
The Roles of the Virome in Cancer


1
Idorsia Pharmaceuticals Ltd., Hegenheimermattweg 91, CH-4123 Allschwil, Switzerland
2
Institute of Medical Microbiology, University of Zurich, Gloriastr. 30, CH-8006 Zurich, Switzerland
3
Max Planck Institute for Molecular Genetics, Ihnestr. 63-73, 14195 Berlin, Germany
*
Authors to whom correspondence should be addressed.
Microorganisms 2021, 9(12), 2538; https://doi.org/10.3390/microorganisms9122538
Submission received: 31 October 2021
/
Revised: 22 November 2021
/
Accepted: 6 December 2021
/
Published: 8 December 2021
(This article belongs to the Special Issue The Role of the Virome in Health and Disease)
Abstract
:Viral infections as well as changes in the composition of the intestinal microbiota and virome have been linked to cancer. Moreover, the success of cancer immunotherapy with checkpoint inhibitors has been correlated with the intestinal microbial composition of patients. The transfer of feces—which contain mainly bacteria and their viruses (phages)—from immunotherapy responders to non-responders, known as fecal microbiota transplantation (FMT), has been shown to be able to convert some non-responders to responders. Since phages may also increase the response to immunotherapy, for example by inducing T cells cross-reacting with cancer antigens, modulating phage populations may provide a new avenue to improve immunotherapy responsiveness. In this review, we summarize the current knowledge on the human virome and its links to cancer, and discuss the potential utility of bacteriophages in increasing the responder rate for cancer immunotherapy.
1. Introduction—The Human Virome
The human body hosts a large number of viruses, an estimated 1012–1014 particles per individual, that collectively are termed the virome [1,2]. Despite recent progress in determining the virome, a large fraction of viral sequence data collected in virome analysis studies remain unidentified ‘dark matter’. This indicates that a large number of currently unknown viruses, and their contributions to health and disease, remain to be characterized. Broadly speaking, the human virome consists of eukaryotic viruses, including pathogens that infect human cells, and viruses that infect bacteria termed bacteriophages or phages. Both human pathogenic viruses and phages have been linked to cancer, as will be discussed in this review. Other viruses that are also present in the virome, such as plant viruses (likely coming from food sources) will not be discussed here since no data are available on a possible connection to cancer. The by far largest component of the human virome are the phages, and most of them are associated with the bacteria they infect, which are most numerous in the intestinal tract, which harbors about 3.8 × 1013 bacteria per individual [3]. Here we summarize the current knowledge on the connection of the human virome to cancer development and discuss the possibility of modulating virus and phage populations to potentially increase the responder rates to cancer immunotherapy.
2. The Roles of the Eukaryotic Virome in Cancer
Various eukaryotic viruses infectious to humans have been implicated in carcinogenesis inside and outside the intestinal tract, mainly those of the families Papillomaviridae and Herpesviridae as well as hepatitis viruses (Table 1). In 2012, 15.4% of new cancers (2.2 million cases) worldwide were attributed to carcinogenic infections [4], most frequently with the bacterium Helicobacter pylori (770,000 cases) followed by 640,000 cases linked to human papillomavirus (HPV), 420,000 and 170,000 cases linked to hepatitis B and C virus (HBV and HCV), respectively, and 120,000 associated with Epstein–Barr virus (EBV, also known as human herpesvirus 4, HHV4). Of note, infections with these viruses does not necessarily lead to cancer. Rather, they often are one of many contributing factors to carcinogenesis. As such, virus-associated cancers typically develop as part of persistent infections over many years [5]. The proposed mechanisms of viral contributions to carcinogenesis also vary widely between viral species and will be briefly discussed in the following subsections. In general, viral infections can contribute to carcinogenesis by any of the following mechanisms: insertional mutation in the host genome, induction of inflammation and modulation of signaling pathways in the infected cells, for example, by viral oncogenes [6,7] (Figure 1).
2.1. Papillomaviridae
Papillomaviridae is a family of non-enveloped viruses with double-stranded DNA genomes [67]. Members infectious to humans, HPV, have been linked to various cancers, most significantly cervical cancer [11,12,13]. However, only a subset of the ~150 types of HPV, the so-called high-risk types such as HPV-16 and-18, are typically linked to cancer [68]. The main proposed mechanism of contribution of high-risk HPV to cervical cancer is through integration of the viral DNA into the host genome and expression of viral oncogenes. It has been shown, for example, that the E6 and E7 early oncogenes degrade tumor suppressors p53 and retinoblastoma protein (pRb), which among other effects cause the cell to arrest in the S-phase, leading to genomic instability, aneuploidy, DNA damage and, consequently, carcinogenesis [69,70] (see Figure 2 for details). It is thought that a similar mechanism is also at work in other HPV-associated cancers such as colorectal cancer [71]. HPV is the most frequently sexually transmitted infectious agent in the United States, and vaccines against HPV are recommended for all girls aged 11 to 12 years, aiming to reduce the burden of cervical cancer [72]. HPV has also been identified as a potential risk factor for various other cancer types, including anal [9,10], bladder [11] colorectal [14,15] esophageal [17,18,19], head and neck [11,20,21], oral [23], prostate [24], renal [25], skin/mucosal [26,27,28] and vulvar cancer [29]. In most of these cases, however, the association with HPV is not as strong as for cervical cancer, or is only seen in subsets of patients, and consequently there is also conflicting data. For example, a study from 2014 found no link between HPV infection and colorectal cancer [16]. In addition, there is an established correlation between HPV infection and head and neck cancer [11,20,21], but also data indicating that the presence of HPV in head and neck tumors predicts better long-term clinical outcome [22]. Moreover, merely associating the presence of specific viral sequences with cancer status does not necessarily indicate a causal role of the virus.
2.2. Herpesviridae
Viruses of the Herpesviridae family are enveloped dsDNA viruses infecting mammals, birds and reptiles [73]. Various members have been implicated in cancer formation, including cytomegalovirus (CMV, also known as human herpesvirus 5, HHV5), Epstein–Barr virus (EBV or HHV4), herpes simplex virus (HSV or HHV1/2) and HHV6 and HHV7 (Table 1). The strongest association with cancer has been reported for EBV, which has been linked to colorectal carcinoma (CRC) [33] (although a later study found no association [16]), esophageal [34] and gastric cancer [35] (interestingly, however, patients with EBV-positive gastric cancer had a better response to chemotherapy and better survival [36]), hepatocellular carcinoma [37], lymphoma (Burkitt lymphoma, diffuse large B-cell lymphoma and peripheral T-cell lymphoma) [38,39,40], oral [41], as well as skin and mucosa associated cancers [26]. Therefore, it has been proposed that vaccination against EBV might be a viable means to prevent EBV-associated cancers [74]. Various EBV vaccine candidates are currently in preclinical or clinical development.
