Trimethyl Lock: A Stable Chromogenic Substrate for Esterases

1
Department of Biochemistry, University of Wisconsin–Madison, 433 Babcock Drive, Madison, WI 53706-1544, USA
2
Department of Chemistry, University of Wisconsin–Madison, 1101 University Avenue, Madison, WI 53706-1322, USA
*
Author to whom correspondence should be addressed.
Molecules 2008, 13(2), 204-211; https://doi.org/10.3390/molecules13020204
Submission received: 19 January 2008
/
Accepted: 30 January 2008
/
Published: 31 January 2008
(This article belongs to the Special Issue Prodrugs)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:p-Nitrophenyl acetate is the most commonly used substrate for detecting the catalytic activity of esterases, including those that activate prodrugs in human cells. This substrate is unstable in aqueous solution, limiting its utility. Here, a stable chromogenic substrate for esterases is produced by the structural isolation of an acetyl ester and p-nitroaniline group using a trimethyl lock moiety. Upon ester hydrolysis, unfavorable steric interactions between the three methyl groups of this o-hydroxycinnamic acid derivative encourage rapid lactonization to form a hydrocoumarin and release p-nitroaniline. This “prochromophore” could find use in a variety of assays.
Introduction
Prodrug strategies have been employed to improve the properties of potential small-molecule chemotherapeutic agents, including their solubility, stability, organ selectivity, duration of action, and bioavailability [1,2]. Recently, our research group reported on the use of the “trimethyl lock” prodrug strategy as the basis for a new class of latent fluorophores [3,4,5]. The trimethyl lock is an o-hydroxycinnamic acid derivative in which severe steric repulsion between three methyl groups leads to rapid lactonization to form a hydrocoumarin with consequent release of an alcohol or amine [6,7,8]. In our latent fluorophores, the trimethyl lock was inserted between a labile functional group and a fluorescent dye. The isolation of the labile group and dye led to a marked improvement in chemical stability with the retention of enzymatic reactivity.
