
Analysis and Ranking of Protein-Protein Docking Models Using Inter-Residue Contacts and Inter-Molecular Contact Maps

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
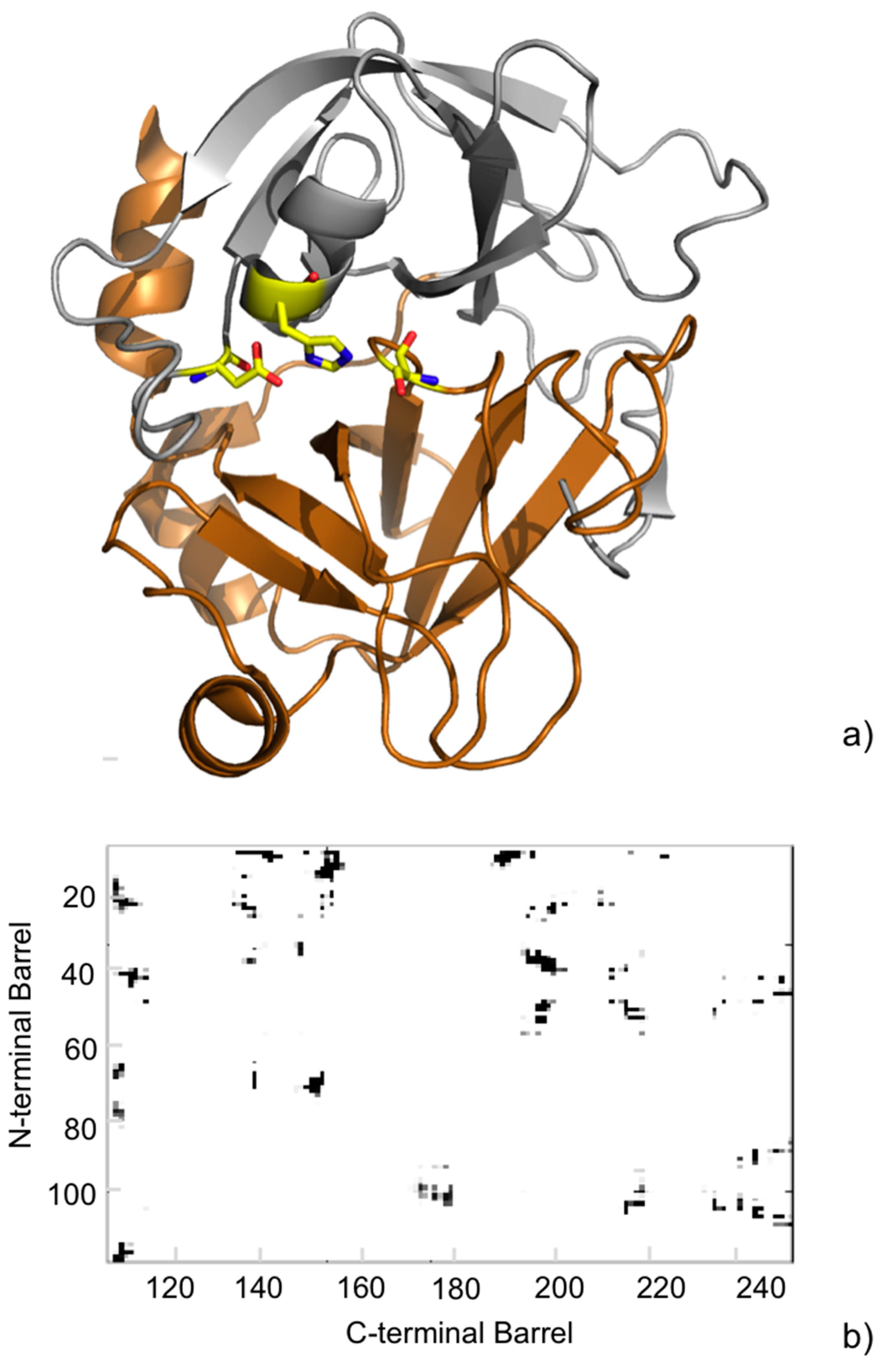
2. Intermolecular Contact Maps to Represent the Interface in Protein-Protein Complexes



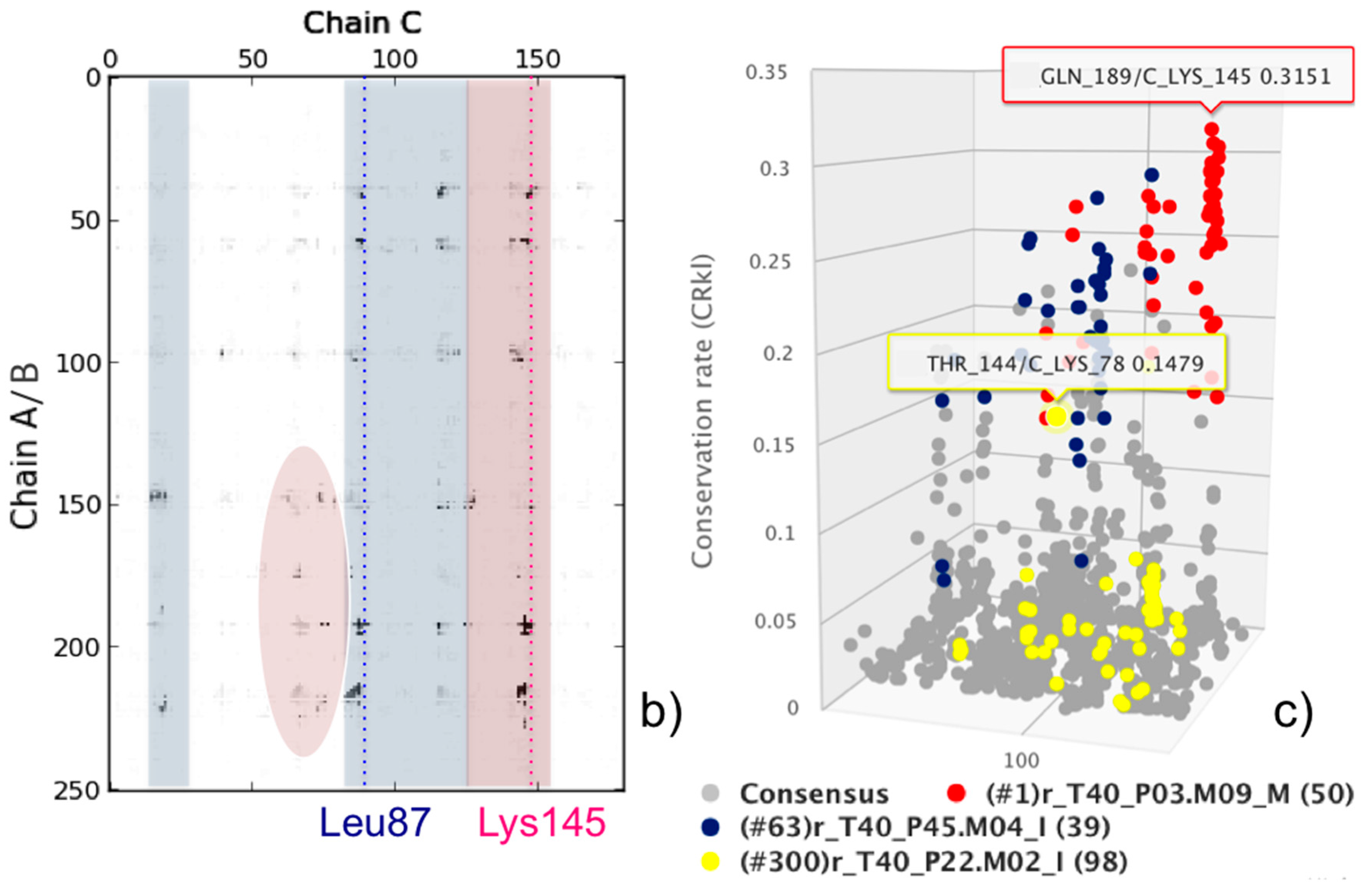
3. A Consensus Approach to Analyze and Select Docking Models in a Real-Life Research Case

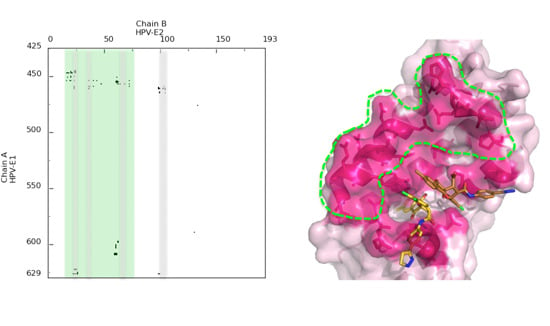
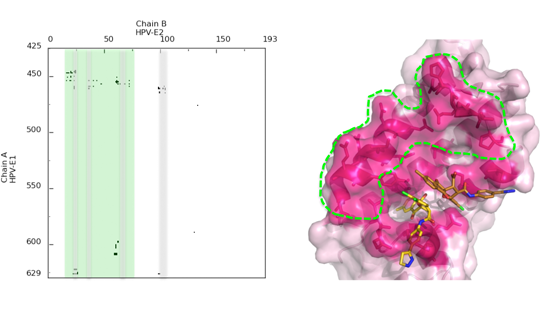
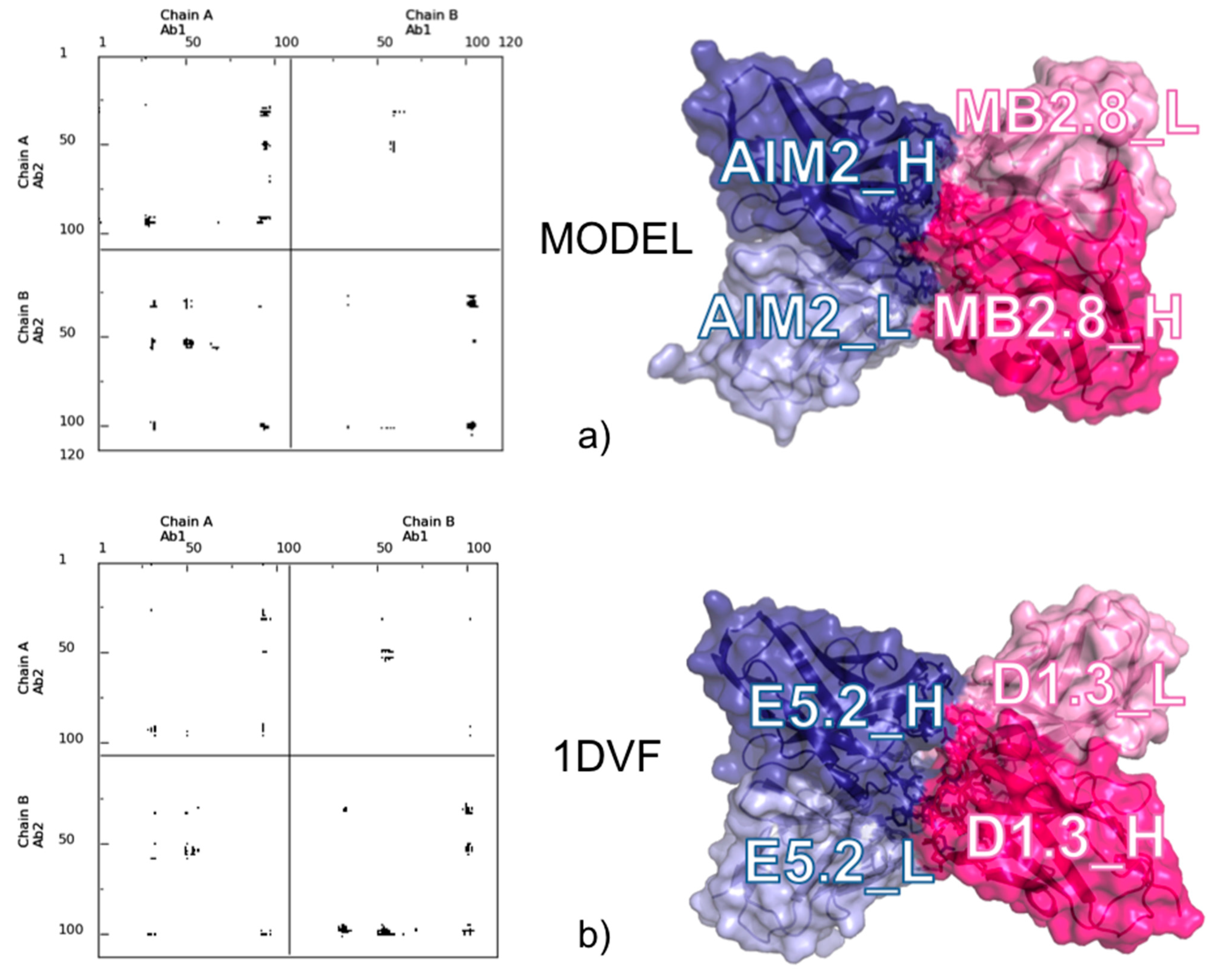
4. A Dynamic View of the Interface


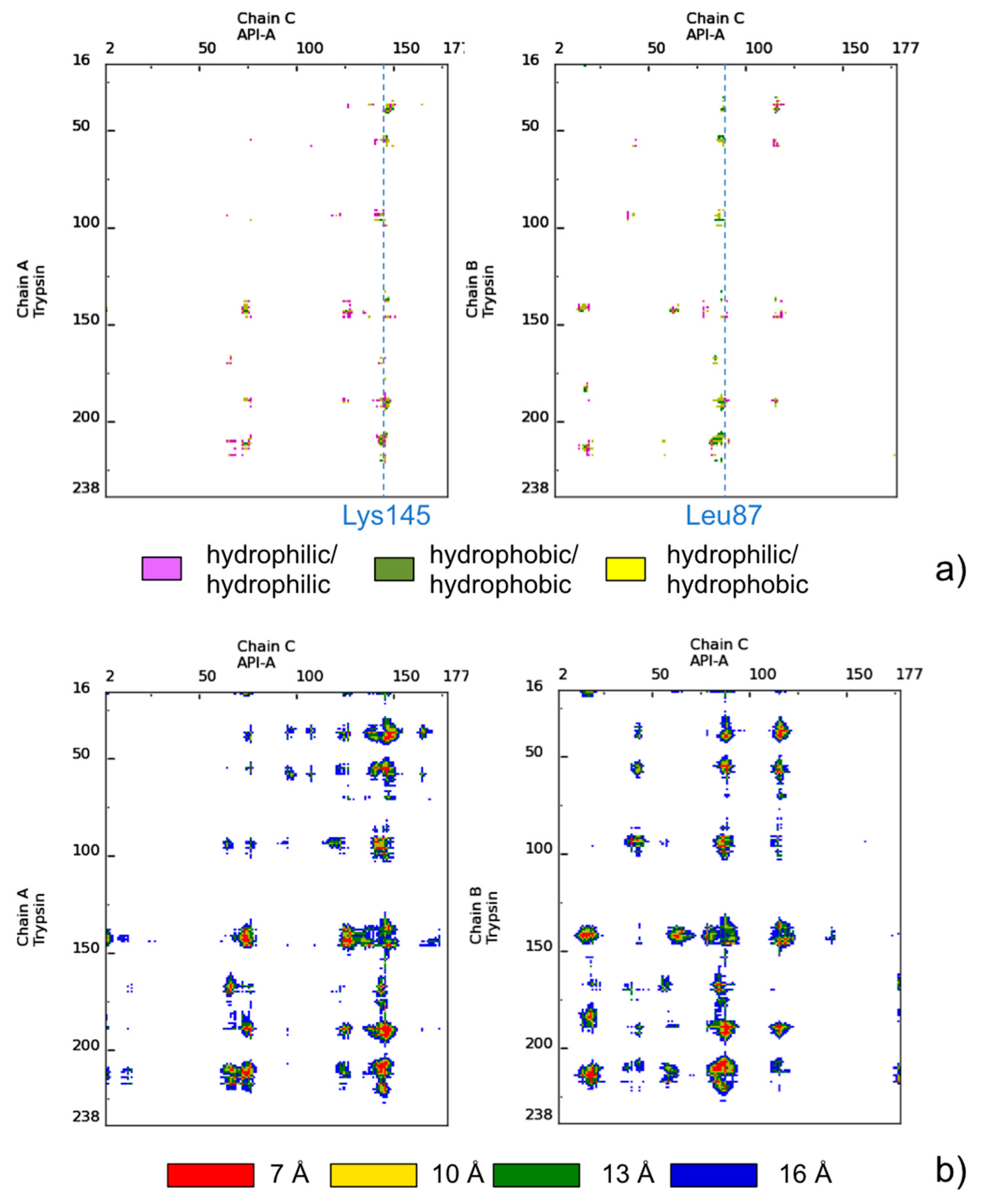
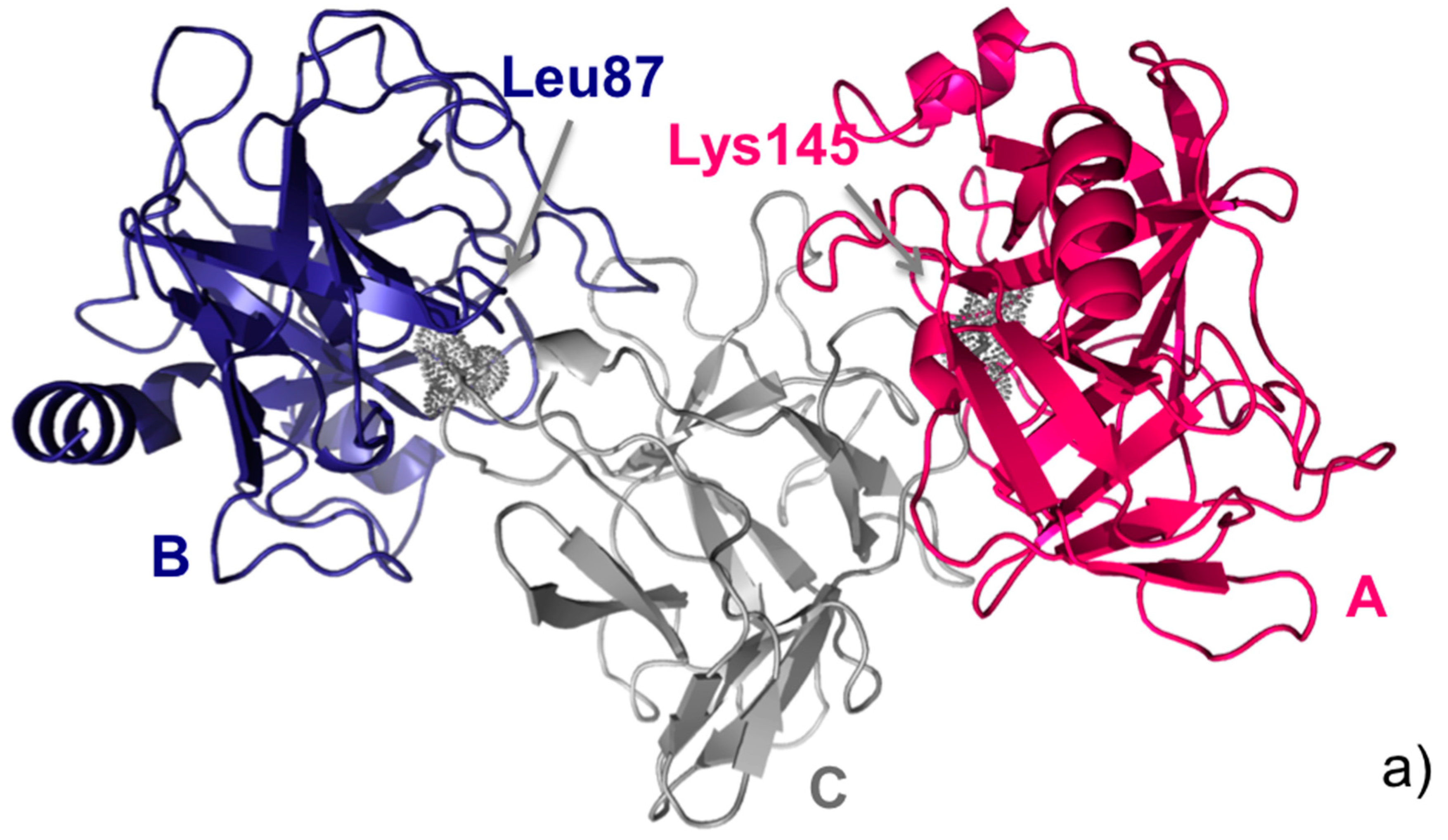
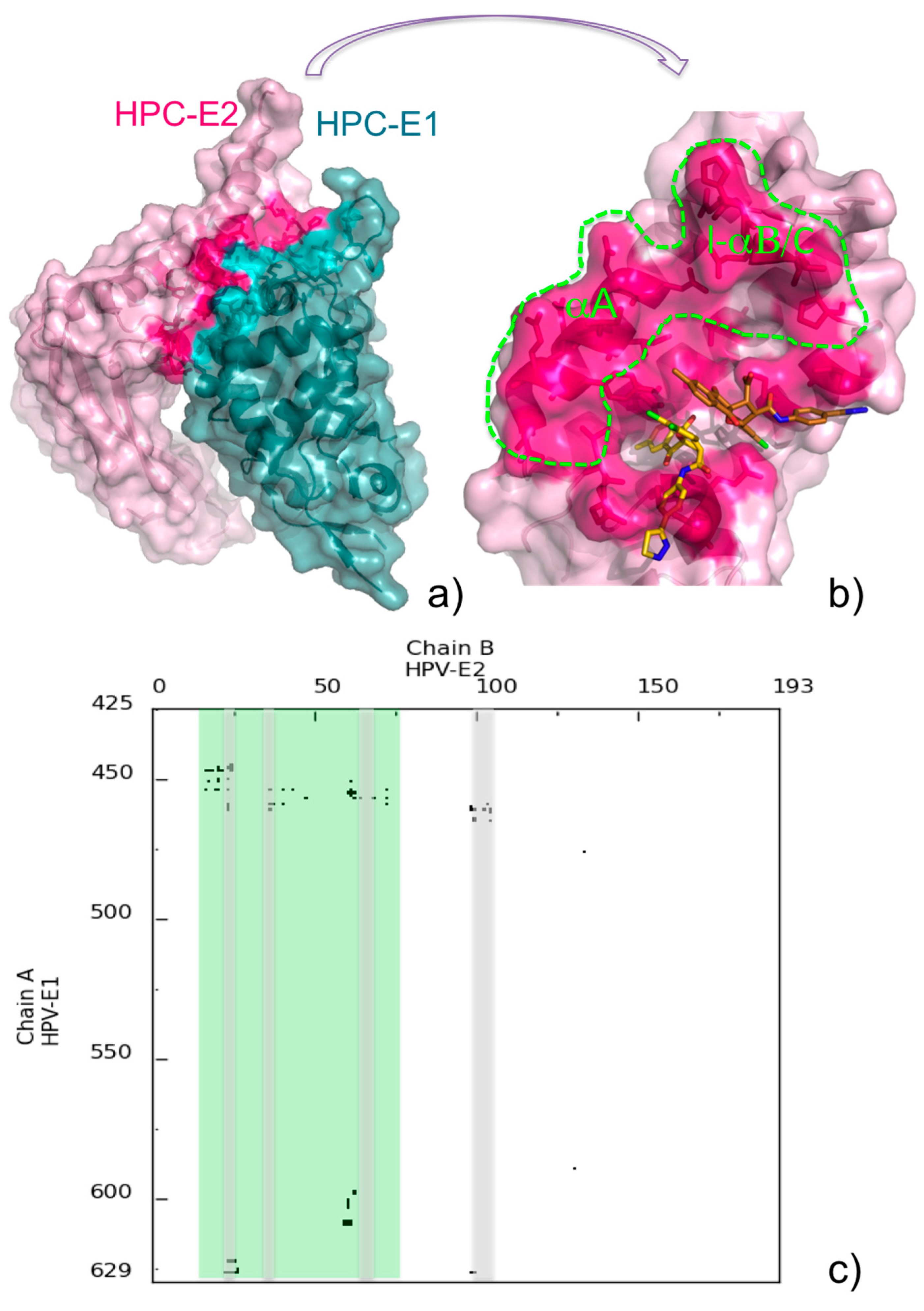
5. Assisting Rational Drug Design Targeting Protein-Protein Interactions

6. Conclusions and Outlook
7. Technical Details
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Metz, A.; Ciglia, E.; Gohlke, H. Modulating protein-protein interactions: From structural determinants of binding to druggability prediction to application. Curr. Pharm. Des. 2012, 18, 4630–4647. [Google Scholar] [CrossRef]
- Gonzalez-Ruiz, D.; Gohlke, H. Targeting protein-protein interactions with small molecules: Challenges and perspectives for computational binding epitope detection and ligand finding. Curr. Med. Chem. 2006, 13, 2607–2625. [Google Scholar] [CrossRef] [PubMed]
- Nisius, B.; Sha, F.; Gohlke, H. Structure-based computational analysis of protein binding sites for function and druggability prediction. J. Biotechnol. 2012, 159, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Vidal, M.; Cusick, M.E.; Barabasi, A.L. Interactome networks and human disease. Cell 2011, 144, 986–998. [Google Scholar] [CrossRef] [PubMed]