CMV has been linked to colorectal cancer [30], however, in non-elderly patients CMV-positive tumors have been associated with increased disease-free survival rate [31]). HHV6 was found to be connected to malignant melanoma [26] and B-cell lymphoma [39], HHV7 to bladder and oral cancer as well as to T-cell lymphoma [26], and HSV has been associated with oral cancer [41].
In HIV-infected individuals, Kaposi sarcoma-associated herpesvirus (KSHV) infection is associated with Kaposi sarcoma [42].
Mechanistically, herpesviruses such as CMV and HSV and others are known to induce DNA synthesis and counteract apoptosis via activation of the rat sarcoma (Ras)/rat fibrosarcoma (Raf)/meiosis-specific kinase (MEK)/extracellular signal-related kinase (ERK) pathway [71,75] (see Figure 3 for details). In addition, KSHV has been shown to inhibit tumor suppressor protein p53 [76].
2.3. Polyomaviridae
Two members of the Polyomaviridae family (non-enveloped double-stranded DNA (dsDNA) viruses), namely BKV and JCV, have been linked to CRC [43,44,46,47,48,49]. However, other studies have challenged this notion and found no link [16,45]. There is also a possible association of BKV with bladder cancer [26]. The clearest evidence, however, has been obtained for Merkel cell polyomavirus (MCV) that has been identified as a major causative factor for Merkel cell carcinoma [50,51]. Consequently, it has been proposed that an anti-MCV vaccine might help to prevent Merkel cell carcinoma [77,78].
It has been hypothesized that MCV contributes to oncogenesis in two steps: integration of the viral genome into the host genome followed by mutational truncation of the large T antigen gene [79]. The truncated T antigen promotes cell cycle progression. The T antigen likely also plays a role in oncogenic transformation mediated by other polyomaviruses such as BKV and JCV. It has been shown to interact with various host proteins, thereby altering the processes of DNA repair, cell cycle regulation and proliferation [80] (see Figure 4 for details).
2.4. Retroviridae
The retroviruses human immunodeficiency virus (HIV) and human T-lymphotropic virus type 1 (HTLV-1) have both been associated with cancer. Three HIV-associated cancers are known, cervical cancer, aggressive B cell non-Hodgkin lymphoma and Kaposi sarcoma [55,56]. In addition, HIV infection has been linked to an increased risk for anal cancer [52,53] and worse overall colostomy-free survival rates [54]. The effect of HIV infection is likely indirect; the virus weakens the immune system and renders infected individuals more susceptible to infections by other viruses, such as Kaposi sarcoma-associated herpesvirus (KSHV), which has been linked to Kaposi sarcoma [42]. For HTLV-1, oncogenesis is thought to involve multiple mechanisms [6,81]. These include: (1) the establishment of a persistent infection that results in chronic inflammation, which attracts phagocytes that release reactive oxygen species, contributing to DNA damage, (2) the genomic integration and expression of viral oncogenes, such as the Tax-1 protein that, among other functions, inhibits tumor suppressor p53, and (3) the suppression of host immune responses. A summary of the functions of Tax-1 is shown in Figure 5.
2.5. Other Viruses
Various other viruses have been linked to cancer. Among these are the hepatitis viruses HBV and HCV. HBV (family Hepadnaviridae with a partially double-stranded DNA genome and an envelope) infects the liver and has been linked to liver cancer [11] but may also be a risk factor for cancers of the bile duct [59], the colon [60] and the pancreas [61,62]. HCV (an enveloped ssRNA virus of the Flaviviridae family) also infects the liver and has been associated with liver and bile duct cancer [11,59]. Liver carcinogenesis by HBV and HCV is multifactorial and has been linked, amongst others, to virus-induced changes in signaling pathways, the inflammatory status and to elevated production of reactive oxygen species (ROS) and reactive nitrogen species (RNS) [82,83,84] (see Figure 6 for details). Anelloviruses (circular single-stranded DNA (ssDNA) viruses) including torque teno virus (TTV) have been associated with hepatic [63] and mucosal [26] cancer as well as leukemias [26]. Human bocavirus (HBoV) and other members of the Parvoviridae family (ssDNA viruses) have been linked to various cancers, including colorectal and lung cancer [64], squamous cell carcinoma [65] and skin cancer [26]. The ssRNA orthobunyaviruses may be linked to colorectal cancer [66]. The mechanisms of oncogenesis of anelloviruses, parvoviruses and orthobunyaviruses remain largely unknown.
3. The Human Intestinal Virome and Its Links to Cancer
The human intestinal microbiota is a complex community of bacteria, archaea, viruses, fungi, and eukaryotic parasites [85]. Collectively, the microbes outnumber human cells. The human body contains about 3 × 1013 human cells and 3.8 × 1013 bacteria [3], and bacterial genes (~2,000,000) are estimated to be about 100 times more numerous than human genes (~20,000) [86]. Viruses, predominantly phages, are likely the most abundant biological entities, with an estimated number of 1015 in the human gastrointestinal tract [87]. Other community members such as archaea of fungi are substantially less abundant. Collectively, one could take the view that we are symbionts of host and microbial cells, each with their own set of genes. The gut microbiota plays important roles in food digestion, development of the immune system and protection against microbial pathogens. Pathological changes in the microbial composition (dysbiosis), induced by infections, chronic diseases, or antibiotic treatment, can lead to failure of control of pathogens and severe damage as in inflammatory bowel disease (IBD). Dysbiosis is also observed in extra-intestinal diseases such as asthma or autism [88].
The composition of the human virome is largely unknown. A recent study described a catalog of tens of thousands of viruses from human metagenomes, which revealed associations with chronic diseases [89]. This study describes 45,000 unique viruses as part of the human microbiota, and about 2000 specific phages were found to correlate with a variety of common chronic diseases such as Parkinson’s disease and obesity. All bacteria are infected by phages, which play a role in maintaining the bacterial population in healthy individuals. Depending on environmental conditions and the genetic composition of the phages, they can lyse their bacterial hosts. In the oceans where bacteria and phages are very abundant lysis occurs in about 20% of the bacteria in about 24 h [90]. The virome may regulate the microbiome and may influence bacterial complexity, whereby the population dynamics likely follow a ‘kill-the-winner’ ecological model. In this model, the most abundant bacteria are killed by their phages, other bacterial taxa will take over the ecological niche and be subsequently killed by their phages [90]. Thereby, phages play a crucial role in shaping the composition of the intestinal bacterial communities. They also facilitate horizontal gene transfer and thereby the functional capacity of the microbiota [90]. The virome is more challenging to study and to characterize than bacterial populations, and many of its contributions to ecosystems are still poorly understood. Many of the viruses are unknown, sometimes designated as ‘viral dark matter’ [91].
