
Preparative Isolation and Purification of Lignans from Justicia procumbens Using High-Speed Counter-Current Chromatography in Stepwise Elution Mode


Abstract
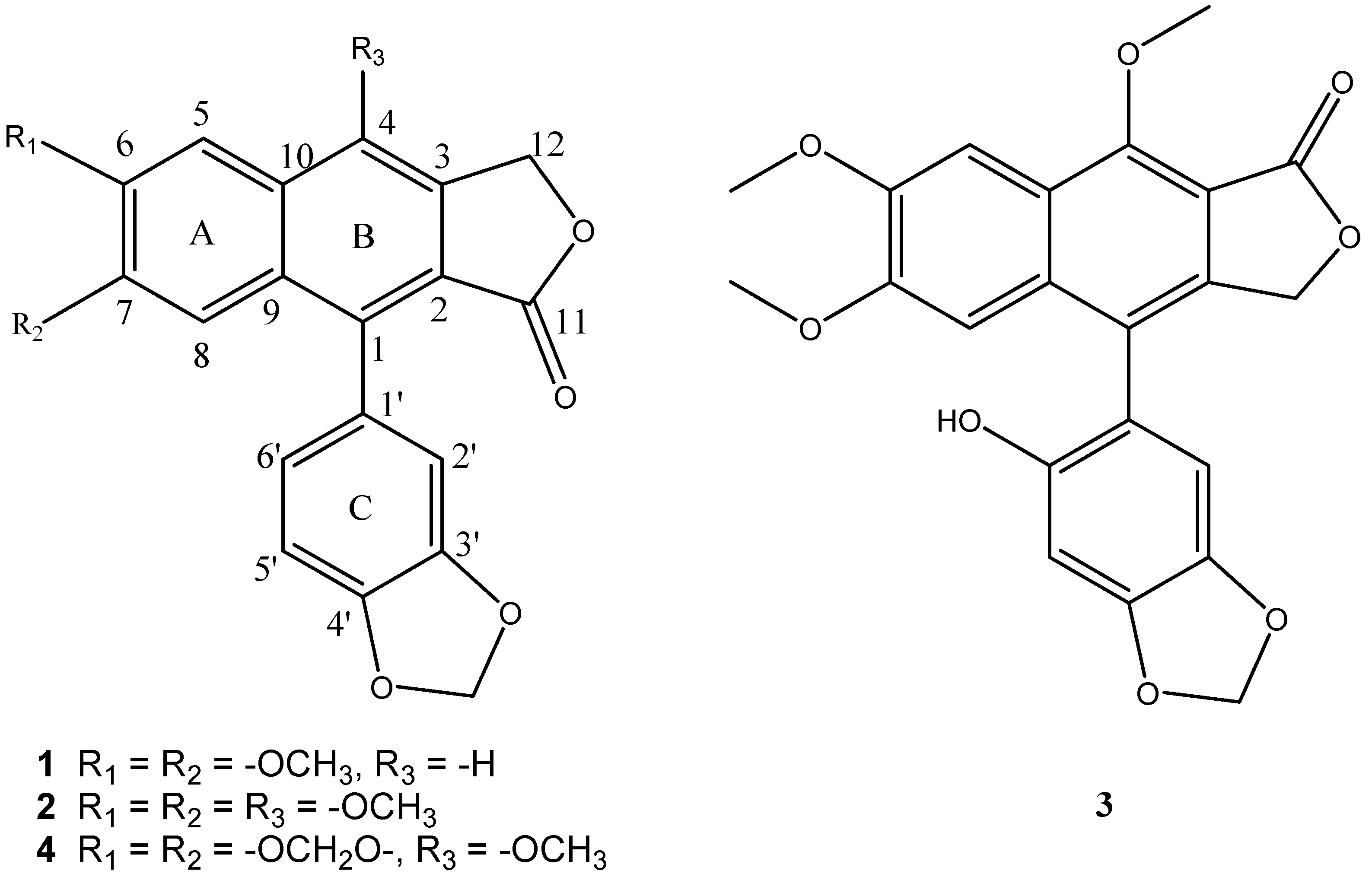
:1. Introduction

2. Results and Discussion
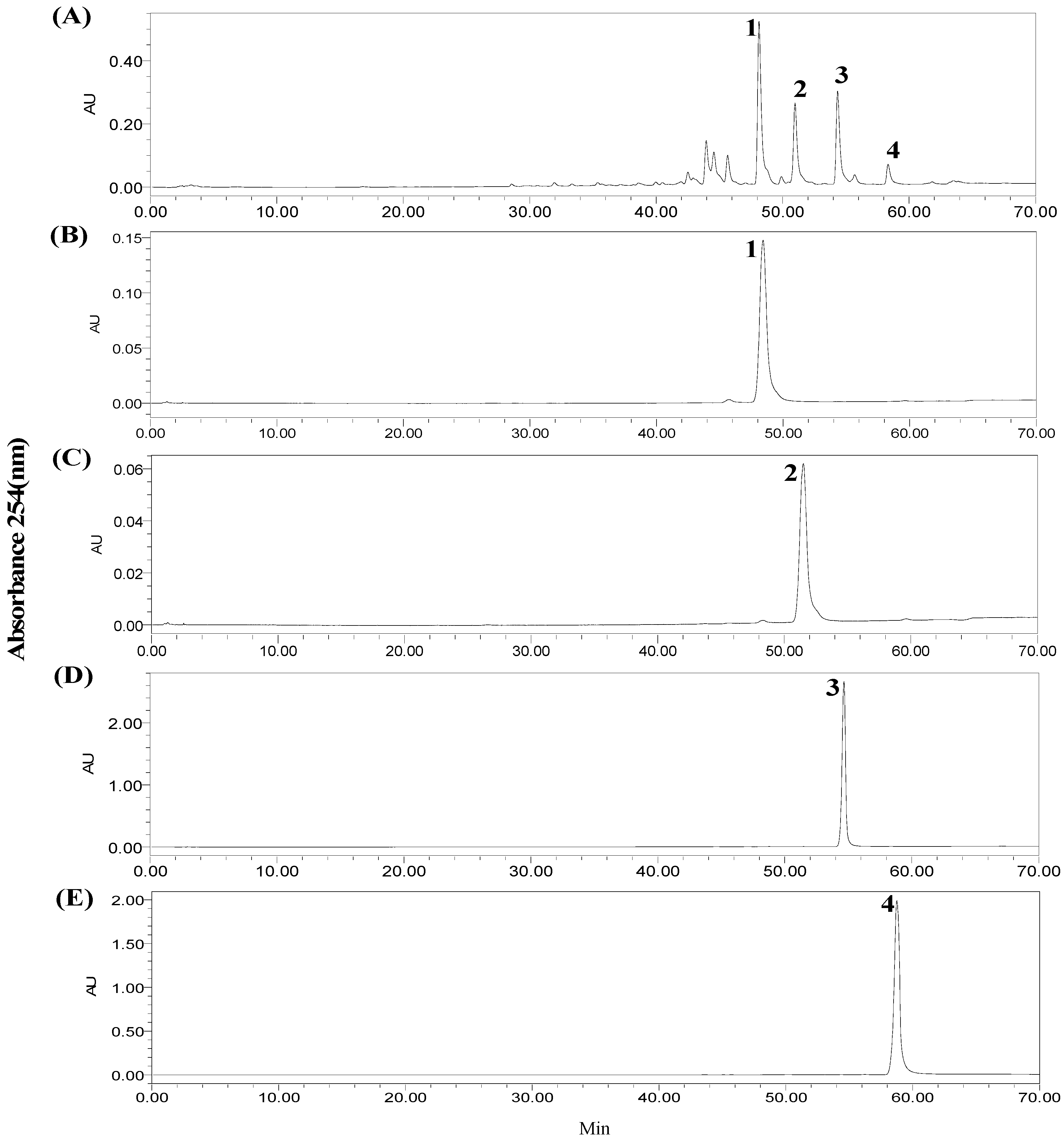
2.1. HPLC Analysis of the Crude Extract

2.2. Selection of the HSCCC Two-Phase Solvent System

{kind=link}
{kind=link}
{kind=link}
| n-Hexane–Ethyl Acetate–Methanol–Water | KD | ||||||
|---|---|---|---|---|---|---|---|
| Compound 1 | Compound 2 | Compound 3 | Compound 4 | ||||
| 1:1:1:1 | >20 | >20 | >20 | >20 | |||
| 1.2:1:1.2:1 | 1.16 | 1.89 | 4.43 | 13.04 | |||
| 1.3:1:1.3:1 | 0.93 | 1.48 | 3.48 | 9.79 | |||
| 1.4:1:1.4:1 | 0.62 | 1.02 | 2.35 | 7.17 | |||
| 1.5:1:1.5:1 | 0.48 | 0.76 | 1.92 | 4.58 | |||
| 1.8:1:1.8:1 | 0.19 | 0.30 | 0.79 | 2.34 | |||
| 2:1:2:1 | 0.12 | 0.20 | 0.54 | 1.49 | |||
| 2.5:1:2.5:1 | 0.07 | 0.16 | 0.31 | 0.88 | |||
| 3:1:3:1 | 0.06 | 0.10 | 0.28 | 0.80 | |||
| 4:1:4:1 | 0.03 | 0.04 | 0.12 | 0.32 | |||
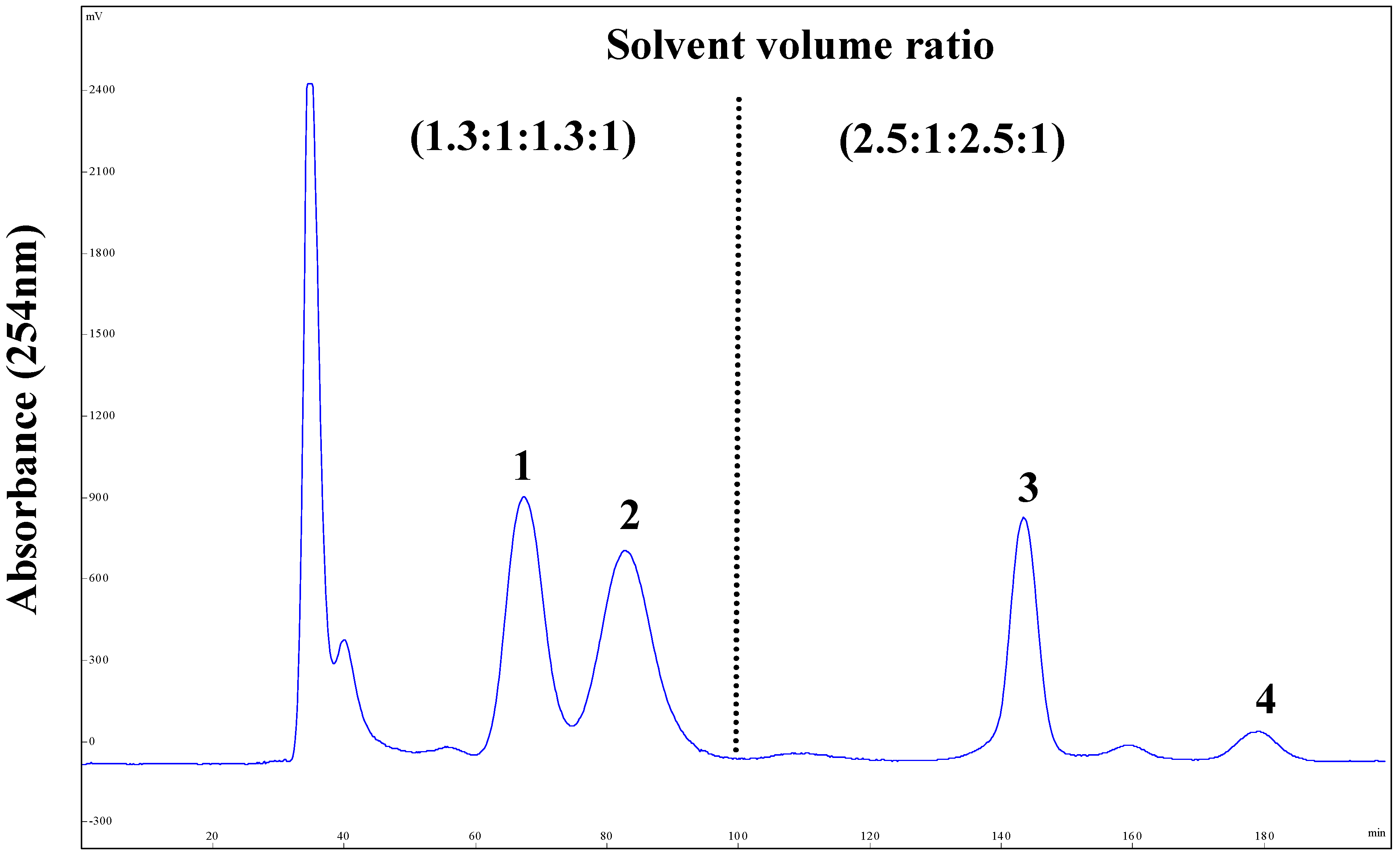
2.3. Stepwise HSCCC Separation

3. Materials and Methods
3.1. Reagents and Materials
3.2. Instruments
3.3. Preparation of Crude Samples
3.4. Measurement of Partition Coefficients (KD)
3.5. Preparation of the Two-Phase Solvent System and Sample Solution
3.6. HSCCC Separation
3.7. HPLC Analysis and Identification of the Peak Fractions
3.8. Identification of Target Compounds
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Luo, Z.; Kong, W.; Qiu, F.; Yang, M.; Li, Q.; Wei, R.; Yang, X.; Qin, J. Simultaneous determination of seven lignans in Justicia procumbens by high performance liquid chromatography-photodiode array detection using relative response factors. J. Sep. Sci. 2013, 36, 699–705. [Google Scholar] [CrossRef] [PubMed]