- Mullard, A. Protein-protein interaction inhibitors get into the groove. Nat. Rev. Drug Discov. 2012, 11, 173–175. [Google Scholar] [CrossRef] [PubMed]
- Reynes, C.; Host, H.; Camproux, A.C.; Laconde, G.; Leroux, F.; Mazars, A.; Deprez, B.; Fahraeus, R.; Villoutreix, B.O.; Sperandio, O. Designing Focused Chemical Libraries Enriched in Protein-Protein Interaction Inhibitors using Machine-Learning Methods. PLoS Comput. Biol. 2010, 6, e1000695. [Google Scholar] [CrossRef] [PubMed]
- Hwang, I.; Park, S. Computational design of protein therapeutics. Drug Discov. Today Technol. 2008, 5, e43–e48. [Google Scholar] [CrossRef] [PubMed]
- Szymkowski, D.E. Creating the next generation of protein therapeutics through rational drug design. Curr. Opin. Drug Discov. Dev. 2005, 8, 590–600. [Google Scholar]
- Zhou, P.; Wang, C.; Ren, Y.; Yang, C.; Tian, F. Computational peptidology: A new and promising approach to therapeutic peptide design. Curr. Med. Chem. 2013, 20, 1985–1996. [Google Scholar] [CrossRef] [PubMed]
- Kijanka, M.; Dorresteijn, B.; Oliveira, S.; van Bergen en Henegouwen, P.M. Nanobody-based cancer therapy of solid tumors. Nanomedicine 2015, 10, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Janin, J. Protein-protein docking tested in blind predictions: The CAPRI experiment. Mol. Biosyst. 2010, 6, 2351–2362. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.Y. Exploring the potential of global protein-protein docking: An overview and critical assessment of current programs for automatic ab initio docking. Drug Discov. Today 2015. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.Y. Search strategies and evaluation in protein-protein docking: Principles, advances and challenges. Drug Discov. Today 2014, 19, 1081–1096. [Google Scholar] [CrossRef] [PubMed]