Changes in the fecal virome, especially the phage population, have been reported recently for CRC patients [7,66,92,93]. It has been suggested that phages may play a causative role in CRC by altering the bacterial populations of the intestine such that pathogenic bacteria can thrive and form biofilms [92,94] (Figure 7). Several phage species have been linked to CRC [66,71]. Comparing the fecal microbiota of CRC patients to healthy controls, specific phages have been found to be more abundant in patients that could play a role in shifting the overall bacterial populations or serve as biomarkers. Phage SpSL1, for example, infects bacteria of the Streptococcus genus and was found to be more abundant in early-stage CRC than in controls but its abundance decreased in later stages [66]. Depletion of commensal Escherichia coli bacteria by various phages whose abundance increases during CRC, including Enterobacteria phage HK544, Punalikevirus, Lambdalikevirus and Mulikevirus, may contribute to the disease-associated dysbiosis [66]. However, it remains unknown if phages have a causal role in the development of dysbiosis, or whether their increased abundance is a consequence of dysbiosis. The observed differences, however, might serve as a starting point for targeted microbiota manipulations aiming to reverse the dysbiosis, which may have a positive impact on treatment.
4. Fecal Microbiota Transplantation—Focus on Viruses and Cancer
In recent years, fecal microbiota transplantation (FMT), has gained attention mostly as a remedy for recurrent Clostridioides difficile infections, with impressive cure rates of about 90% or higher [96]. We described such a case of a patient from Zurich, Switzerland who received fecal material from a healthy donor and recovered without adverse events, meanwhile healthy for almost ten years [97]. An indicator of successful therapy is the diversity of the microbiota, which is higher and different in composition in healthy people than in C. difficile patients where it is diminished due to antibiotic therapy.
Recent data suggest that sterile fecal filtrate in which bacteria have been removed (but phages retained) can also effectively cure C. difficile infections [98,99]. Moreover, successful FMT was associated with the stable transfer of phages from donor to recipient [100,101,102]. We have shown that a core virome comprising the most dominant phage species is transmitted during FMT and stabilizes in the recipient before the bacterial populations do [101]. These studies indicate that phages might be sufficient in conferring the beneficial effects of FMT, although a contribution of metabolites, proteins and bacterial fragments present in the filtrate cannot be excluded.
The composition of the intestinal microbiota has recently been shown to contribute to the success of anticancer therapy with checkpoint inhibitors [103]. The new immunotherapy has a 20–40% response rate, but in the remaining patients the therapy has virtually no effect [104]. How to cure the remaining 60–80% is one of the most urgent research topics. The microbiota likely plays a role. It has been shown recently that FMT using donor stool from responders can boost the response to checkpoint inhibitor therapy of previous non-responder melanoma patients [105,106]. Conversely, antibiotic therapy, which disrupts the intestinal microbiota, correlates with poor outcome of checkpoint inhibitor therapy [107].
Germ-free or antibiotic-treated mice with depleted intestinal bacteria serve as animal models for the effects of the microbiota of responders and non-responders [103]. Indeed, instilling microbiota from responders transferred a “responder” phenotype (i.e., relatively slow tumor growth) to mice, whereas transfer from non-responders rendered mice non-responsive (i.e., relatively fast tumor growth) [108,109,110,111]. The comparison of stool samples from responders and non-responders revealed certain bacterial species whose presence or abundance correlated with successful treatment. This included, among others, Faecalibacterium prausnitzii, Akkermansia muciniphila, Bifidobacterium longum and Bacteroides caccae, depending on the type of cancer [108,109,110,111,112].
Stool transfer from a responder can help to convert a non-responder into a responder. This was shown recently by two studies [105,106]. Davar et al. [105] performed FMT on 15 non-responding patients with malignant melanoma using donor stool from responders. Six of the 15 patients showed objective responses to pembrolizumab, a monoclonal antibody against programmed cell death-1 (PD-1), following the procedure. There were also no severe side effects. Bacteria associated with clinical response were, among others, Bifidobacterium longum, Colinsella aerofaciens and Faecalibacterium prausnitzii, enriched were mostly bacteria of the Firmicutes phylum (Ruminococcaceae and Lachnospiraceae families). In addition, response to therapy correlated with a specific subset of T cells with cytolytic function (CD56+CD8+ T cells) and decreased interleukin-8 expressing myeloid cells. Some of the above-mentioned bacteria were confirming earlier observations [105]. Thus, a single FMT helped to overcome primary resistance to immunotherapy in a subset of melanoma patients. Why still nine out of 15 patients remained non-responsive remains unclear.
In a similar study, ten patients with anti-PD-1 refractory metastatic melanoma received FMT with donor stool from responders followed by reintroduction of anti-PD-1 immunotherapy [106]. Two partial responses and one complete remission were observed. Patients first underwent a microbiota depletion phase with antibiotic therapy for 72 h (vancomycin, neomycin). They received oral stool capsules every two weeks for 90 days and anti-PD-1 therapy with nivolumab. No clear-cut microbiota indicative for responders was determined. The safety of FMT was confirmed. Antibiotic therapy was effective as pre-FMT protocol and was intended to allow for a better engraftment of the transferred microbiota.
Of note, the viromes of responders and non-responders so far have not been analyzed, so it remains unclear if there are certain phage species or eukaryotic viruses that correlate with successful treatment. However, it has been shown recently that a phage-specific T cell epitope is linked to PD-1 responsiveness in mice [113]. This T cell epitope has been proposed to induce memory T cells cross-reacting with a tumor antigen. This opens up the avenue that certain phage species could be used therapeutically to stimulate the immune system to enhance the efficacy of immunotherapy. Moreover, the microbiota of responders may contain such phage species which activate T cells that are cross-reactive to tumor antigens; phages that may be absent in non-responders. Modulating phage populations of non-responders may therefore be a promising avenue to increase responsiveness to immunotherapy. Eukaryotic viruses may also contribute to responsiveness through their known roles in modulating the immune system [114]. Virome analyses of responders and non-responders are warranted to identify such phages and viruses.
