, found 558.1902.
2-Bromo-2′-deoxyadenosine (21a): As described in the general desilylation procedures, compound 21a was prepared from compound 20a (80.0 mg, 0.143 mmol) and KF (33.2 mg, 0.572 mmol) in MeOH (1.4 mL), over 28 h. Chromatography on a silica gel column sequentially eluted with 50% EtOAc in hexanes, EtOAc, and 10% MeOH in EtOAc gave 31.1 mg (66% yield) of compound 21a as a white/off-white solid. Rf (SiO2 and 10% MeOH in EtOAc) = 0.40. 1H-NMR (500 MHz, CD3OD): δ 8.25 (s, 1H, H-8), 6.36 (t, J = 6.8 Hz, 1H, H-1′), 4.57 (m, 1H, H-3′), 4.04 (m, 1H, H-4′), 3.84 (dd, J = 2.9, 12.2 Hz, 1H, H-5′), 3.74 (dd, J = 3.4, 12.2 Hz, 1H, H-5′), 2.76 (app quint, Japp ~ 6.7 Hz, 1H, H-2′), 2.43–2.39 (ddd, J = 2.9, 5.9, 13.2 Hz, 1H, H-2′). 13C-NMR (125 MHz, CD3OD): δ 158.1, 151.4, 145.9, 141.7, 120.3, 89.9, 86.9, 72.9, 63.7, 41.6. HRMS (TOF) calcd for C10H12BrN5O3Na [M + Na]+ 352.0016, found 352.0021.
O6-(Benzotriazol-1-yl)-2-bromo-9-[2,3,5-tri-O-(t-butyldimethylsilyl)-β-d-ribofuranosyl]purine (19b): A mixture of compound 3b (500.0 mg, 0.67 mmol) and SbBr3 (340.8 mg, 0.94 mmol) in dry CH2Br2 (8.3 mL) was cooled to −15 °C using dry ice and acetone, in a nitrogen atmosphere. t-BuONO (280 μL, 2.352 mmol) was added dropwise and the mixture was stirred at −10 °C to −15 °C for 2 h. Because TLC indicated the presence of starting material, another aliquot of t-BuONO (280 μL, 2.352 mmol) was added and the reaction was allowed to progress for 1 h at −15 °C, at which time TLC indicated the reaction to be complete. The reaction mixture was poured into ice-cold, saturated aqueous NaHCO3 (10 mL) with stirring. The mixture was filtered using vacuum (note: use of vacuum for this filtration is critical for maximizing product recovery) and the residue was washed with CH2Cl2 (15 mL). The organic layer was separated and the aqueous layer was back extracted with CH2Cl2 (2 × 15 mL). The combined organic layer was washed with water (10 mL) and brine (10 mL), dried over anhydrous Na2SO4, and evaporated under reduced pressure. Purification of the crude material on a silica gel column sequentially eluted with hexanes, 5% EtOAc in hexanes, and 30% EtOAc in hexanes gave 350 mg (64% yield) of compound 19b as a white foam. Rf (SiO2 and 30% EtOAc in hexanes) = 0.80. 1H-NMR (400 MHz, CDCl3): δ 8.54 (s, 1H, H-8), 8.15 (d, J = 8.4 Hz, 1H, Ar-H), 7.58–7.45 (m, 3H, Ar-H), 6.05 (d, J = 4.0 Hz, 1H, H-1′), 4.57 (t, J = 4.2 Hz, 1H, H-2′), 4.33 (t, J = 4.6 Hz, 1H, H-3′), 4.17 (m, 1H, H-4′), 4.08 (dd, J = 3.6, 11.6 Hz, 1H, H-5′), 3.83 (dd, J = 2.4, 11.6 Hz, 1H, H-5′), 0.96, 0.93, and 0.85 (3s, 27H, t-Bu), 0.16, 0.11, −0.03, and −0.09 (4s, 18H, SiCH3). 13C-NMR (100 MHz, CDCl3): δ 158.5, 154.8, 144.2, 143.4, 142.5, 128.8, 128.7, 124.8, 120.6, 119.4, 108.5, 89.5, 85.2, 76.1, 71.1, 61.9, 29.6, 26.1, 25.8, 25.6, 18.5, 18.0, 17.9, −4.2, −4.6, −4.7, −4.9, −5.3, −5.4. HRMS (TOF) calcd for C34H57BrN7O5Si3 [M + H]+ 806.2907, found 806.2912.
