Original TDAE Strategy Using Propargylic Chloride: Rapid Access to 1,4-Diarylbut-3-ynol Derivatives

Abstract
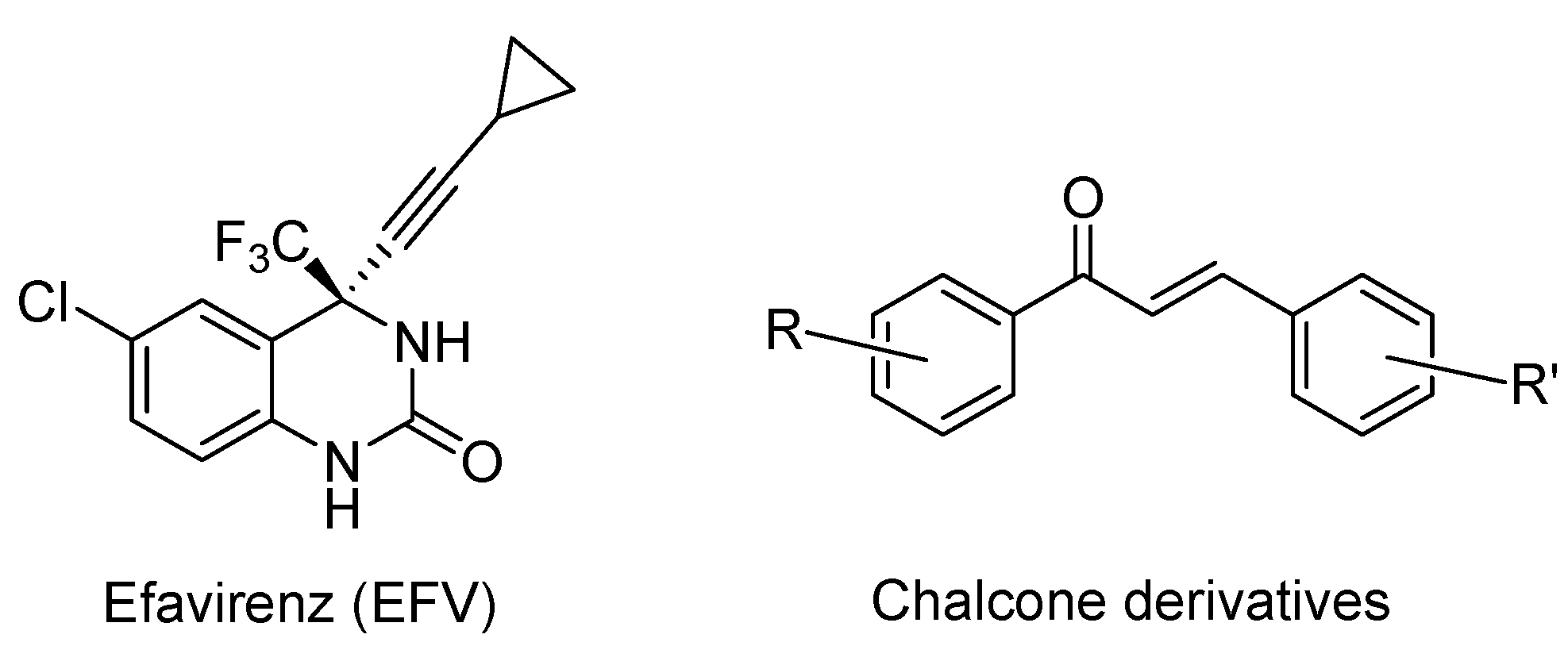
:1. Introduction

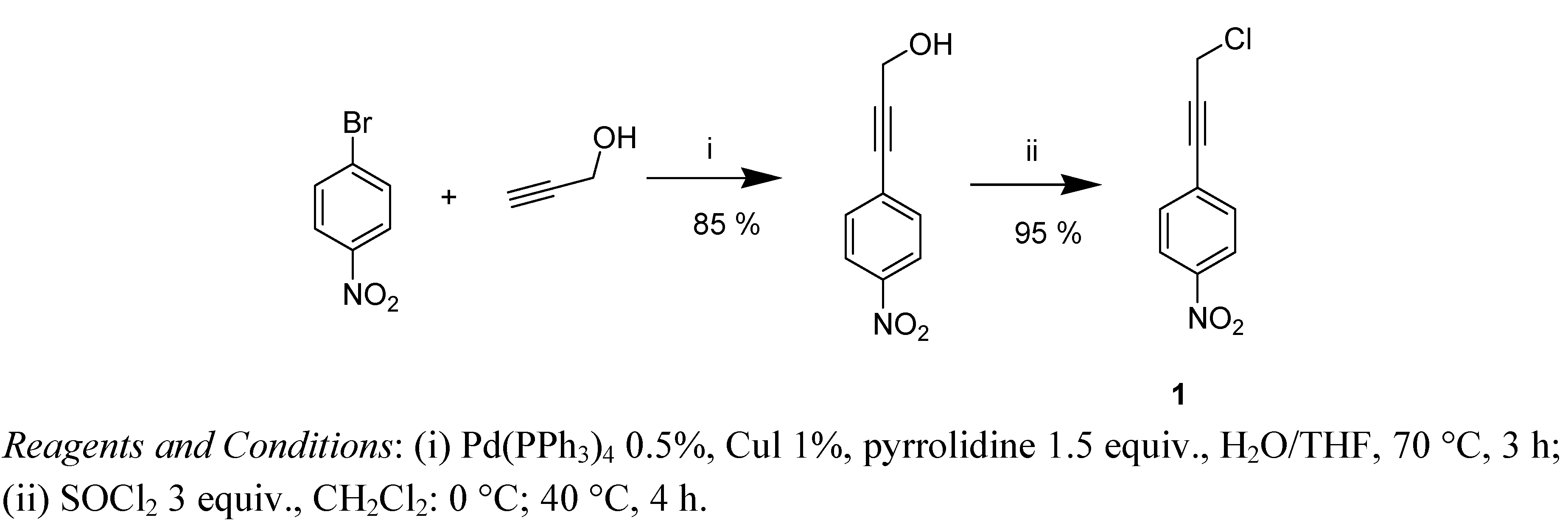
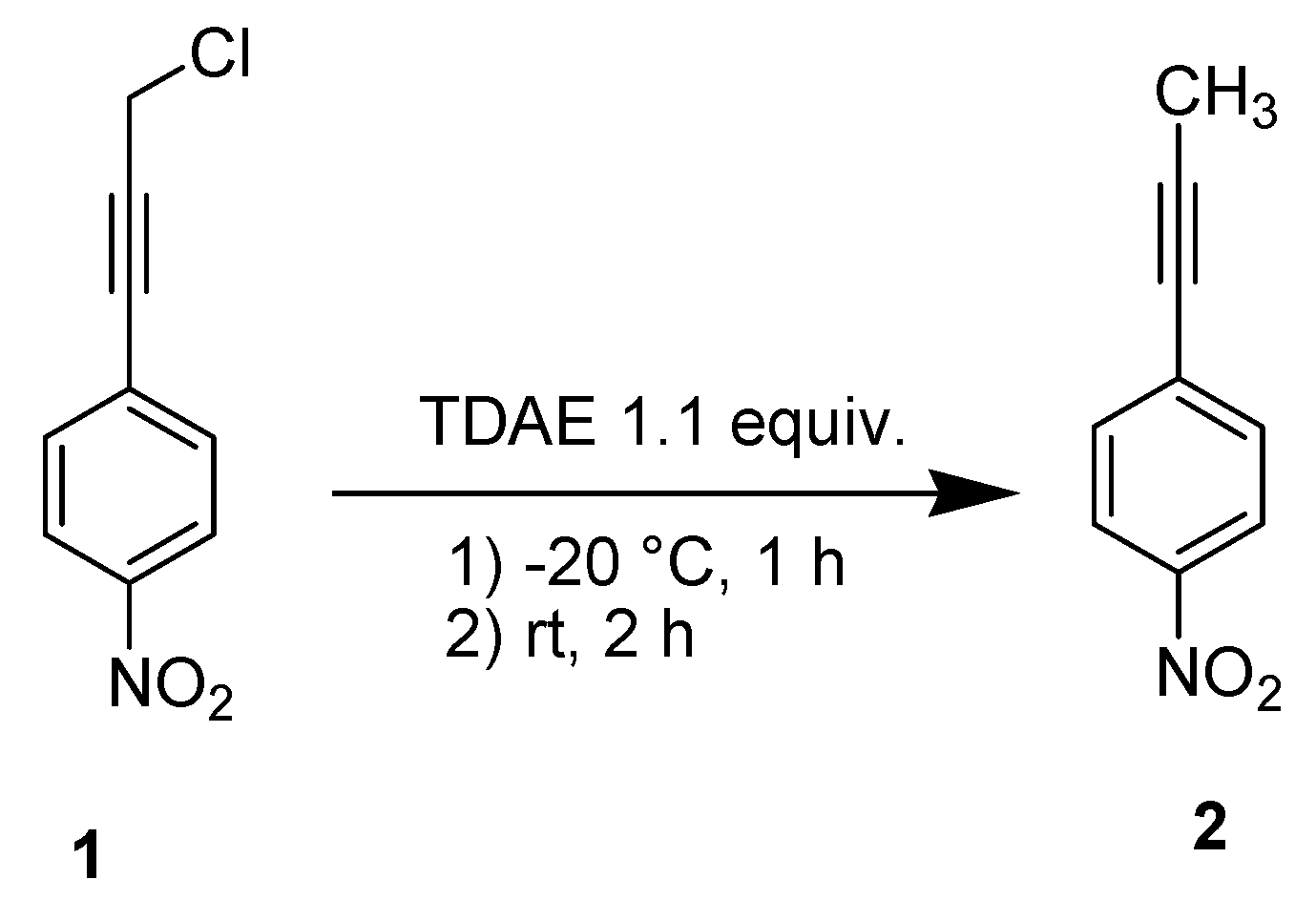
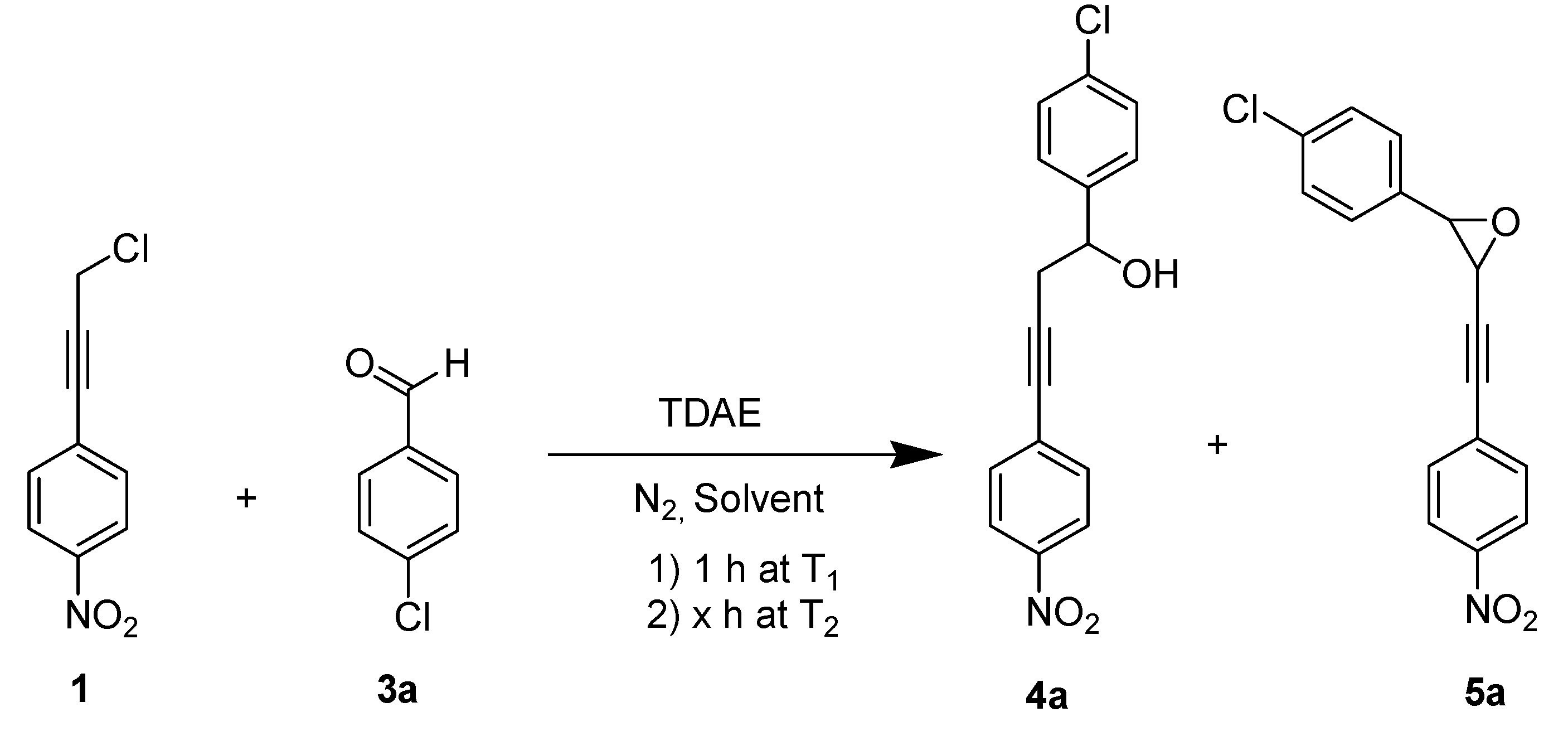
2. Results and Discussion




{kind=link}
{kind=link}
{kind=link}
{kind=link}
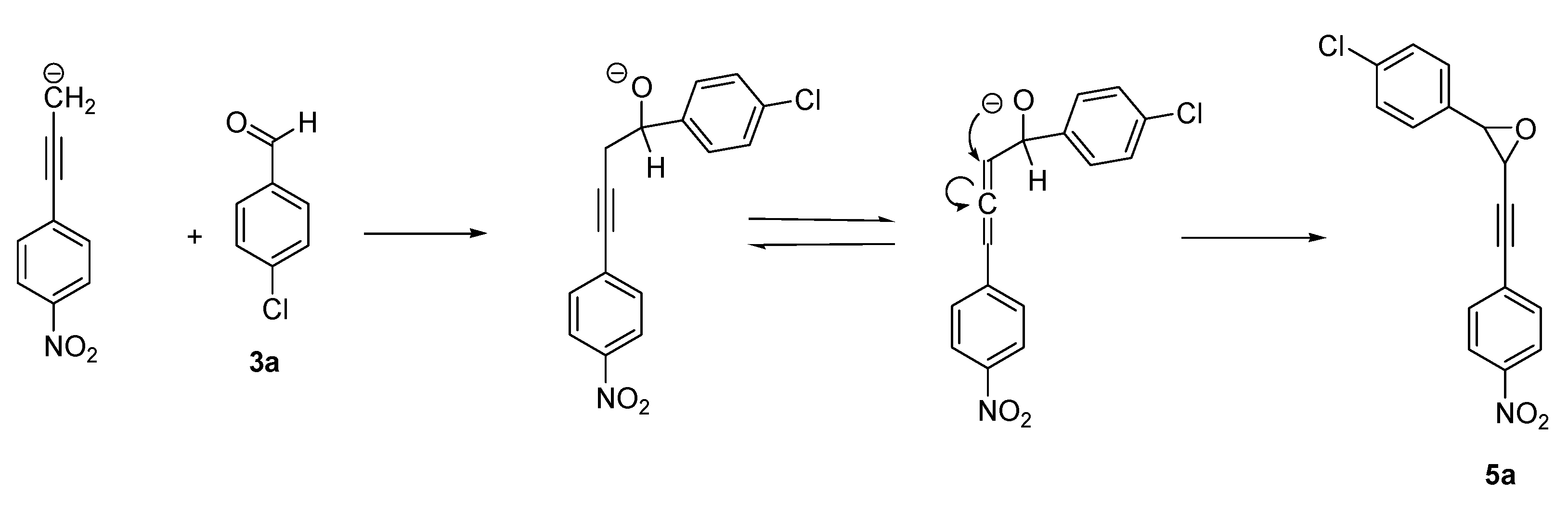{kind=link}
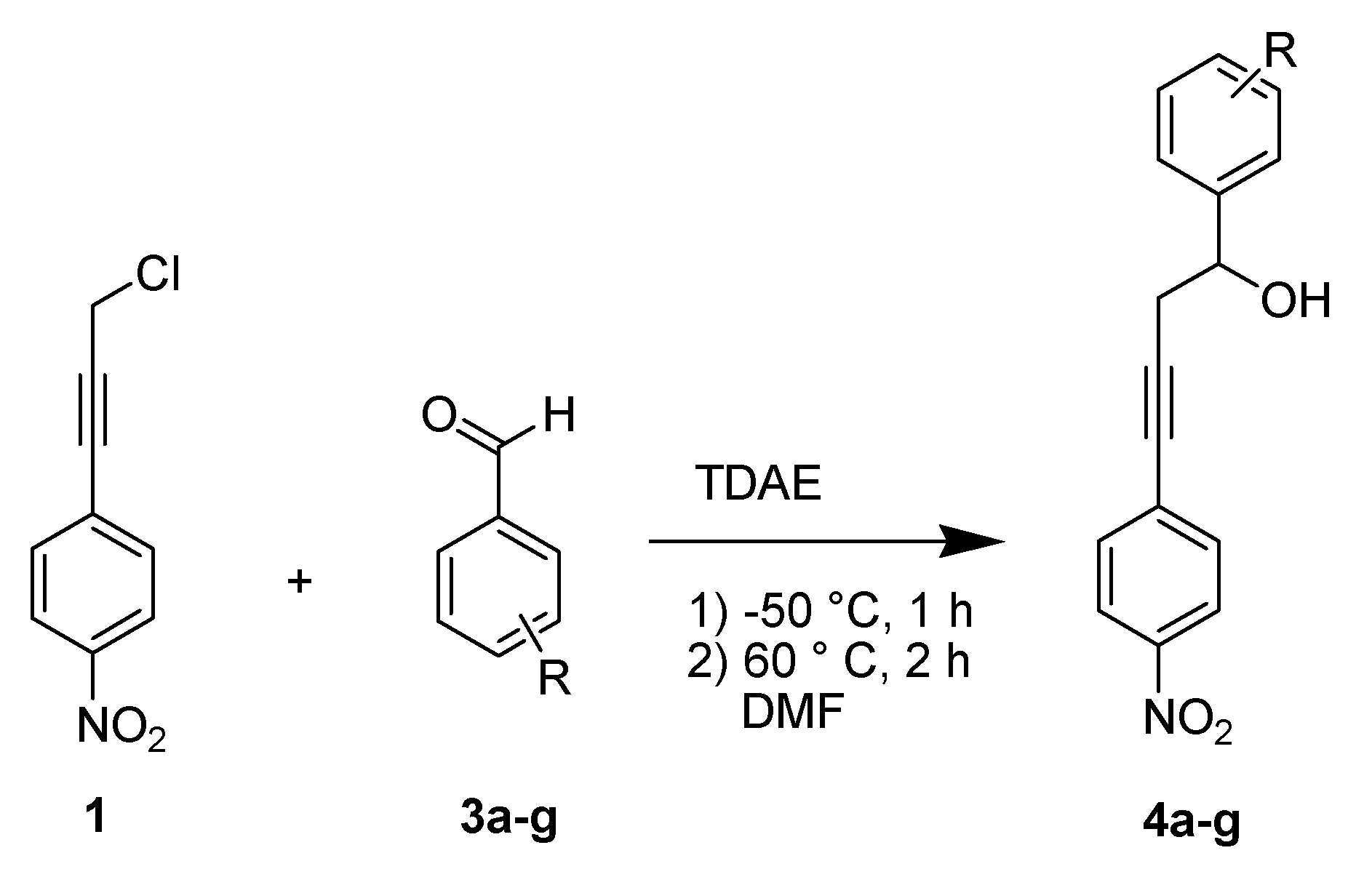{kind=link}
| Entry a | Equiv. of 3a | Solvent | T1d | Time of reaction (stage 2) | T2e | (%) Yield c | ||
|---|---|---|---|---|---|---|---|---|
| 1 | 4a | 5a | ||||||
| 1 | 3 | DMF, 4 mL | −20 °C | 2 h | rt | - | 40 | - |
| 2 b | 3 | THF, 4 mL | −20 °C | 2 h | rt | - | - | - |
| 3 | 3 | MeCN, 4 mL | −20 °C | 2 h | rt | 85 | - | - |
| 4 | 3 | DMF, 4 mL | −20 °C | 6 h | rt | - | 6 | traces |
| 5 | 1 | DMF, 4 mL | −20 °C | 2 h | rt | - | 12 | - |
| 6 | 4 | DMF, 4 mL | −20 °C | 2 h | rt | - | 20 | - |
| 7 | 3 | DMF, 10 mL | −20 °C | 2 h | rt | - | 38 | 20 |
| 8 | 3 | DMF, 4 mL | −50 °C | 2 h | rt | - | 45 | - |
| 9 | 3 | DMF, 4 mL | −50 °C | 2 h | 60 °C | - | 65 | - |
| 10 | 3 | DMF, 10 mL | −50 °C | 2 h | rt | - | 25 | 35 |
| 11 | 3 | DMF, 20 mL | −20 °C | 2 h | rt | - | 30 | traces |