5. Conclusions and Outlook
Carcinogenesis is a multifactorial process that involves genetic and environmental factors, the microbiota, and the virome [115] (Figure 8). Both eukaryotic viruses and phages can contribute to cancer formation and progression, as discussed herein, through various mechanisms. Of note, we are only beginning to understand the complexity of the human virome, and new viruses will be discovered in the ‘dark matter’ of so far unknown virus-like sequences generated during virome analyses. These may include viruses which contribute to cancer that we may, for instance, ingest with our food [116], or the more recently discovered giant viruses including Mimiviridae and Phycodnaviridae whose oncogenic potential may be underestimated [117,118]. Knowing the identity of potential oncogenic viruses would provide the opportunity for intervention and vaccination approaches that have already been implemented for HPV to effectively prevent cervical cancer. The virome may also play a role in the response of patients to cancer immunotherapy with checkpoint inhibitors; similar to beneficial bacterial taxa that correlate with response to treatment, beneficial viruses and phages may be identified. In particular, phages may be crucial in mediating the effects of FMT, a therapeutic intervention that has shown promise to ‘convert’ non-responders to immune therapy to responders [105,106]. We and others have shown that during FMT, phages are transmitted [100,101,102,119]. In some indications, sterile fecal filtrates (containing phages, but not bacteria) may even be more efficient than FMT, as shown recently for necrotizing enterocolitis of preterm infants [120]. The role of phages and eukaryotic viruses in determining the responder status to immunotherapy has not been investigated to date. This knowledge may help to improve response rates for cancer immunotherapy by targeted supplementation and modulation of the patients’ phageomes and viromes.
Author Contributions
K.M. and F.B. wrote the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Shkoporov, A.N.; Hill, C. Bacteriophages of the Human Gut: The “Known Unknown“ of the Microbiome. Cell Host Microbe 2019, 25, 195–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, G.; Bushman, F.D. The human virome: Assembly, composition and host interactions. Nat. Rev. Microbiol. 2021, 19, 514–527. [Google Scholar] [CrossRef] [PubMed]
- Sender, R.; Fuchs, S.; Milo, R. Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLoS Biol. 2016, 14, e1002533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plummer, M.; de Martel, C.; Vignat, J.; Ferlay, J.; Bray, F.; Franceschi, S. Global burden of cancers attributable to infections in 2012: A synthetic analysis. Lancet Glob. Health 2016, 4, e609–e616. [Google Scholar] [CrossRef] [Green Version]
- Mesri, E.A.; Feitelson, M.A.; Munger, K. Human viral oncogenesis: A cancer hallmarks analysis. Cell Host Microbe 2014, 15, 266–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.L.; Wie, J.Y.; Wang, L.; Huang, S.L.; Chen, J.L. Human T-cell lymphotropic virus type 1 and its oncogenesis. Acta Pharmacol. Sin. 2017, 38, 1093–1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marônek, M.; Link, R.; Monteleone, G.; Gardlík, R.; Stolfi, C. Viruses in Cancers of the Digestive System: Active Contributors or Idle Bystanders? Int. J. Mol. Sci. 2020, 21, 8133. [Google Scholar] [CrossRef] [PubMed]
- Bodaghi, S.; Yamanegi, K.; Xiao, S.Y.; Da Costa, M.; Palefsky, J.M.; Zheng, Z.M. Colorectal papillomavirus infection in patients with colorectal cancer. Clin. Cancer Res. 2005, 11, 2862–2867. [Google Scholar] [CrossRef] [Green Version]
- Muresu, N.; Sotgiu, G.; Saderi, L.; Sechi, I.; Cossu, A.; Marras, V.; Meloni, M.; Martinelli, M.; Cocuzza, C.; Tanda, F.; et al. Distribution of HPV Genotypes in Patients with a Diagnosis of Anal Cancer in an Italian Region. Int. J. Environ. Res. Public Health 2020, 17, 4516. [Google Scholar] [CrossRef]
- Kabarriti, R.; Brodin, N.P.; Ohri, N.; Narang, R.; Huang, R.; Chuy, J.W.; Rajdev, L.N.; Kalnicki, S.; Guha, C.; Garg, M.K. Human papillomavirus, radiation dose and survival of patients with anal cancer. Acta Oncol. 2019, 58, 1745–1751. [Google Scholar] [CrossRef] [PubMed]
- Cantalupo, P.G.; Katz, J.P.; Pipas, J.M. Viral sequences in human cancer. Virology 2018, 513, 208–216. [Google Scholar] [CrossRef] [PubMed]
- de Sanjose, S.; Quint, W.G.; Alemany, L.; Geraets, D.T.; Klaustermeier, J.E.; Lloveras, B.; Tous, S.; Felix, A.; Bravo, L.E.; Shin, H.R.; et al. Human papillomavirus genotype attribution in invasive cervical cancer: A retrospective cross-sectional worldwide study. Lancet Oncol. 2010, 11, 1048–1056. [Google Scholar] [CrossRef]
- Clifford, G.; Franceschi, S.; Diaz, M.; Muñoz, N.; Villa, L.L. Chapter 3: HPV type-distribution in women with and without cervical neoplastic diseases. Vaccine 2006, 24 (Suppl. S3), 26–34. [Google Scholar] [CrossRef]
- Kirgan, D.; Manalo, P.; Hall, M.; McGregor, B. Association of human papillomavirus and colon neoplasms. Arch. Surg. 1990, 125, 862–865. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.M.; Leu, S.Y.; Chiang, H.; Fung, C.P.; Liu, W.T. Human papillomavirus type 18 in colorectal cancer. J. Microbiol. Immunol. Infect. 2001, 34, 87–91. [Google Scholar] [PubMed]