To exploit further the utility and modularity of the trimethyl lock system, we have designed a chromogenic esterase substrate, 1. Although chromogenic substrates lack the level of sensitivity of fluorogenic substrates [9], the simplicity and low cost of a spectrophotometer is a notable advantage. The utility of p-nitrophenyl acetate (2, pNPA), the most often-used esterase substrate, is diminished by a number of unfavorable properties, especially the pH-sensitivity of its chromophoric properties and its notorious chemical instability [10,11]. The hydrolysis product of 2, p-nitrophenol (pNP), has a pKa of 7.0 and an absorbance spectrum that exhibits a hypsochromic shift below pH 8 [12]. The pH optimum (pH 6.5–8.0) of pig liver esterase (PLE; EC 3.1.1.1), the most often-used esterase, encompasses the pKa of pNP, such that a small pH change during an assay with pNPA as a substrate leads to inaccurate results [13,14]. A notable improvement to the chemical stability of pNP-based esterase substrates involves using acyloxymethyl ethers in place of the acetate esters [13,14]. Although this substitution endows increased chemical stability, the pH-sensitivity of the pNP chromophore remains problematic. Here, we report on the synthesis and characterization of a novel prochromophore that utilizes the trimethyl lock and overcomes the limitations of pNPA and other esterase substrates.
Results and Discussion
pH-Sensitivity of Chromophores
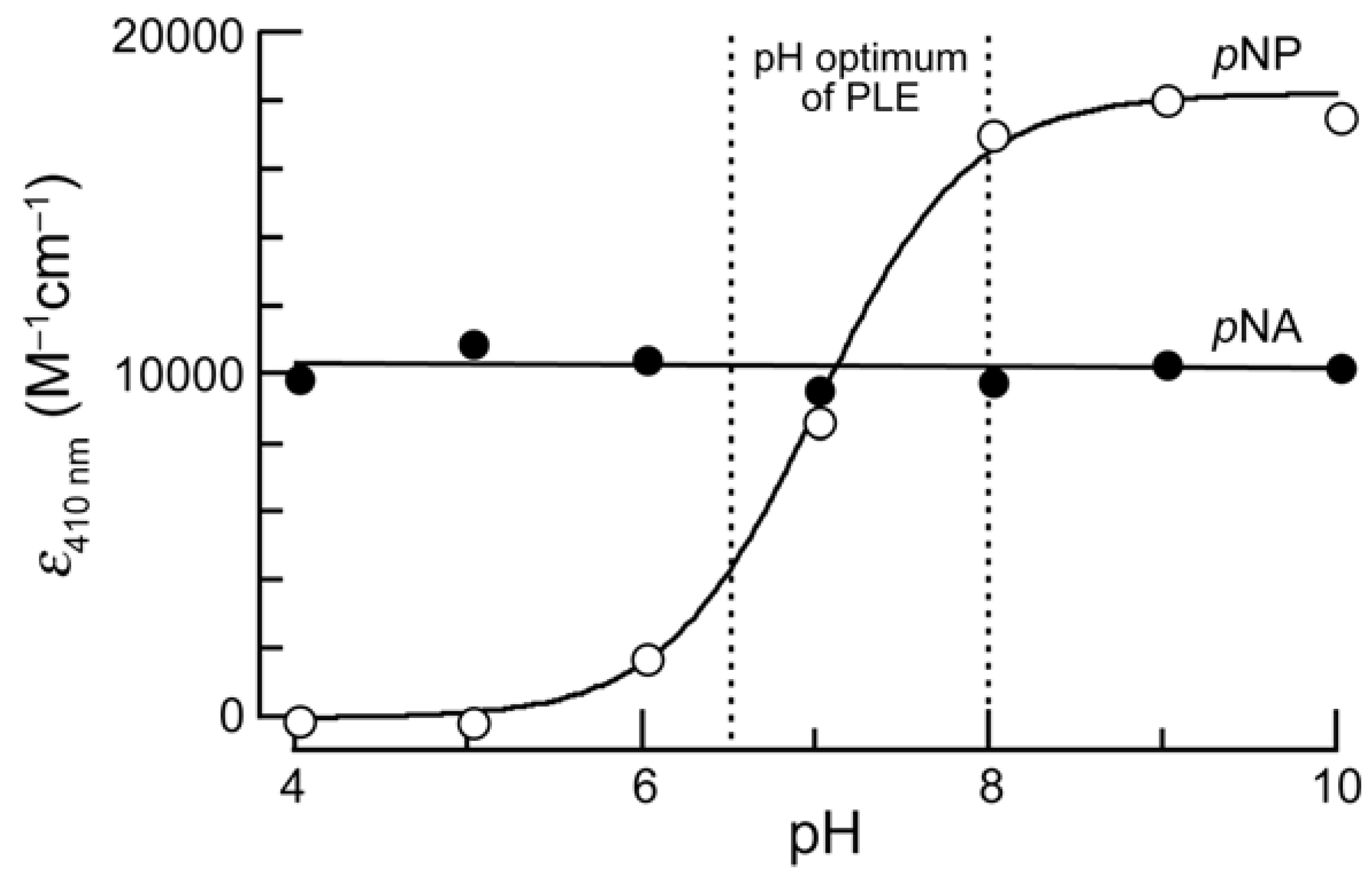
To overcome an intrinsic problem of pNP, we chose to use p-nitroaniline (pNA) as the chromophore in an esterase substrate. In contrast to pNP, pNA does not have a pKa value in the physiologic range, and its absorbance spectrum is constant from pH 4–10 (Figure 1). Derivatization of pNA to make a nitroanilide confers a hypsochromic shift in its absorbance spectrum, enabling it to be used for spectrophotometric assays [15].