| 12 | 3 | DMF, 20 mL | −50 °C | 2 h | rt | - | 38 | 25 |
| 13 | 3 | DMF, 20 mL | −50 °C | 6 h | rt | - | 25 | 25 |


| Aromatic aldehyde | R | (%) Yield b | |
|---|---|---|---|
| 4-Chlorobenzaldehyde | 4-Cl | 4a | 65 |
| 4-Nitrobenzaldehyde | 4-NO2 | 4b | 57 |
| 4-Fluorobenzaldehyde | 4-F | 4c | 34 c |
| 4-Tolualdehyde | 4-CH3 | 4d | traces |
| 4-Trifluoromethylbenzaldehyde | 4-CF3 | 4e | 51 |
| 2-Nitro-4,5-dimethoxybenzaldehyde | 2-NO2-4,5-(OCH3)2 | 4f | 32 |
| Benzaldehyde | H | 4g | 30 d |
3. Experimental
3.1. General
3.2. General Procedure for TDAE Reaction with Aromatic Aldehydes
4. Conclusions
Acknowledgments
- Samples Availability: Samples of the compounds 4a–g, 5a are available from the authors.
References
- Pu, L. Asymmetric alkynylzinc additions to aldehydes and ketones. Tetrahedron 2003, 59, 9873–9886. [Google Scholar] [CrossRef]
- Cozzi, P.G.; Hilgraf, R.; Zimmermann, N. Acetylenes in catalysis: Enantioselective additions to carbonyl groups and imines and applications beyond. Eur. J. Org. Chem. 2004, 2004, 4095–4105. [Google Scholar]
- Qian, Y.; Zhang, H.J.; Lv, P.C.; Zhu, H.L. Synthesis, molecular modeling and biological evaluation of guanidine derivatives as novel antitubulin agents. Bioorg. Med. Chem. 2010, 18, 8218–8225. [Google Scholar] [CrossRef]
