Unstable Simple Volatiles and Gas Chromatography-Tandem Mass Spectrometry Analysis of Essential Oil from the Roots Bark of Oplopanax Horridus Extracted by Supercritical Fluid Extraction

Abstract
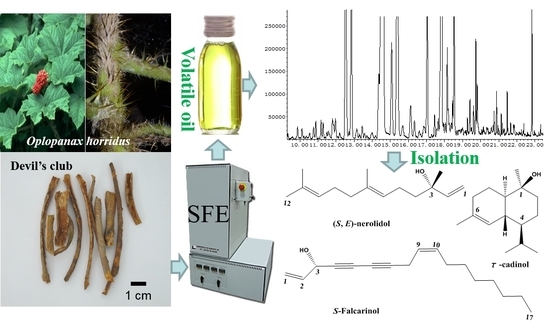
:
1. Introduction

2. Results and Discussion
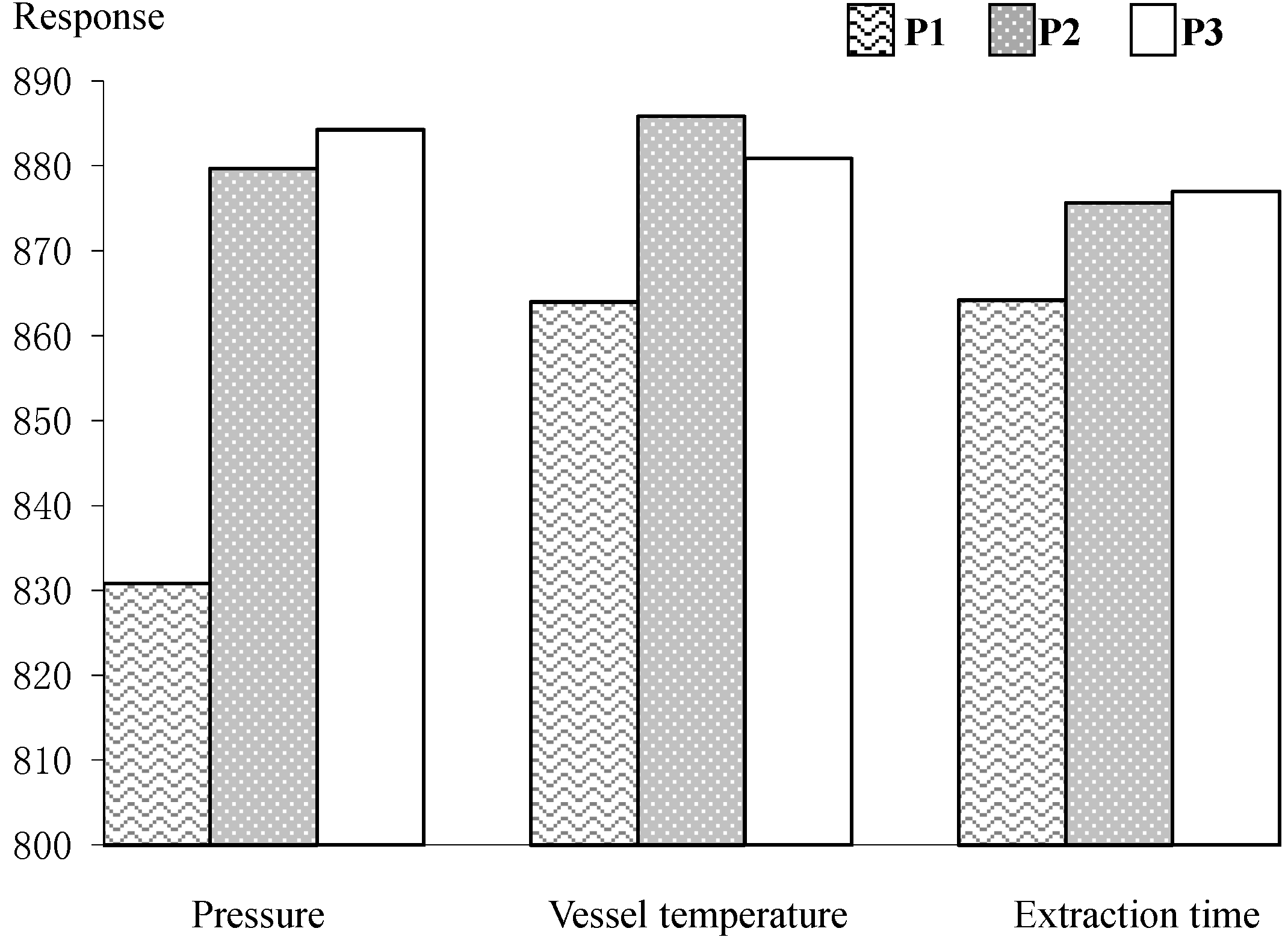
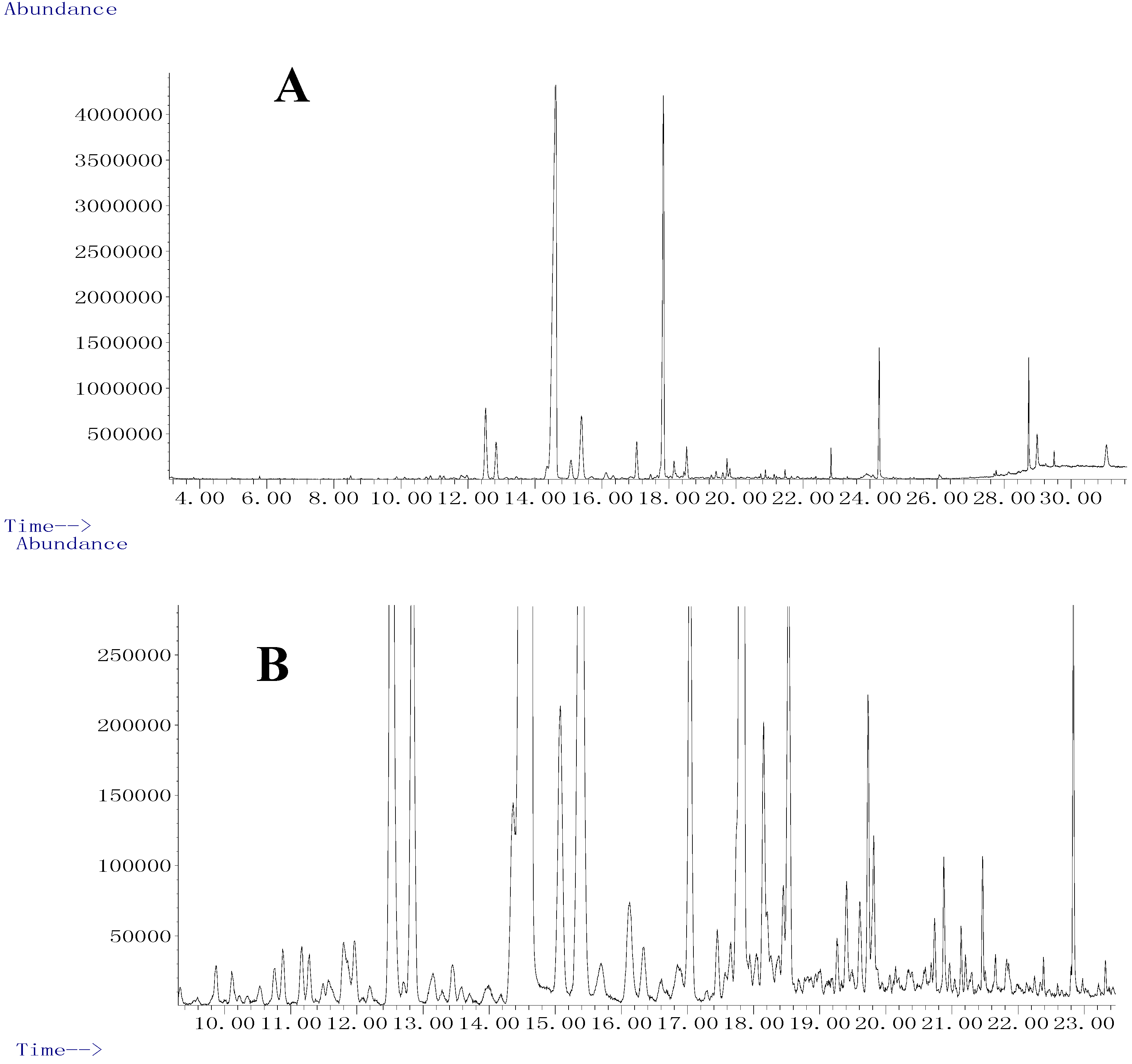
2.1. Development of SFE

2.2. Validation of the Developed Method

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analytes | Intra-day (n = 6) RSD% | Inter-day (n = 6) RSD% | Repeatability (n = 3) RSD% | Recovery (n = 3) (%) | ||
|---|---|---|---|---|---|---|
| HR * | MR * | LR * | ||||
| (S, E)-Nerolidol | 2.9 | 2.1 | 4.0 | 2.3 | 2.2 | 97.6 |
| τ-Cadinol | 1.8 | 2.2 | 3.8 | 3.7 | 2.6 | 103.6 |
| S-Falcarinol | 1.7 | 3.3 | 4.2 | 2.5 | 3.9 | 99.1 |
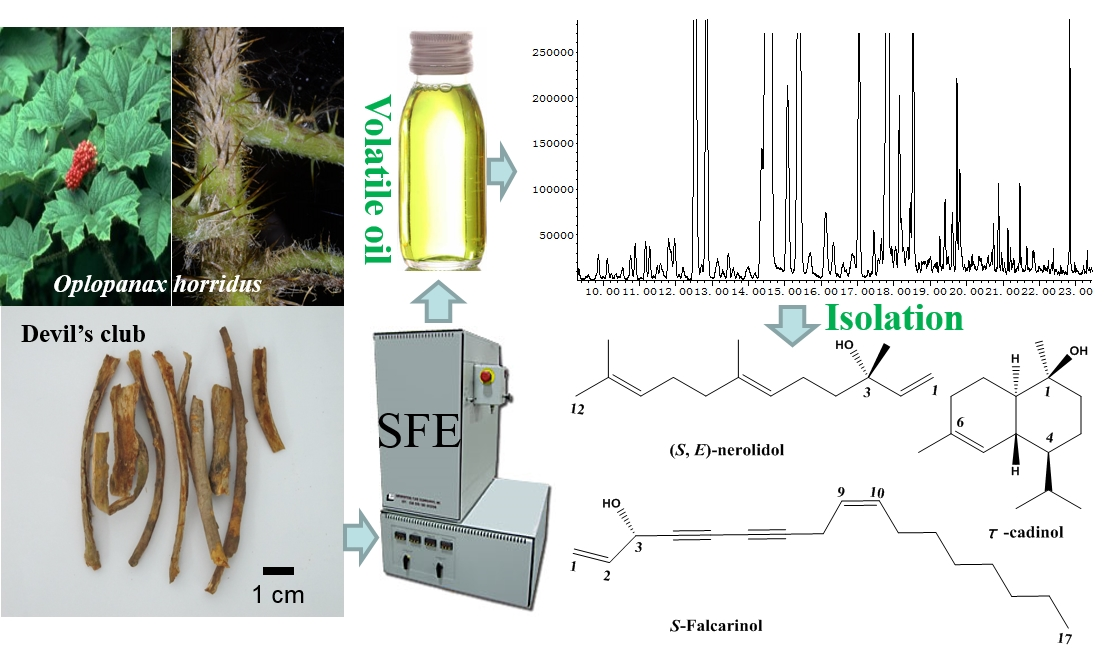
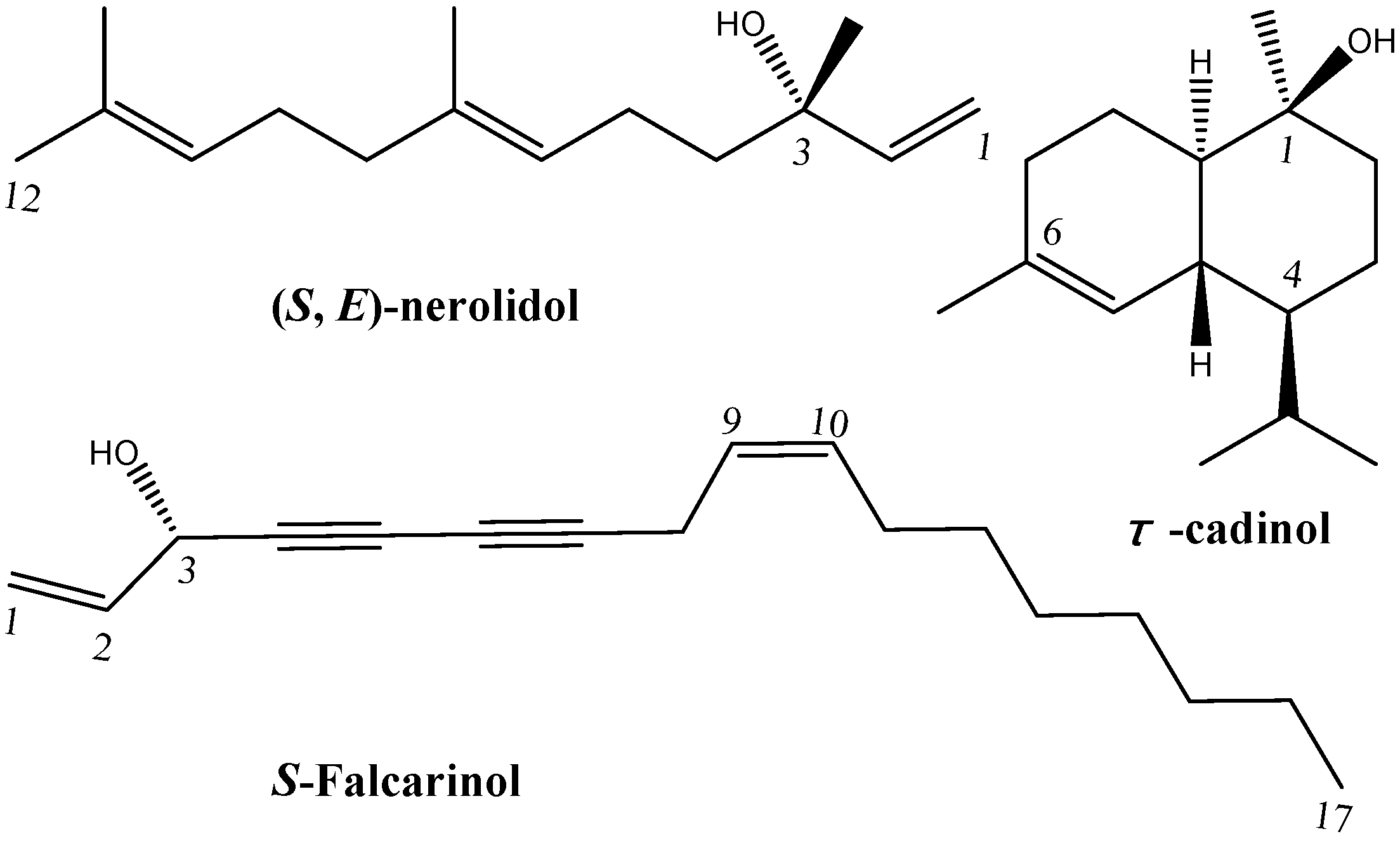
2.3. GC/MS Determination and Identification

| Compound | RI HP-5 a | Molecular Formula | Relative Content (%) |
|---|---|---|---|
| Pentanol | 826 | C5H12O | 0.02 |
| Hexanol | 834 | C6H14O | 0.05 |
| Heptanal | 856 | C7H14O | 0.01 |
| Heptanol | 862 | C7H16O | 0.18 |