Figure 1.
pH-Sensitivity of the extinction coefficients at 410 nm of p-nitroaniline (pNA) and p-nitrophenol (pNP). Measurements were made in triplicate in PBS adjusted to the appropriate pH. The observed pKa of pNP was 7.0.
Figure 1.
pH-Sensitivity of the extinction coefficients at 410 nm of p-nitroaniline (pNA) and p-nitrophenol (pNP). Measurements were made in triplicate in PBS adjusted to the appropriate pH. The observed pKa of pNP was 7.0.

Synthesis of a Prochromophore 1
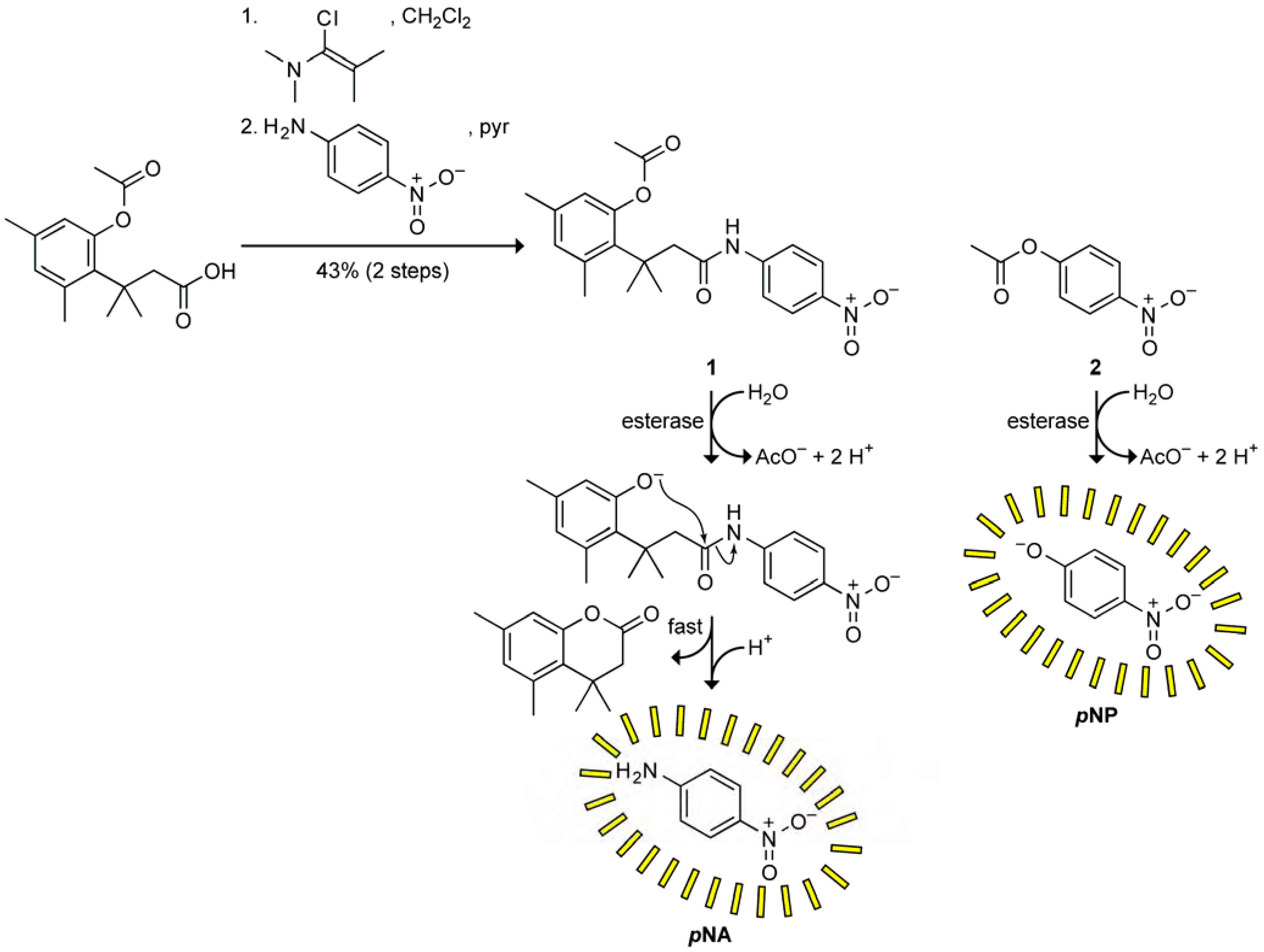
To prepare prochromophore 1, we followed the route used by Amsberry and coworkers to synthesize the trimethyl lock precursor [16]. Condensation with p-nitroaniline to form an amide bond was difficult, presumably due to steric constraints in the trimethyl lock substrate and the low nucleophilicity of the aromatic amine. Traditional peptide coupling reagents such as EDC and DIC led to poor yields, but formation of the acid chloride in situ under neutral conditions using Ghosez’s reagent, which is an α-chloroenamine, followed by the addition of p-nitroaniline and pyridine, was ultimately successful (Scheme 1) [17,18].
Scheme 1.
Route for the synthesis of prochromophore 1, and putative mechanism for the activation of prochromophore 1 and p-nitrophenyl acetate (pNPA; 2) by an esterase.
Scheme 1.
Route for the synthesis of prochromophore 1, and putative mechanism for the activation of prochromophore 1 and p-nitrophenyl acetate (pNPA; 2) by an esterase.

Spectroscopic Properties
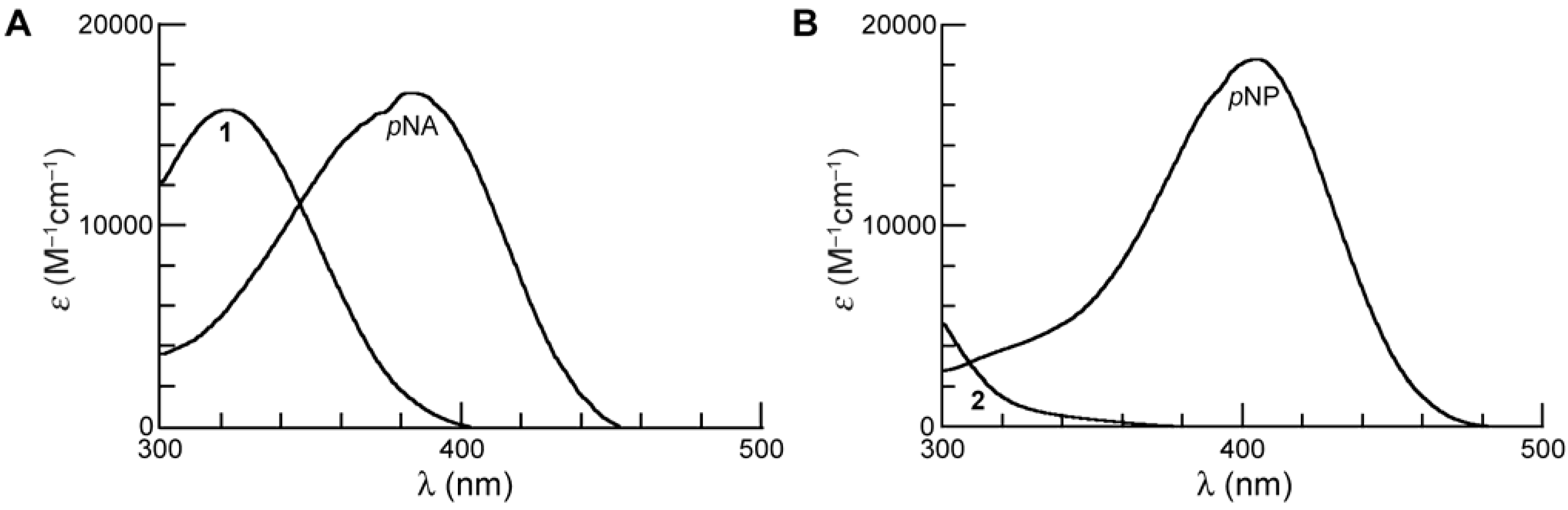
As expected [15], the spectrum of 1 (λmax = 322 nm) displays a hypsochromic shift relative to pNA (λmax = 383 nm). We performed our kinetics assays at 410 nm, where the absorbance of the substrate is negligible (ε410 = ~0 M–1cm–1) but that of the pNA product is high (ε410 = 10,681 M–1cm–1) [19].