- Fiorina, L.; Ricotti, M.; Vanoli, A.; Luinetti, O.; Dallera, E.; Riboni, R.; Paolucci, S.; Brugnatelli, S.; Paulli, M.; Pedrazzoli, P.; et al. Systematic analysis of human oncogenic viruses in colon cancer revealed EBV latency in lymphoid infiltrates. Infect. Agent. Cancer 2014, 9, 18. [Google Scholar] [CrossRef] [Green Version]
- Bjørge, T.; Hakulinen, T.; Engeland, A.; Jellum, E.; Koskela, P.; Lehtinen, M.; Luostarinen, T.; Paavonen, J.; Sapp, M.; Schiller, J.; et al. A prospective, seroepidemiological study of the role of human papillomavirus in esophageal cancer in Norway. Cancer Res. 1997, 57, 3989–3992. [Google Scholar] [PubMed]
- Zhang, S.K.; Guo, L.W.; Chen, Q.; Zhang, M.; Liu, S.Z.; Quan, P.L.; Lu, J.B.; Sun, X.B. The association between human papillomavirus 16 and esophageal cancer in Chinese population: A meta-analysis. BMC Cancer 2015, 15, 1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Tian, X.; Liu, F.; Zhao, Y.; Sun, M.; Chen, D.; Lu, C.; Wang, Z.; Shi, X.; Zhang, Q.; et al. Detection of HPV DNA in esophageal cancer specimens from different regions and ethnic groups: A descriptive study. BMC Cancer 2010, 10, 19. [Google Scholar] [CrossRef] [Green Version]
- Khoury, J.D.; Tannir, N.M.; Williams, M.D.; Chen, Y.; Yao, H.; Zhang, J.; Thompson, E.J.; TCGA Network; Meric-Bernstam, F.; Medeiros, L.J.; et al. Landscape of DNA virus associations across human malignant cancers: Analysis of 3775 cases using RNA-Seq. J. Virol. 2013, 87, 8916–8926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parfenov, M.; Pedamallu, C.S.; Gehlenborg, N.; Freeman, S.S.; Danilova, L.; Bristow, C.A.; Lee, S.; Hadjipanayis, A.G.; Ivanova, E.V.; Wilkerson, M.D.; et al. Characterization of HPV and host genome interactions in primary head and neck cancers. Proc. Natl. Acad. Sci. USA 2014, 111, 15544–15549. [Google Scholar] [CrossRef] [Green Version]
- Curado, M.P.; Hashibe, M. Recent changes in the epidemiology of head and neck cancer. Curr. Opin. Oncol. 2009, 21, 194–200. [Google Scholar] [CrossRef]
- Maden, C.; Beckmann, A.M.; Thomas, D.B.; McKnight, B.; Sherman, K.J.; Ashley, R.L.; Corey, L.; Daling, J.R. Human papillomaviruses, herpes simplex viruses, and the risk of oral cancer in men. Am. J. Epidemiol. 1992, 135, 1093–1102. [Google Scholar] [CrossRef]
- Bae, J.M. Human papillomavirus 16 infection as a potential risk factor for prostate cancer: An adaptive meta-analysis. Epidemiol. Health 2015, 37, e2015005. [Google Scholar] [CrossRef] [Green Version]
- Farhadi, A.; Behzad-Behbahani, A.; Geramizadeh, B.; Sekawi, Z.; Rahsaz, M.; Sharifzadeh, S. High-risk human papillomavirus infection in different histological subtypes of renal cell carcinoma. J. Med. Virol. 2014, 86, 1134–1144. [Google Scholar] [CrossRef]
- Mollerup, S.; Asplund, M.; Friis-Nielsen, J.; Kjartansdóttir, K.R.; Fridholm, H.; Hansen, T.A.; Herrera, J.A.R.; Barnes, C.J.; Jensen, R.H.; Richter, S.R.; et al. High-Throughput Sequencing-Based Investigation of Viruses in Human Cancers by Multienrichment Approach. J. Infect. Dis. 2019, 220, 1312–1324. [Google Scholar] [CrossRef]
- Gewirtzman, A.; Bartlett, B.; Tyring, S. Epidermodysplasia verruciformis and human papilloma virus. Curr. Opin. Infect. Dis. 2008, 21, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Ramoz, N.; Rueda, L.A.; Bouadjar, B.; Montoya, L.S.; Orth, G.; Favre, M. Mutations in two adjacent novel genes are associated with epidermodysplasia verruciformis. Nat. Genet. 2002, 32, 579–581. [Google Scholar] [CrossRef]
- Rakislova, N.; Saco, A.; Sierra, A.; Del Pino, M.; Ordi, J. Role of Human Papillomavirus in Vulvar Cancer. Adv. Anat. Pathol. 2017, 24, 201–214. [Google Scholar] [CrossRef] [PubMed]
- Dimberg, J.; Hong, T.T.; Skarstedt, M.; Löfgren, S.; Zar, N.; Matussek, A. Detection of cytomegalovirus DNA in colorectal tissue from Swedish and Vietnamese patients with colorectal cancer. Anticancer Res. 2013, 33, 4947–4950. [Google Scholar]
- Chen, H.P.; Jiang, J.K.; Chen, C.Y.; Yang, C.Y.; Chen, Y.C.; Lin, C.H.; Chou, T.Y.; Cho, W.L.; Chan, Y.J. Identification of human cytomegalovirus in tumour tissues of colorectal cancer and its association with the outcome of non-elderly patients. J. Gen. Virol. 2016, 97, 2411–2420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.P.; Jiang, J.K.; Chan, C.H.; Teo, W.H.; Yang, C.Y.; Chen, Y.C.; Chou, T.Y.; Lin, C.H.; Chan, Y.J. Genetic polymorphisms of the human cytomegalovirus UL144 gene in colorectal cancer and its association with clinical outcome. J. Gen. Virol. 2015, 96, 3613–3623. [Google Scholar] [CrossRef]
- Song, L.B.; Zhang, X.; Zhang, C.Q.; Zhang, Y.; Pan, Z.Z.; Liao, W.T.; Li, M.Z.; Zeng, M.S. Infection of Epstein-Barr virus in colorectal cancer in Chinese. Ai Zheng 2006, 25, 1356–1360. [Google Scholar]
- Awerkiew, S.; Bollschweiler, E.; Metzger, R.; Schneider, P.M.; Hölscher, A.H.; Pfister, H. Esophageal cancer in Germany is associated with Epstein-Barr-virus but not with papillomaviruses. Med. Microbiol. Immunol. 2003, 192, 137–140. [Google Scholar] [CrossRef] [PubMed]