| Octanol | 868 | C8H16O | 0.21 |
| Benzaldehyde | 876 | C7H6O | 0.06 |
| 2-Octenal | 880 | C8H14O | 0.13 |
| 2-Methylpentenal | 892 | C6H12O | 0.07 |
| α-Pinene | 931 | C10H16 | 0.18 |
| β-Phellandrene | 986 | C10H16 | 0.06 |
| Linalool | 1087 | C10H18O | 0.62 |
| 1,3,5-Undecatriene | 1126 | C11H18 | 0.68 |
| 1,3,5,8-Undecatetraene | 1189 | C11H20 | 0.21 |
| α-Ylangene | 1263 | C15H24 | 0.08 |
| Aromadendrene | 1312 | C15H24 | 0.06 |
| α-Zingiberene | 1389 | C15H24 | 0.28 |
| β-Caryophyllene | 1411 | C15H24 | 0.16 |
| (E)-α-Bergamotene | 1421 | C15H24 | 0.04 |
| α-Copaene | 1433 | C15H24 | 1.01 |
| α-Humulene | 1438 | C15H24 | 0.43 |
| α-Cadinene | 1442 | C15H24 | 0.06 |
| Germacrene A | 1454 | C15H24 | 0.22 |
| (E)-β-Farnesene | 1462 | C15H24 | 0.11 |
| γ-Muurolene | 1482 | C15H24 | 0.07 |
| Germacrene D | 1496 | C15H24 | 0.34 |
| ar-Curcumene | 1510 | C15H22 | 0.04 |
| β-Sesquiphellandrene | 1521 | C15H24 | 0.48 |
| β-Elemene | 1533 | C15H24 | 1.04 |
| α-Muurolene | 1542 | C15H24 | 0.53 |
| allo-Aromadendrene | 1544 | C15H24 | 0.49 |
| γ -Cadinene | 1551 | C15H24 | 1.02 |
| α-Farnesene | 1557 | C15H24 | 0.35 |