- Moal, I.H.; Moretti, R.; Baker, D.; Fernandez-Recio, J. Scoring functions for protein-protein interactions. Curr. Opin. Struct. Biol. 2013, 23, 862–867. [Google Scholar] [CrossRef] [PubMed]
- Moal, I.H.; Torchala, M.; Bates, P.A.; Fernandez-Recio, J. The scoring of poses in protein-protein docking: Current capabilities and future directions. BMC Bioinform. 2013, 14, 286. [Google Scholar] [CrossRef] [PubMed]
- Vangone, A.; Spinelli, R.; Scarano, V.; Cavallo, L.; Oliva, R. COCOMAPS: A web application to analyse and visualize contacts at the interface of biomolecular complexes. Bioinformatics 2011, 27, 2915–2916. [Google Scholar] [CrossRef] [PubMed]
- Holm, L.; Sander, C. Mapping the protein universe. Science 1996, 273, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Pulim, V.; Berger, B.; Bienkowska, J. Optimal contact map alignment of protein-protein interfaces. Bioinformatics 2008, 24, 2324–2328. [Google Scholar] [CrossRef] [PubMed]
- Vangone, A.; Oliva, R.; Cavallo, L. CONS-COCOMAPS: A novel tool to measure and visualize the conservation of inter-residue contacts in multiple docking solutions. BMC Bioinform. 2012, 13 (Suppl. 4), S19. [Google Scholar] [CrossRef] [PubMed]