- Martínez-López, J.L.; Torres, J.; Camorlinga-Ponce, M.; Mantilla, A.; Leal, Y.A.; Fuentes-Pananá, E.M. Evidence of Epstein-Barr virus association with gastric cancer and non-atrophic gastritis. Viruses 2014, 6, 301–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corallo, S.; Fucà, G.; Morano, F.; Salati, M.; Spallanzani, A.; Gloghini, A.; Volpi, C.C.; Trupia, D.V.; Lobefaro, R.; Guarini, V.; et al. Clinical Behavior and Treatment Response of Epstein-Barr Virus-Positive Metastatic Gastric Cancer: Implications for the Development of Future Trials. Oncologist 2020, 25, 780–786. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Wu, B.A.; Zeng, Y.M.; Chen, G.C.; Li, X.X.; Chen, J.T.; Guo, Y.W.; Li, M.H.; Zeng, Y. Epstein-Barr virus in hepatocellular carcinogenesis. World J. Gastroenterol. 2004, 10, 3409–3413. [Google Scholar] [CrossRef]
- Brady, G.; MacArthur, G.J.; Farrell, P.J. Epstein-Barr virus and Burkitt lymphoma. J. Clin. Pathol. 2007, 60, 1397–1402. [Google Scholar] [CrossRef] [PubMed]
- Strong, M.J.; O’Grady, T.; Lin, Z.; Xu, G.; Baddoo, M.; Parsons, C.; Zhang, K.; Taylor, C.M.; Flemington, E.K. Epstein-Barr virus and human herpesvirus 6 detection in a non-Hodgkin’s diffuse large B-cell lymphoma cohort by using RNA sequencing. J. Virol. 2013, 87, 13059–13062. [Google Scholar] [CrossRef] [Green Version]
- Nakhoul, H.; Lin, Z.; Wang, X.; Roberts, C.; Dong, Y.; Flemington, E. High-Throughput Sequence Analysis of Peripheral T-Cell Lymphomas Indicates Subtype-Specific Viral Gene Expression Patterns and Immune Cell Microenvironments. mSphere 2019, 4, e00248-19. [Google Scholar] [CrossRef] [Green Version]
- Jalouli, J.; Jalouli, M.M.; Sapkota, D.; Ibrahim, S.O.; Larsson, P.A.; Sand, L. Human papilloma virus, herpes simplex virus and epstein barr virus in oral squamous cell carcinoma from eight different countries. Anticancer Res. 2012, 32, 571–580. [Google Scholar]
- Goncalves, P.H.; Montezuma-Rusca, J.M.; Yarchoan, R.; Uldrick, T.S. Cancer prevention in HIV-infected populations. Semin. Oncol. 2016, 43, 173–188. [Google Scholar] [CrossRef] [Green Version]
- Narayanan, M.; Szymanski, J.; Slavcheva, E.; Rao, A.; Kelly, A.; Jones, K.; Jaffers, G. BK virus associated renal cell carcinoma: Case presentation with optimized PCR and other diagnostic tests. Am. J. Transplant. 2007, 7, 1666–1671. [Google Scholar] [CrossRef] [PubMed]
- Abend, J.R.; Jiang, M.; Imperiale, M.J. BK virus and human cancer: Innocent until proven guilty. Semin. Cancer Biol. 2009, 19, 252–260. [Google Scholar] [CrossRef] [Green Version]
- Shoraka, H.R.; Abobakri, O.; Naghibzade Tahami, A.; Mollaei, H.R.; Bagherinajad, Z.; Malekpour Afshar, R.; Shahesmaeili, A. Prevalence of JC and BK viruses in Patients with Colorectal Cancer: A Systematic Review and Meta- Analysis. Asian Pac. J. Cancer Prev. 2020, 21, 1499–1509. [Google Scholar] [CrossRef]
- Coelho, T.R.; Gaspar, R.; Figueiredo, P.; Mendonça, C.; Lazo, P.A.; Almeida, L. Human JC polyomavirus in normal colorectal mucosa, hyperplastic polyps, sporadic adenomas, and adenocarcinomas in Portugal. J. Med. Virol. 2013, 85, 2119–2127. [Google Scholar] [CrossRef]
- Hori, R.; Murai, Y.; Tsuneyama, K.; Abdel-Aziz, H.O.; Nomoto, K.; Takahashi, H.; Cheng, C.M.; Kuchina, T.; Harman, B.V.; Takano, Y. Detection of JC virus DNA sequences in colorectal cancers in Japan. Virchows Arch. 2005, 447, 723–730. [Google Scholar] [CrossRef]
- Laghi, L.; Randolph, A.E.; Chauhan, D.P.; Marra, G.; Major, E.O.; Neel, J.V.; Boland, C.R. JC virus DNA is present in the mucosa of the human colon and in colorectal cancers. Proc. Natl. Acad. Sci. USA 1999, 96, 7484–7489. [Google Scholar] [CrossRef] [Green Version]
- Jung, W.T.; Li, M.S.; Goel, A.; Boland, C.R. JC virus T-antigen expression in sporadic adenomatous polyps of the colon. Cancer 2008, 112, 1028–1036. [Google Scholar] [CrossRef] [Green Version]
- Hashida, Y.; Nakajima, K.; Nakajima, H.; Shiga, T.; Tanaka, M.; Murakami, M.; Matsuzaki, S.; Naganuma, S.; Kuroda, N.; Seki, Y.; et al. High load of Merkel cell polyomavirus DNA detected in the normal skin of Japanese patients with Merkel cell carcinoma. J. Clin. Virol. 2016, 82, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Moore, P.S.; Chang, Y. Why do viruses cause cancer? Highlights of the first century of human tumour virology. Nat. Rev. Cancer 2010, 10, 878–889. [Google Scholar] [CrossRef] [Green Version]
- Jin, F.; Vajdic, C.M.; Law, M.; Amin, J.; van Leeuwen, M.; McGregor, S.; Poynten, I.M.; Templeton, D.J.; Grulich, A.E. Incidence and time trends of anal cancer among people living with HIV in Australia. AIDS 2019, 33, 1361–1368. [Google Scholar] [CrossRef] [PubMed]
- Colón-López, V.; Shiels, M.S.; Machin, M.; Ortiz, A.P.; Strickler, H.; Castle, P.E.; Pfeiffer, R.M.; Engels, E.A. Anal Cancer Risk Among People with HIV Infection in the United States. J. Clin. Oncol. 2018, 36, 68–75. [Google Scholar] [CrossRef]
- Grew, D.; Bitterman, D.; Leichman, C.G.; Leichman, L.; Sanfilippo, N.; Moore, H.G.; Du, K. HIV Infection Is Associated With Poor Outcomes for Patients With Anal Cancer in the Highly Active Antiretroviral Therapy Era. Dis. Colon Rectum 2015, 58, 1130–1136. [Google Scholar] [CrossRef]
- Grulich, A.E.; van Leeuwen, M.T.; Falster, M.O.; Vajdic, C.M. Incidence of cancers in people with HIV/AIDS compared with immunosuppressed transplant recipients: A meta-analysis. Lancet 2007, 370, 59–67. [Google Scholar] [CrossRef]