| 1,10-Di-epi-cubenol | 1562 | C15H24O | 0.25 |
| δ-Cadinene | 1568 | C15H24 | 0.28 |
| Bicyclogermacrene | 1572 | C15H24 | 4.51 |
| Ishwarane | 1577 | C15H24 | 0.38 |
| Germacrene B | 1577 | C15H24 | 1.14 |
| (S,E)-Nerolidol | 1589 | C15H26O | 52.52 |
| Spathulenol | 1592 | C15H24O | 0.78 |
| Germacrene D-4-ol | 1610 | C15H26O | 0.63 |
| Gleenol | 1621 | C15H26O | 0.47 |
| Guaiol | 1630 | C15H26O | 0.52 |
| Endo-1-bourbonanol | 1638 | C15H26O | 0.54 |
| τ-Cadinol | 1644 | C15H26O | 21.62 |
| τ-Muurolol | 1655 | C15H26O | 1.03 |
| S-Falcarinol | 1660 | C17H24O | 3.63 |
| α-Eudesmol | 1670 | C15H26O | 0.09 |
| Bulnesol | 1677 | C15H26O | 0.11 |
3. Experimental Section
3.1. General Procedures
3.2. Plant Material
3.3. Supercritical Fluid Extraction of Volatiles from O. horridus
3.4. GC/MS Analysis
3.5. Method Validation
3.6. Chemical Isolation of Volatile Compounds
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lantz, T.C.; Antos, J.A. Clonal expansion in the deciduous understory shrub, Devil’s club (Oplopanax horridus; Araliaceae). Can. J. Bot. 2002, 80, 1052–1062. [Google Scholar] [CrossRef]