Figure 2.
(A) Absorbance spectra of prochromophore 1 and its hydrolysis product, p-nitroaniline (pNA), in PBS (pH 7.4). (B) Absorbance spectra of p-nitrophenyl acetate (pNPA; 2) and its hydrolysis product, p-nitrophenol (pNP) in PBS (pH 7.4).
Figure 2.
(A) Absorbance spectra of prochromophore 1 and its hydrolysis product, p-nitroaniline (pNA), in PBS (pH 7.4). (B) Absorbance spectra of p-nitrophenyl acetate (pNPA; 2) and its hydrolysis product, p-nitrophenol (pNP) in PBS (pH 7.4).

Chemical Stability
Stability in aqueous solution is an important property for enzymatic substrates, particularly those for hydrolases. It is well established that pNPA (2) hydrolyzes spontaneously in neutral buffer [10,11], and that the rate of its hydrolysis increases in the presence of proteins or amino acids, especially those with free amino or phenolic groups [10]. pNPA has even been used as an acetylating agent to modify cysteine and lysine residues in proteins [20]. Accordingly, the use of pNPA as an esterase substrate requires the performance of numerous control experiments to ensure that pNP is being produced by the putative catalyst in a truly catalytic manner [21]. Moreover, the use of pNPA is not amenable to any assay requiring long incubation times or low enzyme concentrations [22].
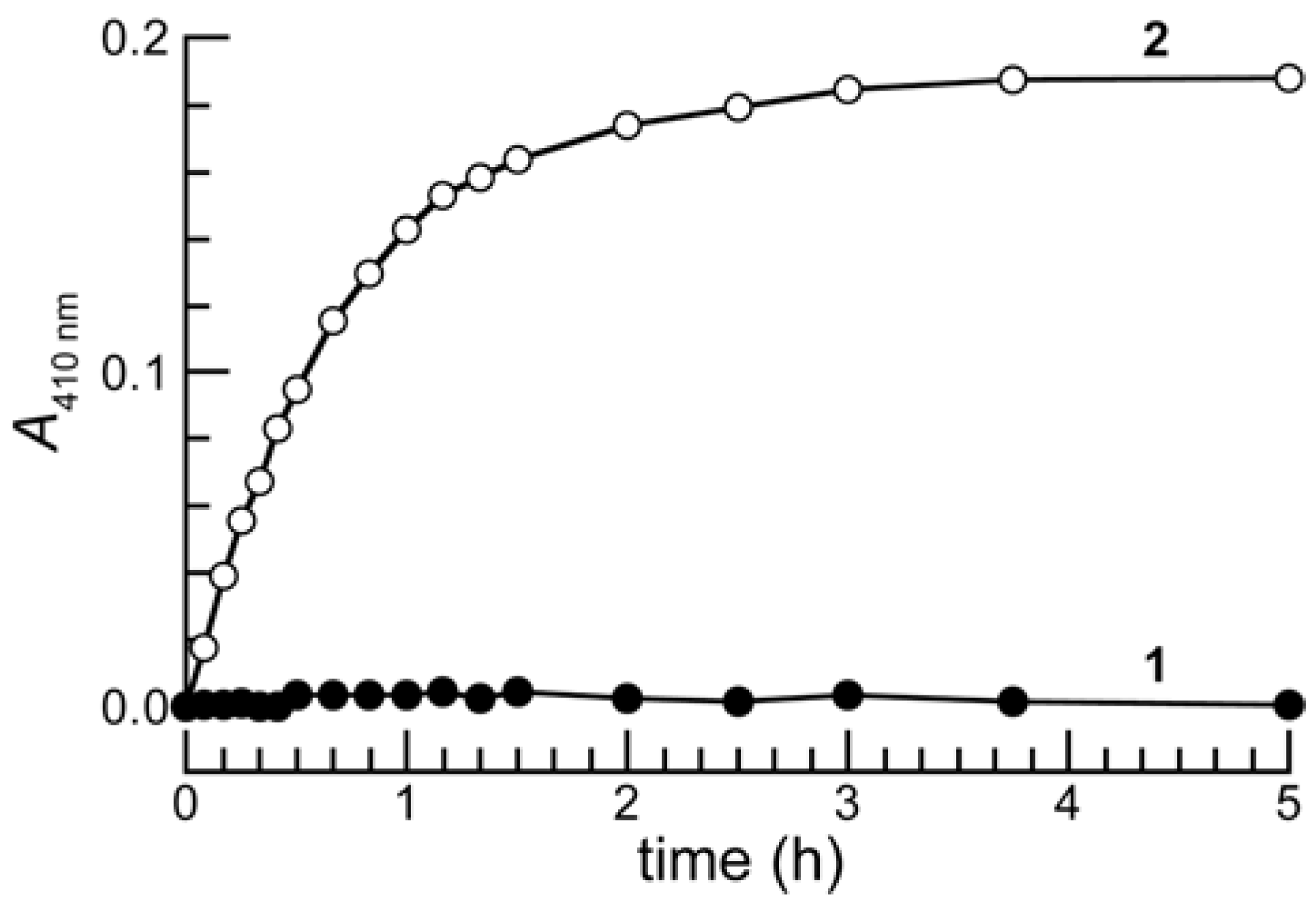
As shown in Figure 3, prochromophore 1 was remarkably stable in phosphate-buffered saline (PBS) containing bovine serum albumin (BSA) whereas 2 was hydrolyzed completely in a few hours. This large difference in stability is likely due, in part, to the difference in the pKa of the leaving groups, as pNP is a much better leaving group (pKa 7.0) than is the electron-rich phenol of the trimethyl lock (e.g., o-methylphenol has a pKa of 10.28) [7,22].