- Hernández-Ramírez, R.U.; Shiels, M.S.; Dubrow, R.; Engels, E.A. Cancer risk in HIV-infected people in the USA from 1996 to 2012: A population-based, registry-linkage study. Lancet HIV 2017, 4, e495–e504. [Google Scholar] [CrossRef]
- Panfil, A.R.; Martinez, M.P.; Ratner, L.; Green, P.L. Human T-cell leukemia virus-associated malignancy. Curr. Opin. Virol. 2016, 20, 40–46. [Google Scholar] [CrossRef] [Green Version]
- Giam, C.Z.; Semmes, O.J. HTLV-1 Infection and Adult T-Cell Leukemia/Lymphoma—A Tale of Two Proteins: Tax and HBZ. Viruses 2016, 8, 161. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhao, Y.; Li, B.; Huang, J.; Wu, L.; Xu, D.; Yang, J.; He, J. Hepatitis viruses infection and risk of intrahepatic cholangiocarcinoma: Evidence from a meta-analysis. BMC Cancer 2012, 12, 289. [Google Scholar] [CrossRef] [Green Version]
- Su, F.H.; Le, T.N.; Muo, C.H.; Te, S.A.; Sung, F.C.; Yeh, C.C. Chronic Hepatitis B Virus Infection Associated with Increased Colorectal Cancer Risk in Taiwanese Population. Viruses 2020, 12, 97. [Google Scholar] [CrossRef] [Green Version]
- Iloeje, U.H.; Yang, H.I.; Jen, C.L.; Su, J.; Wang, L.Y.; You, S.L.; Lu, S.N.; Chen, C.J. Risk of pancreatic cancer in chronic hepatitis B virus infection: Data from the REVEAL-HBV cohort study. Liver Int. 2010, 30, 423–429. [Google Scholar] [CrossRef]
- Hassan, M.M.; Li, D.; El-Deeb, A.S.; Wolff, R.A.; Bondy, M.L.; Davila, M.; Abbruzzese, J.L. Association between hepatitis B virus and pancreatic cancer. J. Clin. Oncol. 2008, 26, 4557–4562. [Google Scholar] [CrossRef]
- Tokita, H.; Murai, S.; Kamitsukasa, H.; Yagura, M.; Harada, H.; Takahashi, M.; Okamoto, H. High TT virus load as an independent factor associated with the occurrence of hepatocellular carcinoma among patients with hepatitis C virus-related chronic liver disease. J. Med. Virol. 2002, 67, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Schildgen, V.; Malecki, M.; Tillmann, R.L.; Brockmann, M.; Schildgen, O. The Human Bocavirus Is Associated with Some Lung and Colorectal Cancers and Persists in Solid Tumors. PLoS ONE 2013, 8, e68020. [Google Scholar]
- Höpken, M.; Förster, I.; Maune, S.; Brockmann, M.; Schildgen, O.; Schildgen, V. Association of the Human Bocavirus With Tonsil Squamous Cell Carcinomas. Front. Microbiol. 2018, 9, 2450. [Google Scholar] [CrossRef] [Green Version]
- Nakatsu, G.; Zhou, H.; Wu, W.K.K.; Wong, S.H.; Coker, O.O.; Dai, Z.; Li, X.; Szeto, C.H.; Sugimura, N.; Lam, T.Y.; et al. Alterations in Enteric Virome Are Associated with Colorectal Cancer and Survival Outcomes. Gastroenterology 2018, 155, 529–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Villiers, E.M.; Fauquet, C.; Broker, T.R.; Bernard, H.U.; zur Hausen, H. Classification of papillomaviruses. Virology 2004, 324, 17–27. [Google Scholar] [CrossRef] [Green Version]
- Doorbar, J.; Egawa, N.; Griffin, H.; Kranjec, C.; Murakami, I. Human papillomavirus molecular biology and disease association. Rev. Med. Virol. 2015, 25 (Suppl. S1), 2–23. [Google Scholar] [CrossRef] [Green Version]
- Buitrago-Pérez, A.; Garaulet, G.; Vázquez-Carballo, A.; Paramio, J.M.; García-Escudero, R. Molecular Signature of HPV-Induced Carcinogenesis: pRb, p53 and Gene Expression Profiling. Curr. Genomics. 2009, 10, 26–34. [Google Scholar] [CrossRef] [Green Version]
- Lehoux, M.; D’Abramo, C.M.; Archambault, J. Molecular Mechanisms of Human Papillomavirus-Induced Carcinogenesis. Public Health Genom. 2009, 12, 268–280. [Google Scholar] [CrossRef] [Green Version]
- Emlet, C.; Ruffin, M.; Lamendella, R. Enteric Virome and Carcinogenesis in the Gut. Dig. Dis. Sci. 2020, 65, 852–864. [Google Scholar] [CrossRef] [Green Version]
- Braaten, K.P.; Laufer, M.R. Human Papillomavirus (HPV), HPV-Related Disease, and the HPV Vaccine. Rev. Obstet. Gynecol. 2008, 1, 2–10. [Google Scholar]
- Davison, A.J.; Eberle, R.; Ehlers, B.; Hayward, G.S.; McGeoch, D.J.; Minson, A.C.; Pellett, P.E.; Roizman, B.; Studdert, M.J.; Thiry, E. The order Herpesvirales. Arch. Virol. 2009, 154, 171–177. [Google Scholar] [CrossRef]
- Van Zyl, D.G.; Mautner, J.; Delecluse, H.J. Progress in EBV Vaccines. Front. Oncol. 2019, 9, 104. [Google Scholar] [CrossRef] [Green Version]
- Filippakis, H.; Spandidos, D.A.; Sourvinos, G. Herpesviruses: Hijacking the Ras signaling pathway. Biochim. Biophys. Acta 2010, 1803, 777–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, Q.; Xiao, B.; Si, H.; Cervini, A.; Gao, J.; Lu, J.; Upadhyay, S.K.; Verma, S.C.; Robertson, E.S. Kaposi’s sarcoma herpesvirus upregulates Aurora A expression to promote p53 phosphorylation and ubiquitylation. PLoS Pathog. 2012, 8, e1002566. [Google Scholar] [CrossRef] [Green Version]
- Frazer, I.H. The actinic keratosis virome: Can we prevent squamous cell carcinoma with a vaccine? Curr. Probl. Dermatol. 2015, 46, 28–35. [Google Scholar]
- Cassler, N.M.; Merrill, D.; Bichakjian, C.K.; Brownell, I. Merkel Cell Carcinoma Therapeutic Update. Curr. Treat. Options Oncol. 2016, 17, 36. [Google Scholar] [CrossRef] [Green Version]