2-Bromo-2′,3′,5′-tri-O-(t-butyldimethylsilyl)-adenosine (20b): To a solution of compound 19b (200 mg, 0.247 mmol) in 1,2-DME (4 mL), 28%–30% aqueous ammonia (60 μL) was added, and the mixture was stirred at room temperature for 45 min. The mixture was diluted with EtOAc (20 mL) and washed with 5% aqueous NaCl (20 mL). The organic layer was separated and the aqueous layer was back extracted with EtOAc (25 mL). The combined organic layer was dried over anhydrous Na2SO4 and evaporated under reduced pressure. The crude material was chromatographed on a silica gel column by sequential elution with hexanes, 20% EtOAc in hexanes, and 40% EtOAc in hexanes to afford 140 mg (82% yield) of compound 20b as a white foam. Rf (SiO2 and EtOAc) = 0.55. 1H-NMR (400 MHz, CDCl3): δ 8.10 (s, 1H, H-8), 5.92 (d, J = 4.8 Hz, 1H, H-1′), 5.77 (br s, 2H, Ar-NH2), 4.69 (t, J = 4.6 Hz, 1H, H-2′), 4.32 (t, J = 4.2 Hz, 1H, H-3′), 4.13 (m, 1H, H-4′), 4.06 (dd, J = 4.8, 11.2 Hz, 1H, H-5′), 3.80 (dd, J = 2.8, 11.2 Hz, 1H, H-5′), 0.95, 0.93, and 0.83 (3s, 27H, t-Bu), 0.14, 0.11, −0.00, and −0.15 (4s, 18H, SiCH3). 13C-NMR (100 MHz, CDCl3): δ 155.8, 150.5, 144.6, 140.0, 119.4, 110.0, 89.0, 85.3, 75.4, 71.7, 62.3, 26.0, 25.8, 25.7, 18.4, 18.0, 17.9, −4.3, −4.7, −5.0, −5.3, −5.4. HRMS (TOF) calcd for C28H55BrN5O4Si3 [M + H]+ 688.2740, found 688.2728.
2-Bromoadenosine (21b): As described in the general desilylation procedures, compound 21b was prepared from compound 20b (100 mg, 0.145 mmol) and KF (50.5 mg, 0.870 mmol) in MeOH (1.9 mL), over 24 h. Chromatography on a silica gel column sequentially eluted with 50% EtOAc in hexanes, EtOAc, and 10% MeOH in EtOAc which gave 34.9 mg (69% yield) of compound 21b as a pale yellow solid. Rf (SiO2 and 10% MeOH in EtOAc) = 0.39. 1H-NMR (400 MHz, CD3OD): δ 8.25 (s, 1H, H-8), 5.92 (d, J = 6.4 Hz, 1H, H-1′), 4.67 (d, J = 5.6 Hz, 1H, H-2′), 4.32 (dd, J = 2.8, 4.8 Hz, 1H, H-3′), 4.15 (m, 1H, H-4′), 3.90 (dd, J = 2.8, 12.4 Hz, 1H, H-5′), 3.76 (dd, J = 2.8, 12.4 Hz, 1H, H-5′). 13C-NMR (100 MHz, CD3OD): δ 156.5, 150.2, 144.1, 139.8, 118.4, 87.2, 85.7, 73.6, 70.3, 61.3. HRMS (TOF) calcd for C10H13BrN5O4 [M + H]+ 346.0145, found 346.0154.
3.6. Protocols for Tests against HCL, TCL, and CCL
Blood was obtained in sodium heparin tubes from patients as part of protocols with consent forms approved by the investigators review board (IRB) of the National Cancer Institute. The blood was diluted 1:1 with PBS without calcium or magnesium, layered over 15 mL Ficoll in 50 mL tubes, and centrifuged to obtain mononuclear cells. Patients with high lymphocytosis had leukemic cells >80%–90% pure after Ficoll. The cells were viably frozen in 7.5% DMSO in leucine-poor media (LPM, 88% leucine-free RPMI, 2% RPMI, and 10% FBS) in cryovials and stored under liquid nitrogen. LPM also contained penicillin, streptomycin, glutamine, gentamycin, and doxycycline. To assay, thawed cells were washed, suspended in LPM, added to 96-well round-bottom plates (15 μL/well), and treated with 15 μL of purine analogues diluted in LPM. The aliquots were incubated 3 days, then treated with 10 μL of either ATP (CellTiter-Glo, Promega, Madison, WI, USA) or {3H}-leucine (Perkin-Elmer, Waltham, MA, USA) diluted in leucine-free RPMI. After 30 min of ATP, the plate is read for bioluminescence. After 6 h of {3H}-leucine, the cells were liberated by freeze-thaw, harvested on to glass-fiber filters, counted either by a beta scintillation counter. The number of cells cultured in 30 μL aliquots for ATP assay was 20,000, 20,000, and 100,000 for HCL, TCL, and CLL, respectively. The cell number for {3H}-leucine assay was 60,000, 60,000 and 200,000 for HCL, TCL, and CLL, respectively. HCL and TCL cells were pulsed with 1 μCi of {3H}-leucine, while CLL cells were pulsed with 1.5–2 μCi/well. The IC50 was the calculated concentration needed for 50% inhibition, defined as the ATP uptake or {3H}-leucine incorporation corresponding to halfway between that of control (cells with LPM alone) and that of cycloheximide 10 μg/mL. Reported IC50 values were the means of 3 triplicate experiments.