- Ohwi, J.; Meyer, F.G.; Walker, E.H. Flora of Japan; Smithsonian Institution Press: Washington, DC, USA, 1965; p. 666. [Google Scholar]
- Wu, C.Y. Flora of China; Science Press: Beijing, China, 1988; Volume 54, p. 16. [Google Scholar]
- Lantz, T.C.; Swerhun, K.; Turner, N.J. Devil’s club (Oplopanax horridus): An ethnobotanical review. HerbalGram 2004, 62, 33–48. [Google Scholar]
- Huang, W.H.; Zhang, Q.W.; Yuan, C.S.; Wang, C.Z.; Li, S.P.; Zhou, H.H. Chemical constituents of the plants from the genus Oplopanax. Chem. Biodivers. 2014, 11, 181–196. [Google Scholar] [CrossRef] [PubMed]
- Calway, T.; Du, G.J.; Wang, C.Z.; Huang, W.H.; Zhao, J.; Li, S.P.; Yuan, C.S. Chemical and pharmacological studies of Oplopanax horridus, a North American botanical. J. Nat. Med. 2012, 66, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Garneau, F.X.; Collin, G.; Gagnon, H.; Jean, F.I.; Strobl, H.; Pichette, A. The essential oil composition of devil’s club, Oplopanax horridus J.E. Smith Miq. Flavour Fragr. J. 2006, 21, 792–794. [Google Scholar] [CrossRef]
- Zhao, Y.R.; Kang, T.G.; Dou, D.Q.; Smith, C.D. Study on chemical constituents of volatile oil from stem bark of O. horridus by GC-MS. Liaoning Zhongyiyao Daxue Xuebao 2008, 10, 114–115. [Google Scholar]