- Tu, H.Y.; Huang, A.M.; Hour, T.C.; Yang, S.C.; Pu, Y.S. Synthesis and biological evaluation of 2',5'-dimethoxychalcone derivatives as microtubule-targeted anticancer agents. Bioorg. Med. Chem. 2010, 18, 2089–2098. [Google Scholar] [CrossRef]
- Álvarez, C.; Corchete, P.; Pérez-Melero, C.; Peláez, R.; Menarde, M. Exploring the effect of 2,3,4-trimethoxy-phenyl moiety as a component of indolephenstatins. Eur. J. Med. Chem. 2010, 45, 588–597. [Google Scholar] [CrossRef]
- Mahesh, M.; Murphy, J.A.; LeStrat, F.; Wessel, H.P. Reduction of arenediazonium salts by tetrakis(dimethylamino)ethylene (TDAE): Efficient formation of products derived from aryl radicals. Beilstein J. Org. Chem. 2009, 5. [Google Scholar] [CrossRef]
- Takechi, N.; Ait-Mohand, S.; Medebielle, M.; Dolbier, W.R., Jr. Nucleophilic trifluoromethylation of acyl chlorides using the trifluoromethyl iodide/TDAE reagent. Tetrahedron Lett. 2002, 43, 4317–4319. [Google Scholar] [CrossRef]
- Medebielle, M.; Dolbier, W.R., Jr. Nucleophilic difluoromethylation and trifluoromethylation using tetrakis(dimethylamino)ethylene (TDAE) reagent. J. Fluorine Chem. 2008, 129, 930–942. [Google Scholar] [CrossRef]