Figure 3.
Time course for the mean change in the absorbance at 410 nm of prochromophore 1 (13 μM) and p-nitrophenyl acetate (2) (13 μM) in PBS containing BSA (1.0 mg/mL). Three separate experiments were performed, and absorbance measurements were made in triplicate.
Figure 3.
Time course for the mean change in the absorbance at 410 nm of prochromophore 1 (13 μM) and p-nitrophenyl acetate (2) (13 μM) in PBS containing BSA (1.0 mg/mL). Three separate experiments were performed, and absorbance measurements were made in triplicate.

Enzymatic Reactivity
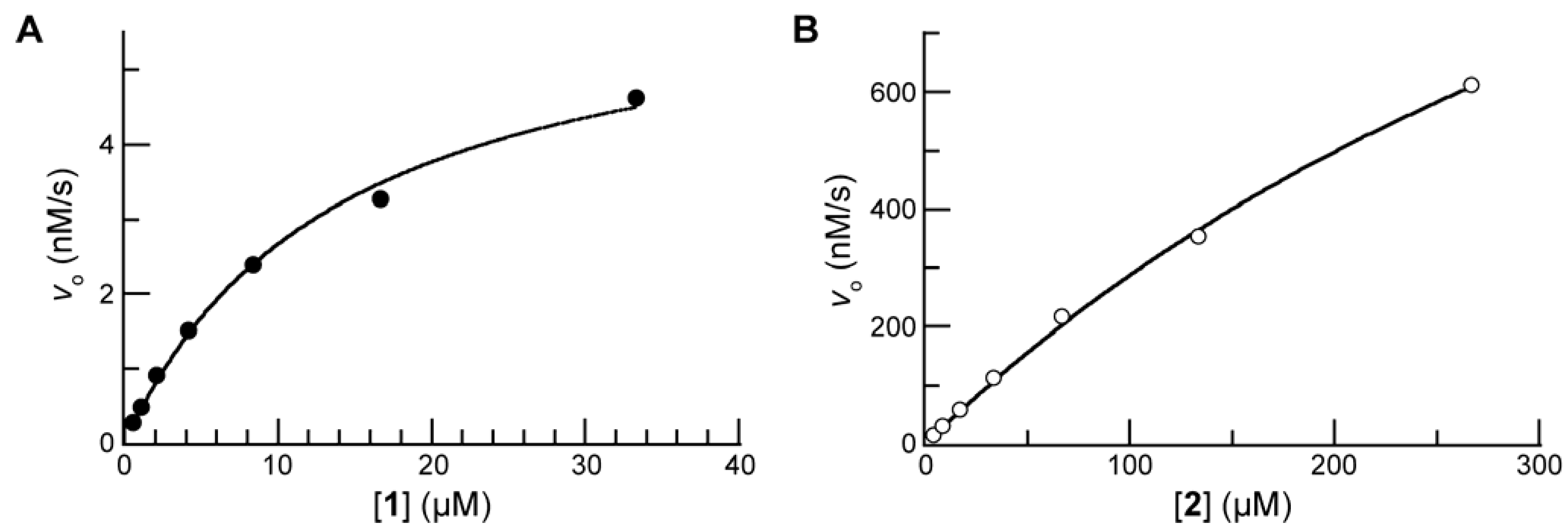
Next, we compared prochromophore 1 and 2 as substrates for PLE. The addition of the trimethyl lock moiety rendered 1 to be less soluble than pNA, and 10% v/v DMSO was used as a cosolvent for all kinetic assays. The value of the Michaelis constant for 1 (KM = 14 μM) was substantially lower than that for 2 (KM = 0.54 mM). The active site of the human homologue of PLE is 10–15 Å deep within a hydrophobic pocket, and the increased size and hydrophobicity of 1 could contribute to the decreased KM value [23]. We have noticed a similar inverse relationship between the molecular size and KM value of our fluorogenic esterase substrates [5,6,7]. The value of the second-order rate constant for the turnover of 1 (kcat/KM = 3.0 × 105 M–1s–1) was within an order of magnitude of that for 2 (kcat/KM = 2.2 × 106 M–1s–1).