- Donelian, A.; Carlson, L.H.C.; Lopes, T.J.; Machado, R.A.F. Comparison of extraction of patchouli (Pogostemon cablin) essential oil with supercritical CO2 and by steam distillation. J. Supercrit. Fluids 2009, 48, 15–20. [Google Scholar] [CrossRef]
- Fornari, T.; Vicente, G.; Vazquez, E.; Garcia-Risco, M.R.; Reglero, G. Isolation of essential oil from different plants and herbs by supercritical fluid extraction. J. Chromatogr. A 2012, 1250, 34–48. [Google Scholar] [CrossRef] [PubMed]
- Capuzzo, A.; Maffei, M.E.; Occhipinti, A. Supercritical fluid extraction of plant flavors and fragrances. Molecules 2013, 18, 7194–7238. [Google Scholar] [CrossRef] [PubMed]
- Calza, P.; Medana, C.; Baiocchi, C.; Pelizzetti, E. Identification of degradation products by adopting GC or HPLC/MS techniques. Curr. Anal. Chem. 2005, 1, 267–287. [Google Scholar] [CrossRef]
- Jalali-Heravi, M.; Parastar, H. Recent trends in application of multivariate curve resolution approaches for improving gas chromatography-mass spectrometry analysis of essential oils. Talanta 2011, 85, 835–849. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds are available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shao, L.; Bao, M.-H.; Ouyang, D.-S.; Wang, C.-Z.; Yuan, C.-S.; Zhou, H.-H.; Huang, W.-H. Unstable Simple Volatiles and Gas Chromatography-Tandem Mass Spectrometry Analysis of Essential Oil from the Roots Bark of Oplopanax Horridus Extracted by Supercritical Fluid Extraction. Molecules 2014, 19, 19708-19717. https://doi.org/10.3390/molecules191219708
Shao L, Bao M-H, Ouyang D-S, Wang C-Z, Yuan C-S, Zhou H-H, Huang W-H. Unstable Simple Volatiles and Gas Chromatography-Tandem Mass Spectrometry Analysis of Essential Oil from the Roots Bark of Oplopanax Horridus Extracted by Supercritical Fluid Extraction. Molecules. 2014; 19(12):19708-19717. https://doi.org/10.3390/molecules191219708
Chicago/Turabian StyleShao, Li, Mei-Hua Bao, Dong-Sheng Ouyang, Chong-Zhi Wang, Chun-Su Yuan, Hong-Hao Zhou, and Wei-Hua Huang. 2014. "Unstable Simple Volatiles and Gas Chromatography-Tandem Mass Spectrometry Analysis of Essential Oil from the Roots Bark of Oplopanax Horridus Extracted by Supercritical Fluid Extraction" Molecules 19, no. 12: 19708-19717. https://doi.org/10.3390/molecules191219708