- Oliva, R.; Vangone, A.; Cavallo, L. Ranking multiple docking solutions based on the conservation of inter-residue contacts. Proteins 2013, 81, 1571–1584. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.M.; Blundell, T.L.; Fernandez-Recio, J. pyDock: Electrostatics and desolvation for effective scoring of rigid-body protein-protein docking. Proteins 2007, 68, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Gong, X.; Wang, P.; Yang, F.; Chang, S.; Liu, B.; He, H.; Cao, L.; Xu, X.; Li, C.; Chen, W.; et al. Protein-protein docking with binding site patch prediction and network-based terms enhanced combinatorial scoring. Proteins 2010, 78, 3150–3155. [Google Scholar] [CrossRef] [PubMed]
- Gray, J.J.; Moughon, S.; Wang, C.; Schueler-Furman, O.; Kuhlman, B.; Rohl, C.A.; Baker, D. Protein-protein docking with simultaneous optimization of rigid-body displacement and side-chain conformations. J. Mol. Biol. 2003, 331, 281–299. [Google Scholar] [CrossRef]
- Comeau, S.R.; Gatchell, D.W.; Vajda, S.; Camacho, C.J. ClusPro: An automated docking and discrimination method for the prediction of protein complexes. Bioinformatics 2004, 20, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Pierce, B.; Weng, Z. ZRANK: Reranking protein docking predictions with an optimized energy function. Proteins 2007, 67, 1078–1086. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.Y.; Zou, X. An iterative knowledge-based scoring function for protein-protein recognition. Proteins 2008, 72, 557–579. [Google Scholar] [CrossRef] [PubMed]
- Khashan, R.; Zheng, W.; Tropsha, A. Scoring protein interaction decoys using exposed residues (SPIDER): A novel multibody interaction scoring function based on frequent geometric patterns of interfacial residues. Proteins 2012, 80, 2207–2217. [Google Scholar] [CrossRef] [PubMed]