- Giuglio-Tonolo, G.; Terme, T.; Médebielle, M.; Vanelle, P. Original reaction of p-nitrobenzyl chloride with aldehydes using tetrakis(dimethylamino)ethylene (TDAE). Tetrahedron Lett. 2003, 44, 6433–6435. [Google Scholar] [CrossRef]
- Amiri-Attou, O.; Terme, T.; Vanelle, P. Functionalization of 6-nitrobenzo[1,3]dioxole with carbonyl compounds via TDAE methodology. Molecules 2005, 10, 545–551. [Google Scholar] [CrossRef]
- Montana, M.; Terme, T.; Vanelle, P. Original synthesis of oxiranes via TDAE methodology: Reaction of 2,2-dibromomethylquinoxaline with aromatic aldehydes. Tetrahedron Lett. 2005, 46, 8373–8376. [Google Scholar] [CrossRef]
- Khoumeri, O.; Montana, M.; Terme, T.; Vanelle, P. First TDAE approach in quinonic series: Synthesis of new 2-substituted 1,4-dimethoxy-9,10-anthraquinones. Tetrahedron 2008, 64, 11237–11242. [Google Scholar] [CrossRef]
- Giuglio-Tonolo, G.; Terme, T.; Medebielle, M.; Vanelle, P. Nitrobenzylation of α-carbonyl ester derivatives using TDAE approach. Tetrahedron Lett. 2004, 45, 5121–5124. [Google Scholar] [CrossRef]
- Montana, M.; Crozet, M.D.; Castera-Ducros, C.; Terme, T.; Vanelle, P. Rapid synthesis of new azaheterocyclic hydroxy-malonate derivatives using TDAE approach. Heterocycles 2008, 75, 925–932. [Google Scholar] [CrossRef]