- Su, C.L.; Huang, L.L.; Huang, L.M.; Lee, J.C.; Lin, C.N.; Won, S.J. Caspase-8 acts as a key upstream executor of mitochondria during justicidin A-induced apoptosis in human hepatoma cells. FEBS Lett. 2006, 580, 3185–3191. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Pan, J.; Yang, M.; Wu, J.; Yang, J. Chromatographic fingerprint analysis and simultaneous determination of eight lignans in Justicia procumbens and its compound preparation by HPLC-DAD. J. Sep. Sci. 2011, 34, 667–674. [Google Scholar] [CrossRef] [PubMed]
- Weng, J.R.; Ko, H.H.; Yeh, T.L.; Lin, H.C.; Lin, C.N. Two new arylnaphthalide lignans and antiplatelet constituents from Justicia procumbens. Arch. Pharm. 2004, 337, 207–212. [Google Scholar] [CrossRef]
- Fukamiya, N.; Lee, K.H. Antitumor agents, Justicidin-A and diphyllin, two cytotoxic principles from Justicia procumbens. J. Nat. Prod. 1986, 49, 348–350. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.C.; Hsin, W.C.; Ko, F.N.; Huang, Y.L.; Ou, J.C.; Teng, C.M. Antiplatelet arylnaphthalide lignans from Justicia procumbens. J. Nat. Prod. 1996, 59, 1149–1150. [Google Scholar] [CrossRef] [PubMed]
- Willfor, S.M.; Smeds, A.I.; Holmbom, B.R. Chromatographic analysis of lignans. J. Chromatogr. A 2006, 1112, 64–77. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Wu, J.; Xu, X.; Jin, Y.; Guo, Y.; Chen, J. A new lignan from the Jian-er syrup and its content determination by RP-HPLC. J. Pharm. Biomed. 2006, 41, 662–666. [Google Scholar] [CrossRef]