Figure 4.
(A) Saturation curve for the hydrolysis of prochromophore 1 (33→0.5 μM) by PLE (0.25 μg/mL) in PBS (pH 7.7) containing DMSO (10% v/v) and monitored at λ = 410 nm; kcat/KM = 3.0 × 105 M–1s–1 and KM = 14 μM. Data points are the mean of triplicate determinations. (B) Saturation curve for the hydrolysis of pNPA (2; 266→4 μM) by PLE (0.25 μg/mL) in PBS (pH 7.7) containing DMSO (10% v/v) and monitored at λ = 410 nm; kcat/KM = 2.2 × 106 M–1s–1 and KM = 0.54 mM. Data points are the mean of triplicate determinations.
Figure 4.
(A) Saturation curve for the hydrolysis of prochromophore 1 (33→0.5 μM) by PLE (0.25 μg/mL) in PBS (pH 7.7) containing DMSO (10% v/v) and monitored at λ = 410 nm; kcat/KM = 3.0 × 105 M–1s–1 and KM = 14 μM. Data points are the mean of triplicate determinations. (B) Saturation curve for the hydrolysis of pNPA (2; 266→4 μM) by PLE (0.25 μg/mL) in PBS (pH 7.7) containing DMSO (10% v/v) and monitored at λ = 410 nm; kcat/KM = 2.2 × 106 M–1s–1 and KM = 0.54 mM. Data points are the mean of triplicate determinations.

Conclusions
We have synthesized an improved chromogenic esterase substrate by incorporating the trimethyl lock. Adopting this prodrug strategy led to superior chemical stability while maintaining high enzymatic reactivity. These attributes could be useful in numerous contexts, including high-throughput screens and directed evolution of esterases and lipases [23,24], and the de novo discovery of new catalysts for ester-bond hydrolysis [25,26]. The modular design of this substrate enables its tailoring to confer reactivity with other enzymes. For example, replacing the acetyl group with a phosphoryl group could yield a substrate superior to p-nitrophenyl phosphate, which like pNPA suffers from deleterious chemical instability.
Experimental
General
Dichloromethane was drawn from a Baker CYCLE-TAINER solvent-delivery system. All other reagents were from Aldrich Chemical (Milwaukee, WI) or Fisher Scientific (Hanover Park, IL), and were used without further purification.
Thin-layer chromatography was performed by using aluminum-backed plates coated with silica gel containing F254 phosphor, and was visualized by UV illumination or developed with ceric ammonium molybdate stain. Flash chromatography was performed on open columns with silica gel-60 (230–400 mesh).
PBS contained (in 1.00 L) KCl (0.20 g), KH2PO4 (0.20 g), NaCl (8.0 g), and Na2HPO4·7H2O (2.16 g) and had pH 7.4.
Spectroscopy
NMR spectra were obtained with a Bruker DMX-400 Avance spectrometer at the National Magnetic Resonance Facility at Madison (NMRFAM). Electron-impact mass spectra were obtained using a Waters (Micromass) AutoSpec™ (Beverly, MA) mass spectrometer at the Mass Spectrometry Facility in the Department of Chemistry.
UV–visible spectrophotometric measurements were made at the University of Wisconsin–Madison Biophysics Instrumentation Facility (BIF) with a Cary 400 UV–visible spectrophotometer from Varian (Palo Alto, CA) equipped with sample stirring and a thermostatted cuvette holder set at 25 °C, using a circulating water bath. Cuvettes were glass or quartz from Starna Cells (Atascadero, CA). Compounds were prepared as stock solutions in DMSO. PLE (163 kDa) was from Sigma Chemical (St. Louis, MO; product number E2884) as a suspension in (NH4)2SO4 (3.2 M), and was diluted to appropriate concentrations in PBS before use. Kinetic parameters were calculated with the programs Microsoft Excel 2000 and GraphPad Prism 4.