- Juspin, T.; Laget, M.; Terme, T.; Azas, N.; Vanelle, P. TDAE-assisted synthesis of new imidazo[2,1-b]thiazole derivatives as anti-infectious agents. Eur. J. Med. Chem. 2010, 45, 840–845. [Google Scholar] [CrossRef]
- Amiri-Attou, O.; Terme, T.; Vanelle, P. Original and rapid access to new alkaloid analogues of neocryptolepine: Synthesis of substituted 6-methyl-6H-indolo[2,3-b]quinolines via TDAE strategy. Synlett 2005, 3047–3050. [Google Scholar]
- Juspin, T.; Terme, P.; Vanelle, P. TDAE strategy using α-diketones: Rapid access to 2,3-diphenylquinoline and acenaphtho[1,2-b]quinoline derivatives. Synlett 2009, 1485–1489. [Google Scholar]
- Montana, M.; Terme, T.; Vanelle, P. TDAE-initiated synthesis of oxiranes in heterocyclic series: Reaction of 2-(dibromomethyl)quinoxaline with α-dicarbonyl derivatives. Lett. Org. Chem. 2010, 7, 453–456. [Google Scholar] [CrossRef]
- Khoumeri, O.; Giuglio-Tonolo, G.; Crozet, M.D.; Terme, T.; Vanelle, P. Original synthesis of 2-substituted-4,11-dimethoxy-1-(phenylsulfonyl)-2,3-dihydro-1H-naphtho[2,3-f]indole-5,10-diones using TDAE and Cu-catalyzed reaction strategy. Tetrahedron 2011, 67, 6173–6180. [Google Scholar] [CrossRef]