- Kozakov, D.; Schueler-Furman, O.; Vajda, S. Discrimination of near-native structures in protein-protein docking by testing the stability of local minima. Proteins 2008, 72, 993–1004. [Google Scholar] [CrossRef] [PubMed]
- Venkatraman, V.; Yang, Y.D.; Sael, L.; Kihara, D. Protein-protein docking using region-based 3D Zernike descriptors. BMC Bioinform. 2009, 10, 407. [Google Scholar] [CrossRef] [PubMed]
- Champ, P.C.; Camacho, C.J. FastContact: A free energy scoring tool for protein-protein complex structures. Nucleic Acids Res. 2007, 35, W556–W560. [Google Scholar] [CrossRef] [PubMed]
- Andrusier, N.; Nussinov, R.; Wolfson, H.J. FireDock: Fast interaction refinement in molecular docking. Proteins 2007, 69, 139–159. [Google Scholar] [CrossRef] [PubMed]
- Torchala, M.; Moal, I.H.; Chaleil, R.A.; Agius, R.; Bates, P.A. A Markov-chain model description of binding funnels to enhance the ranking of docked solutions. Proteins 2013, 81, 2143–2149. [Google Scholar] [CrossRef] [PubMed]
- De Vries, S.J.; van Dijk, A.D.; Krzeminski, M.; van Dijk, M.; Thureau, A.; Hsu, V.; Wassenaar, T.; Bonvin, A.M. HADDOCK versus HADDOCK: New features and performance of HADDOCK2.0 on the CAPRI targets. Proteins 2007, 69, 726–733. [Google Scholar] [CrossRef] [PubMed]
- Moreira, I.S.; Martins, J.M.; Coimbra, J.T.; Ramos, M.J.; Fernandes, P.A. A new scoring function for protein-protein docking that identifies native structures with unprecedented accuracy. Phys. Chem. Chem. Phys. PCCP 2015, 17, 2378–2387. [Google Scholar] [CrossRef] [PubMed]
- Vangone, A.; Cavallo, L.; Oliva, R. Using a consensus approach based on the conservation of inter-residue contacts to rank CAPRI models. Proteins 2013, 81, 2210–2220. [Google Scholar] [CrossRef] [PubMed]