- Gordaliza, M.; Garcia, P.A.; del Corral, J.M.; Castro, M.A.; Gómez-Zurita, M.A. Podophyllotoxin: Distribution, sources, applications and new cytotoxic derivatives. Toxicon 2004, 44, 441–459. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.Y.; Chen, S.L.; Yang, M.H.; Wu, J.; Sinkkonen, J.; Zou, K. An update on lignans: Natural products and synthesis. Nat. Prod. Rep. 2009, 26, 1251–1292. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.C.; Hsin, W.C.; Huang, Y.L. Six new diarylbutane lignans from Justicia procumbens. J. Nat. Prod. 1998, 61, 227–229. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Chen, J.; Liang, Z.; Zhao, C. New lignans and their biological activities. Chem. Biodivers. 2014, 11, 51–54. [Google Scholar]
- Qu, H.; Madl, R.L.; Takemoto, D.J.; Baybutt, R.C.; Wang, W. Lignans are involved in the antitumor activity of wheat bran in colon cancer SW480 cells. J. Nutr. 2005, 135, 598–602. [Google Scholar] [PubMed]
- Wu, M.D.; Huang, R.L.; Kuo, L.M.; Hung, C.C.; Ong, C.W.; Kuo, Y.H. The anti-HBsAg (human type B hepatitis, surface antigen) and anti-HBeAg (human type B hepatitis, e antigen) C18 dibenzocyclooctadiene lignans from Kadsura matsudai and Schizandra arisanensis. Chem. Pharm. Bull. 2003, 51, 1233–1236. [Google Scholar] [CrossRef] [PubMed]
- Day, S.H.; Lin, Y.C.; Tsai, M.L.; Tsao, L.T.; Ko, H.H.; Chung, M.I.; Lee, J.C.; Wang, J.P.; Won, S.J.; Lin, C.N. Potent cytotoxic lignans from Justicia procumbens and their effects on nitric oxide and tumor necrosis factor-alpha production in mouse macrophages. J. Nat. Prod. 2002, 65, 379–381. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Fan, D.; Xiong, B.; Kong, L.; Zhu, X. Isolation of new flavan-3-ol and lignan glucoside from Loropetalum chinense and their antimicrobial activities. Fitoterapia 2013, 90, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Asano, J.; Chiba, K.; Tada, M.; Yoshii, T. Antiviral activity of lignans and their glycosides from Justicia procumbens. Phytochemistry 1996, 42, 713–717. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Liu, Z.; Huang, J.A.; Dong, X.; Song, L.; Pan, Y.; Liu, F. Preparative isolation and purification of theaflavins and catechins by high-speed countercurrent chromatography. J. Chromatogr. B 2008, 867, 282–286. [Google Scholar] [CrossRef]
- Ito, Y. Golden rules and pitfalls in selecting optimum conditions for high-speed counter-current chromatography. J. Chromatogr. A 2005, 1065, 145–168. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Ji, L.; Boakye-Yiadom, M.; Li, W.; Song, X.; Gao, X. Preparative isolation and purification of four compounds from Cistanches deserticola Y.C. Ma by high-speed counter-current chromatography. Molecules 2012, 17, 8276–8284. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, I.A.; Fisher, D. Role of counter-current chromatography in the modernisation of Chinese herbal medicines. J. Chromatogr. A 2009, 1216, 740–753. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Liang, J. Counter-current chromatography for high throughput analysis of natural products. Comb. Chem. High Throughput Screen. 2010, 13, 932–942. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Dong, H.; Liu, Y.; Yang, B.; Wang, X.; Huang, L. Application of high-speed counter-current chromatography for preparative separation of cyclic peptides from Vaccaria segetalis. J. Chromatogr. B 2011, 879, 811–814. [Google Scholar] [CrossRef]