- Khoumeri, O.; Crozet, M.D.; Terme, T.; Vanelle, P. Original TDAE application: Synthesis of 2-substituted-4,11-dimethoxy-anthra[2,3-b]furan-5,10-diones via intramolecular Buchwald reaction. Tetrahedron Lett. 2009, 50, 6372–6376. [Google Scholar] [CrossRef]
- Dunn, L.A.; Tan, K.S.W.; Vanelle, P.; Juspin, T.; Crozet, M.D.; Terme, T.; Upcroft, P.; Upcroft, J.A. Development of metronidazole-resistant lines of Blastocystis sp. Parasitol. Res. 2012, 111, 441–450. [Google Scholar] [CrossRef]
- Roche, M.; Terme, T.; Vanelle, P. First Long-Distance SRN1 on a propargylic chloride. Tetrahedron Lett. 2012, 53, 4184–4187. [Google Scholar] [CrossRef]
- Gellis, A.; Kovacic, H.; Boufatah, N.; Vanelle, P. Synthesis and cytotoxicity evaluation of some benzimidazole-4,7-diones as bioreductive anticancer agents. Eur. J. Med. Chem. 2008, 43, 1858–1864. [Google Scholar] [CrossRef]
- Delmas, F.; Gasquet, M.; Timon-David, P.; Madadi, N.; Vanelle, P.; Vaille, A.; Maldonado, J. Synthesis and in vitro anti-protozoan activity of new 5-nitrothiophene oxime ether derivatives. Eur. J. Med. Chem. 1993, 28, 23–27. [Google Scholar] [CrossRef]
- Boufatah, N.; Gellis, A.; Maldonado, J.; Vanelle, P. Efficient microwave-assisted synthesis of new sulfonylbenzimidazole-4,7-diones: Heterocyclic quinones with potential antitumor activity. Tetrahedron 2004, 60, 9131–9137. [Google Scholar] [CrossRef]