- Chermak, E.; Petta, A.; Serra, L.; Vangone, A.; Scarano, V.; Cavallo, L.; Oliva, R. CONSRANK: A server for the analysis, comparison and ranking of docking models based on inter-residue contacts. Bioinformatics 2015, 31, 1481–1483. [Google Scholar] [CrossRef] [PubMed]
- Bao, R.; Zhou, C.Z.; Jiang, C.; Lin, S.X.; Chi, C.W.; Chen, Y. The ternary structure of the double-headed arrowhead protease inhibitor API-A complexed with two trypsins reveals a novel reactive site conformation. J. Biol. Chem. 2009, 284, 26676–26684. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ruan, K.C.; Chi, C.W. The assignment of the reactive sites of the double-headed arrowhead proteinase inhibitor A and B. Acta Biochim. Biophys. Sin. 2002, 34, 662–666. [Google Scholar] [PubMed]
- Vangone, A.; Abdel-Azeim, S.; Caputo, I.; Sblattero, D.; di Niro, R.; Cavallo, L.; Oliva, R. Structural basis for the recognition in an idiotype-anti-idiotype antibody complex related to celiac disease. PLoS ONE 2014, 9, e102839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braden, B.C.; Fields, B.A.; Ysern, X.; Dall’Acqua, W.; Goldbaum, F.A.; Poljak, R.J.; Mariuzza, R.A. Crystal structure of an Fv-Fv idiotope-anti-idiotope complex at 1.9 A resolution. J. Mol. Biol. 1996, 264, 137–151. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Azeim, S.; Chermak, E.; Vangone, A.; Oliva, R.; Cavallo, L. MDcons: Intermolecular contact maps as a tool to analyze the interface of protein complexes from molecular dynamics trajectories. BMC Bioinform. 2014, 15 (Suppl. 5), S1. [Google Scholar] [CrossRef] [PubMed]