- Jiang, W.; Wu, X.; Wu, B. Isolation and identification of two novel attractant compounds from Chinese cockroach (Eupolyphaga sinensis Walker) by combination of HSCCC, NMR and CD techniques. Molecules 2013, 18, 11299–11310. [Google Scholar] [CrossRef] [PubMed]
- Du, Q.; Jiang, Z.; Wang, D. Excellent combination of counter-current chromatography and preparative high-performance liquid chromatography to separate galactolipids from pumpkin. J. Chromatogr. A 2009, 1216, 4176–4180. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, I.; Hewitson, P.; Ignatova, S. Scale-up of counter-current chromatography: Demonstration of predictable isocratic and quasi-continuous operating modes from the test tube to pilot/process scale. J. Chromatogr. A 2009, 1216, 8787–8792. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Wang, H.; Yan, X.; Zhu, P.; Chen, J.; Yang, R. Preparative isolation and purification of macrolactin antibiotics from marine bacterium Bacillus amyloliquefaciens using high-speed counter-current chromatography in stepwise elution mode. J. Chromatogr. A 2013, 1272, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Su, Z.; Yang, Y.; Ba, H.; Aisa, H.A. Isolation of three sesquiterpene lactones from the roots of Cichorium glandulosum Boiss. et Huet. by high-speed counter-current chromatography. J. Chromatogr. A 2007, 1176, 217–222. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Lu, Y.; Jiang, L.; Wu, B.; Zhang, F.; Pan, Y. Preparative isolation and purification of antioxidative stilbene oligomers from Vitis chunganeniss using high-speed counter-current chromatography in stepwise elution mode. J. Sep. Sci. 2009, 32, 2339–2345. [Google Scholar] [CrossRef] [PubMed]