- Crozet, M.P.; Archaimbault, G.; Vanelle, P.; Nouguier, R. SRN1 Reactions in the heterocyclic series 4. Reactivity of 2,2-dimethyl-5-nitro-1,3-dioxane salts. Tetrahedron Lett. 1985, 26, 5133–5134. [Google Scholar]
- El-Kashef, H.S.; El-Emary, T.I.; Gasquet, M.; Timon-David, P.; Maldonado, J.; Vanelle, P. New pyrazolo[3,4-b]pyrazines: Synthesis and biological activity. Pharmazie 2000, 55, 572–576. [Google Scholar]
- Verhaeghe, P.; Azas, N.; Hutter, S.; Castera-Ducros, C.; Laget, M.; Dumetre, A.; Gasquet, M.; Reboul, J.-P.; Rault, S.; Rathelot, P.; et al. Synthesis and in vitro antiplasmodial evaluation of 4-anilino-2-trichloromethylquinazolines. Bioorg. Med. Chem. 2009, 17, 4313–4322. [Google Scholar]
- Chinchilla, R.; Najera, C. The Sonogashira Reaction: A booming methodology in synthetic organic chemistry. Chem. Rev. 2007, 107, 874–922. [Google Scholar] [CrossRef]
- Since, M.; Terme, T.; Vanelle, P. Original TDAE strategy using α-halocarbonyl derivatives. Tetrahedron 2009, 65, 6128–6134. [Google Scholar] [CrossRef]
- Price, C.C.; Carmelite, D.D. Reactions of epoxides in dimethyl sulfoxide catalyzed by potassium t-butoxide. J. Am. Chem. Soc. 1966, 88, 4039–4044. [Google Scholar] [CrossRef]