- Ko, T.P.; Liao, C.C.; Ku, W.Y.; Chak, K.F.; Yuan, H.S. The crystal structure of the DNase domain of colicin E7 in complex with its inhibitor Im7 protein. Structure 1999, 7, 91–102. [Google Scholar] [CrossRef]
- Abdel-Azeim, S.; Oliva, R.; Chermak, E.; De Cristofaro, R.; Cavallo, L. Molecular dynamics characterization of five pathogenic Factor X mutants associated with decreased catalytic activity. Biochemistry 2014, 53, 6992–7001. [Google Scholar] [CrossRef] [PubMed]
- Nazare, M.; Will, D.W.; Matter, H.; Schreuder, H.; Ritter, K.; Urmann, M.; Essrich, M.; Bauer, A.; Wagner, M.; Czech, J.; et al. Probing the subpockets of factor Xa reveals two binding modes for inhibitors based on a 2-carboxyindole scaffold: A study combining structure-activity relationship and X-ray crystallography. J. Med. Chem. 2005, 48, 4511–4525. [Google Scholar] [CrossRef] [PubMed]
- Lancellotti, S.; Peyvandi, F.; Pagliari, M.T.; Cairo, A.; Abdel-Azeim, S.; Chermak, E.; Lazzareschi, I.; Mastrangelo, S.; Cavallo, L.; Oliva, R.; et al. The D173G mutation in ADAMTS-13 causes a severe form of congenital thrombotic thrombocytopenic purpura: A clinical, biochemical and in silico study. Thromb. Haemost 2015. [Google Scholar] [CrossRef]
- Abbate, E.A.; Berger, J.M.; Botchan, M.R. The X-ray structure of the papillomavirus helicase in complex with its molecular matchmaker E2. Genes Dev. 2004, 18, 1981–1996. [Google Scholar] [CrossRef] [PubMed]
- White, P.W.; Titolo, S.; Brault, K.; Thauvette, L.; Pelletier, A.; Welchner, E.; Bourgon, L.; Doyon, L.; Ogilvie, W.W.; Yoakim, C.; et al. Inhibition of human papillomavirus DNA replication by small molecule antagonists of the E1-E2 protein interaction. J. Biol. Chem. 2003, 278, 26765–26772. [Google Scholar] [CrossRef] [PubMed]