Prochromophore 1
3-(2'-Acetoxy-4',6'-dimethylphenyl)-3,3-dimethylpropanoic acid [25] (100 mg, 0.38 mmol) was dissolved in anhydrous CH2Cl2 (1.0 mL). 1-Chloro-N,N,2-trimethylpropenylamine (55 µL, 0.42 mmol) in CH2Cl2 (0.2 mL) was added quickly, and the reaction mixture was stirred under Ar(g) for 3 h at 0 °C. The reaction progress was followed by TLC (30% v/v EtOAc in hexanes) after quenching a small aliquot with MeOH to generate the methyl ester. 4-Nitroaniline (105 mg, 0.76 mmol) and anhydrous pyridine (50 μL) were dissolved in CH2Cl2 (1.0 mL), and the resulting solution was added to the reaction mixture, which was then stirred at ambient temperature overnight. The reaction mixture was partitioned between CH2Cl2 and water. The layers were separated, and the aqueous phase was extracted with CH2Cl2. The combined organic layers were washed with saturated brine, and dried over anhydrous MgSO4(s). Solvent was removed under reduced pressure, and the residue was purified by column chromatography (silica gel, 20% v/v EtOAc in hexanes) to yield 1 as a white crystalline solid (62 mg; 43%, 2 steps). 1H NMR (400 MHz, CDCl3) δ: 8.06 (d, J = 9.2 Hz, 2H), 7.82 (br s, 1H), 7.34 (d, J = 9.2, 2H), 6.74 (s, 1H), 6.65 (s, 1H), 2.52 (s, 2H), 2.32 (s, 3H), 2.36 (s, 3H), 2.23 (s, 3H), 1.72 ppm (s, 6H). 13C NMR (400 MHz, CDCl3) δ: 173.2, 170.3, 150.4, 144.3, 143.2, 139.4, 137.6, 133.4, 132.9, 125.0, 123.6, 118.6, 51.3, 40.8, 32.5, 25.7, 22.1, 20.3 ppm. HRMS (EI): m/z 405.1577 [M+Na]+ ([C21H24N2O5Na] = 405.1578).
Acknowledgements
We are grateful to S. S. Chandran, E. L. Myers, and R. F. Turcotte for contributive discussions. This work was supported by Grant CA073808 (NIH). L.D.L. was supported by Biotechnology Training Grant 08349 (NIH) and an ACS Division of Organic Chemistry Graduate Fellowship sponsored by the Genentech Foundation. NMRFAM was supported by Grant P41RR02301 (NIH). The BIF was established with Grants BIR-9512577 (NSF) and RR13790 (NIH).
References and Notes
- Amsberry, K. L.; Borchardt, R. T. The lactonization of 2'-hydroxyhydrocinnamic acid amides: A potential prodrug for amines. J. Org. Chem. 1990, 55, 5867–5877. [Google Scholar] [CrossRef]
- Testa, B.; Mayer, J. M. Hydrolysis in Drug and Prodrug Metabolism: Chemistry, Biochemistry and Enzymology; Verlag Helvetica Chimica Acta: Zurich, Switzerland, 2003. [Google Scholar]
- Chandran, S. S.; Dickson, K. A.; Raines, R. T. Latent fluorophore based on the trimethyl lock. J. Am. Chem. Soc. 2005, 127, 1652–1653. [Google Scholar] [CrossRef] [PubMed]
- Lavis, L. D.; Chao, T. Y.; Raines, R. T. Fluorogenic label for biomolecular imaging. ACS Chem. Biol. 2006, 1, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Lavis, L. D.; Chao, T. Y.; Raines, R. T. Latent blue and red fluorophores based on the trimethyl lock. ChemBioChem 2006, 7, 1151–1154. [Google Scholar] [CrossRef] [PubMed]
- Milstein, S.; Cohen, L. A. Stereopopulation control. I. Rate enhancement in the lactonizations of o-hydroxyhydrocinnamic acids. J. Am. Chem. Soc. 1972, 94, 9158–9165. [Google Scholar]
- Borchardt, R. T.; Cohen, L. A. Stereopopulation control. II. Rate enhancement of intramolecular nucleophilic displacement. J. Am. Chem. Soc. 1972, 94, 9166–9174. [Google Scholar]
- Dillon, M. P.; Cai, H.; Maag, H. Application of the “trimethyl lock” to Ganciclovir, a pro-prodrug with increased oral bioavailability. Bioorg. Med. Chem. Lett. 1996, 6, 1653–1656. [Google Scholar] [CrossRef]
- Lavis, L.D.; Raines, R.T. Bright ideas for chemical biology. ACS Chem. Biol. 2008, 3. in press. [Google Scholar]
- Hartley, B. S.; Kilby, B. A. The reaction of p-nitrophenyl esters with chymotrypsin and insulin. Biochem. J. 1954, 56, 288–297. [Google Scholar] [PubMed]
- Menger, F. M.; Ladika, M. Origin of rate accelerations in an enzyme model: The p-nitrophenyl ester syndrome. J. Am. Chem. Soc. 1987, 109, 3145–3146. [Google Scholar] [CrossRef]