3.7. Protocols for Tests against Breast Cancer Cells
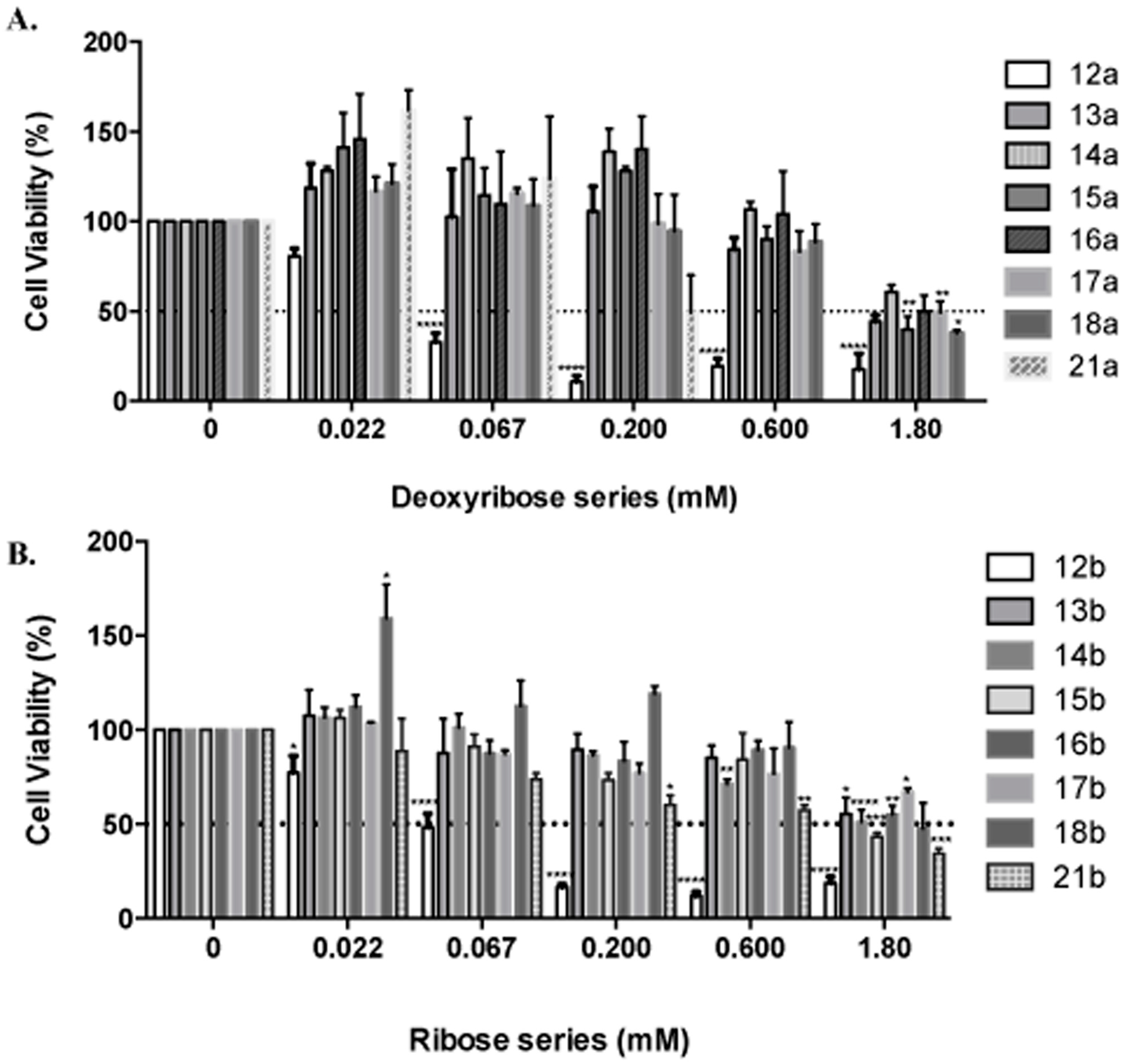
MDA-MB-231 breast cancer (BC) cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA), and were cultured at 37 °C in 5% CO
2 using culture medium recommended by the supplier. Each tested compound was dissolved in sterile DMSO, then diluted (4.8–57.7 mM) in sterile 1X PBS for a final DMSO concentration of 0.1%. Then, 2 × 10
5 cells/well, were seeded and cultured for 24 h as described [
51,
52]. Afterwards, the media was changed to 5% FBS for 1 h and cells were treated in duplicate with dilutions of each treatment (0–1.8 mM) for 24 h. Cells were fixed (cold methanol), and nuclei stained (0.4% propidium iodide, (PI)) (Sigma-Aldrich, St Louis, MO, USA), and measured using a GloMax
® Microplate Reader (Promega, Madison, WI, USA). Cell viability was calculated as percent of surviving cells after treatment relative to vehicle wells. The IC
50 was obtained from dose response curve fittings using the non-linear regression function of GraphPad Prism
® version 6.0b for Mac (GraphPad Software, San Diego, CA, USA) [
53].














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