- Yoakim, C.; Ogilvie, W.W.; Goudreau, N.; Naud, J.; Hache, B.; O’Meara, J.A.; Cordingley, M.G.; Archambault, J.; White, P.W. Discovery of the first series of inhibitors of human papillomavirus type 11: Inhibition of the assembly of the E1-E2-Origin DNA complex. Bioorg. Med. Chem. Lett. 2003, 13, 2539–2541. [Google Scholar] [CrossRef]
- Wang, Y.; Coulombe, R.; Cameron, D.R.; Thauvette, L.; Massariol, M.J.; Amon, L.M.; Fink, D.; Titolo, S.; Welchner, E.; Yoakim, C.; et al. Crystal structure of the E2 transactivation domain of human papillomavirus type 11 bound to a protein interaction inhibitor. J. Biol. Chem. 2004, 279, 6976–6985. [Google Scholar] [CrossRef] [PubMed]
- Lensink, M.F.; Mendez, R.; Wodak, S.J. Docking and scoring protein complexes: CAPRI 3rd Edition. Proteins 2007, 69, 704–718. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oliva, R.; Chermak, E.; Cavallo, L. Analysis and Ranking of Protein-Protein Docking Models Using Inter-Residue Contacts and Inter-Molecular Contact Maps. Molecules 2015, 20, 12045-12060. https://doi.org/10.3390/molecules200712045
Oliva R, Chermak E, Cavallo L. Analysis and Ranking of Protein-Protein Docking Models Using Inter-Residue Contacts and Inter-Molecular Contact Maps. Molecules. 2015; 20(7):12045-12060. https://doi.org/10.3390/molecules200712045
Chicago/Turabian StyleOliva, Romina, Edrisse Chermak, and Luigi Cavallo. 2015. "Analysis and Ranking of Protein-Protein Docking Models Using Inter-Residue Contacts and Inter-Molecular Contact Maps" Molecules 20, no. 7: 12045-12060. https://doi.org/10.3390/molecules200712045