- Oldfield, C.; Robinson, B. H.; Freedman, R. B. Acid–base behaviour of 4-nitrophenol and 4-nitrophenyl-2-sulphonate in water-in-oil microemulsions stabilized by aerosol-OT. J. Chem. Soc. Faraday Trans. 1990, 86, 833–841. [Google Scholar] [CrossRef]
- Leroy, E.; Bensel, N.; Reymond, J.-L. A low background high-throughput screening (HTS) fluorescence assay for lipases and esterases using acyloxymethylethers of umbelliferone. Bioorg. Med. Chem. Lett. 2003, 13, 2105–2108. [Google Scholar] [CrossRef] [PubMed]
- Sicart, R.; Collin, M.-P.; Reymond, J.-L. Fluorogenic substrates for lipases, esterases, and acylases using a TIM-mechanism for signal release. Biotechnol. J. 2007, 2, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Lowe, G.; Yuthavong, Y. pH-Dependence and structure–activity relationships in the papain-catalysed hydrolysis of anilides. Biochem. J. 1971, 124, 117–122. [Google Scholar] [PubMed]
- Amsberry, K. L.; Gerstenberger, A. E.; Borchardt, R. T. Amine prodrugs which utilize hydroxy amide lactonization. II. A potential esterase-sensitive amide prodrug. Pharm. Res. 1991, 8, 455–461. [Google Scholar]
- Haveaux, B.; Dekoker, A.; Rens, M.; Sidani, A. R.; Toye, J.; Ghosez, L. Chloroenamines, reactive intermediates for synthesis: 1-Chloro-N,N,2-trimethylpropenylamine. Org. Synth. 1979, 59, 26–34. [Google Scholar] [CrossRef]
- Furstner, A.; Weintritt, H. Total synthesis of roseophilin. J. Am. Chem. Soc. 1998, 120, 2817–2825. [Google Scholar] [CrossRef]
- Lyublinskaya, L. A.; Belyaev, S. V.; Strongin, A. Y.; Matyash, L. F.; Levin, E. D.; Stepanov, V. M. A new chromogenic substrate for subtilisin. Anal. Biochem. 1974, 62, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Park, J. H.; Agnello, C. F.; Mathew, E. S→N Transfer and dual acetylation in the S-acetylation and N-acetylation of 3-phosphoglyceraldehyde dehydrogenase by substrates. J. Biol. Chem. 1966, 241, 769–771. [Google Scholar] [PubMed]
- Park, J. H.; Meriwether, B. P.; Clodfelder, P.; Cunningham, L. W. The hydrolysis of p-nitrophenyl acetate catalyzed by 3-phosphoglyceraldehyde dehydrogenase. J. Biol. Chem. 1961, 236, 136–141. [Google Scholar] [PubMed]
- Wolf, N. M.; Morisseau, C.; Jones, P. D.; Hock, B.; Hammock, B. D. Development of a high-throughput screen for soluble epoxide hydrolase inhibition. Anal. Biochem. 2006, 355, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Baumann, H.; Henke, E.; Knoarzycka-Bessler, M.; Bornscheuer, U. T. Directed evolution of lipases and esterases. Methods Enzymol. 2004, 388, 199–207. [Google Scholar] [PubMed]
- Schmidt, M.; Bornscheuer, U. T. High-throughput assays for lipases and esterases. Biomol. Eng. 2005, 22, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Hecht, M. H. Enzyme-like proteins from an unselected library of designed amino acid sequences. Protein Eng. Des. Sel. 2004, 17, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Hecht, M. H.; Das, A.; Go, A.; Bradley, L. H.; Wei, Y. De novo proteins from designed combinatorial libraries. Protein. Sci. 2004, 13, 1711–1723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sample Availability: Contact authors.
© 2008 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
MDPI and ACS Style
Levine, M.N.; Lavis, L.D.; Raines, R.T. Trimethyl Lock: A Stable Chromogenic Substrate for Esterases. Molecules 2008, 13, 204-211. https://doi.org/10.3390/molecules13020204
AMA Style
Levine MN, Lavis LD, Raines RT. Trimethyl Lock: A Stable Chromogenic Substrate for Esterases. Molecules. 2008; 13(2):204-211. https://doi.org/10.3390/molecules13020204
Chicago/Turabian StyleLevine, Michael N., Luke D. Lavis, and Ronald T. Raines. 2008. "Trimethyl Lock: A Stable Chromogenic Substrate for Esterases" Molecules 13, no. 2: 204-211. https://doi.org/10.3390/molecules13020204