- Williams, D.H.; Flemming, I. Spectroscopic Problems in Organic Chemistry. In Spectroscopic Methods in Organic Chemistry, 4th ed; Mc Graw-Hill: NewYork, NY, USA, 1991; pp. 76–101. [Google Scholar]
- Montana, M.; Terme, T.; Vanelle, P. Original synthesis of alpha-chloroketones in azaheterocyclic series using TDAE approach. Tetrahedron Lett. 2006, 47, 6573–6576. [Google Scholar] [CrossRef]
- Nadji-Boukrouche, A.R.; Khoumeri, O.; Terme, T.; Liacha, M.; Vanelle, P. Original TDAE reactivity in benzoxa- and benzothiazolone series. ARKIVOC 2010, X, 358–370. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Roche, M.; Terme, T.; Vanelle, P. Original TDAE Strategy Using Propargylic Chloride: Rapid Access to 1,4-Diarylbut-3-ynol Derivatives. Molecules 2013, 18, 1540-1548. https://doi.org/10.3390/molecules18021540
Roche M, Terme T, Vanelle P. Original TDAE Strategy Using Propargylic Chloride: Rapid Access to 1,4-Diarylbut-3-ynol Derivatives. Molecules. 2013; 18(2):1540-1548. https://doi.org/10.3390/molecules18021540
Chicago/Turabian StyleRoche, Manon, Thierry Terme, and Patrice Vanelle. 2013. "Original TDAE Strategy Using Propargylic Chloride: Rapid Access to 1,4-Diarylbut-3-ynol Derivatives" Molecules 18, no. 2: 1540-1548. https://doi.org/10.3390/molecules18021540