- Charlton, J.L.; Oleschuk, C.J.; Si, J.; Chee, G.L.; Yang, M. Hindered rotation in arylnaphthalene lignans. J. Org. Chem. 1996, 61, 3452–3457. [Google Scholar] [CrossRef]
- Yang, M.; Wu, J.; Cheng, F.; Zhou, Y. Complete assignments of 1H and 13C-NMR data for seven arylnaphthalide lignans from Justicia procumbens. Magn. Reson. Chem. 2006, 44, 727–730. [Google Scholar] [CrossRef] [PubMed]
- Gopalaiah, K.; Kavitha, J.; Kanumuri, R.V.; Rajasekhar, D.; Subbaraju, G.V. Justicia lignans: Part 9—Two new lignans from Justicia neesii Ramamoorthy (white flower variety). Indian J. Chem. 2001, 40, 596–600. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are not available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, P.; Luo, Q.; Ding, L.; Fang, F.; Yuan, Y.; Chen, J.; Zhang, J.; Jin, H.; He, S. Preparative Isolation and Purification of Lignans from Justicia procumbens Using High-Speed Counter-Current Chromatography in Stepwise Elution Mode. Molecules 2015, 20, 7048-7058. https://doi.org/10.3390/molecules20047048
Zhou P, Luo Q, Ding L, Fang F, Yuan Y, Chen J, Zhang J, Jin H, He S. Preparative Isolation and Purification of Lignans from Justicia procumbens Using High-Speed Counter-Current Chromatography in Stepwise Elution Mode. Molecules. 2015; 20(4):7048-7058. https://doi.org/10.3390/molecules20047048
Chicago/Turabian StyleZhou, Peijuan, Qijun Luo, Lijian Ding, Fang Fang, Ye Yuan, Juanjuan Chen, Jinrong Zhang, Haixiao Jin, and Shan He. 2015. "Preparative Isolation and Purification of Lignans from Justicia procumbens Using High-Speed Counter-Current Chromatography in Stepwise Elution Mode" Molecules 20, no. 4: 7048-7058. https://doi.org/10.3390/molecules20047048