- Harms, P.W.; Harms, K.L.; Moore, P.S.; DeCaprio, J.A.; Nghiem, P.; Wong, M.K.K.; Brownell, I.; International Workshop on Merkel Cell Carcinoma Research (IWMCC) Working Group. The biology and treatment of Merkel cell carcinoma: Current understanding and research priorities. Nat. Rev. Clin. Oncol. 2018, 15, 763–776. [Google Scholar] [CrossRef] [Green Version]
- White, M.K.; Khalili, K. Polyomaviruses and human cancer: Molecular mechanisms underlying patterns of tumorigenesis. Virology 2004, 324, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Mohanty, S.; Harhaj, E.W. Mechanisms of Oncogenesis by HTLV-1 Tax. Pathogens 2020, 9, 543. [Google Scholar] [CrossRef]
- Torresi, J.; Tran, B.M.; Christiansen, D.; Earnest-Silveira, L.; Schwab, R.H.M.; Vincan, E. HBV-related hepatocarcinogenesis: The role of signalling pathways and innovative ex vivo research models. BMC Cancer 2019, 19, 707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGivern, D.R.; Lemon, S.M. Virus-specific mechanisms of carcinogenesis in hepatitis C virus associated liver cancer. Oncogene 2011, 30, 1969–1983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karpiński, T.M. The Microbiota and Pancreatic Cancer. Gastroenterol. Clin. North Am. 2019, 48, 447–464. [Google Scholar] [CrossRef] [PubMed]
- Matijašić, M.; Meštrović, T.; Paljetak, H.Č.; Perić, M.; Barešić, A.; Verbanac, D. Gut Microbiota beyond Bacteria-Mycobiome, Virome, Archaeome, and Eukaryotic Parasites in IBD. Int. J. Mol. Sci. 2020, 21, 2668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, J.A.; Blaser, M.J.; Caporaso, J.G.; Jansson, J.K.; Lynch, S.V.; Knight, R. Current understanding of the human microbiome. Nat. Med. 2018, 24, 392–400. [Google Scholar] [CrossRef]
- Carding, S.R.; Davis, N.; Hoyles, L. Review article: The human intestinal virome in health and disease. Aliment Pharmacol. Ther. 2017, 46, 800–815. [Google Scholar] [CrossRef] [PubMed]
- Carding, S.; Verbeke, K.; Vipond, D.T.; Corfe, B.M.; Owen, L.J. Dysbiosis of the gut microbiota in disease. Microb. Ecol. Health Dis. 2015, 26, 26191. [Google Scholar] [CrossRef] [PubMed]
- Tisza, M.J.; Buck, C.B. A catalog of tens of thousands of viruses from human metagenomes reveals hidden associations with chronic diseases. Proc. Natl. Acad. Sci. USA 2021, 118, e2023202118. [Google Scholar] [CrossRef] [PubMed]
- Suttle, C.A. Marine viruses—Major players in the global ecosystem. Nat. Rev. Microbiol. 2007, 5, 801–812. [Google Scholar] [CrossRef]
- Roux, S.; Hallam, S.J.; Woyke, T.; Sullivan, M.B. Viral dark matter and virus-host interactions resolved from publicly available microbial genomes. eLife 2015, 4, e08490. [Google Scholar] [CrossRef]
- Hannigan, G.D.; Duhaime, M.B.; Ruffin, M.T., 4th; Koumpouras, C.C.; Schloss, P.D. Diagnostic Potential and Interactive Dynamics of the Colorectal Cancer Virome. mBio 2018, 9, e02248-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Handley, S.A.; Devkota, S. Going Viral: A Novel Role for Bacteriophage in Colorectal Cancer. mBio 2019, 10, e02626-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flynn, K.J.; Baxter, N.T.; Schloss, P.D. Metabolic and Community Synergy of Oral Bacteria in Colorectal Cancer. mSphere 2016, 1, e00102-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norman, J.M.; Handley, S.A.; Baldridge, M.T.; Droit, L.; Liu, C.Y.; Keller, B.C.; Kambal, A.; Monaco, C.L.; Zhao, G.; Fleshner, P.; et al. Disease-specific alterations in the enteric virome in inflammatory bowel disease. Cell 2015, 160, 447–460. [Google Scholar] [CrossRef] [Green Version]
- Stripling, J.; Rodriguez, M. Current Evidence in Delivery and Therapeutic Uses of Fecal Microbiota Transplantation in Human Diseases-Clostridium difficile Disease and Beyond. Am. J. Med. Sci. 2018, 356, 424–432. [Google Scholar] [CrossRef] [PubMed]
- Broecker, F.; Kube, M.; Klumpp, J.; Schuppler, M.; Biedermann, L.; Hecht, J.; Hombach, M.; Keller, P.M.; Rogler, G.; Moelling, K. Analysis of the intestinal microbiome of a recovered Clostridium difficile patient after fecal transplantation. Digestion 2013, 88, 243–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ott, S.J.; Waetzig, G.H.; Rehman, A.; Moltzau-Anderson, J.; Bharti, R.; Grasis, J.A.; Cassidy, L.; Tholey, A.; Fickenscher, H.; Seegert, D.; et al. Efficacy of Sterile Fecal Filtrate Transfer for Treating Patients with Clostridium difficile Infection. Gastroenterology 2017, 152, 799–811.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kao, D.H.; Roach, B.; Walter, J.; Lobenberg, R.; Wong, K. A51 Effect of lyophilized sterile fecal filtrate vs lyophilized donor stool on recurrent Clostridium difficile infection (RCDI): Preliminary results from a randomized, double-blind pilot study. J. Can. Assoc. Gastroenterol. 2019, 2 (Suppl. 2), 101–102. [Google Scholar] [CrossRef]
- Zuo, T.; Wong, S.H.; Lam, K.; Lui, R.; Cheung, K.; Tang, W.; Ching, J.Y.L.; Chan, P.K.S.; Chan, M.C.W.; Wu, J.C.Y.; et al. Bacteriophage transfer during faecal microbiota transplantation in Clostridium difficile infection is associated with treatment outcome. Gut 2018, 67, 634–643. [Google Scholar]
- Broecker, F.; Russo, G.; Klumpp, J.; Moelling, K. Stable core virome despite variable microbiome after fecal transfer. Gut Microbes 2017, 8, 214–220. [Google Scholar] [CrossRef] [Green Version]
- Draper, L.A.; Ryan, F.J.; Smith, M.K.; Jalanka, J.; Mattila, E.; Arkkila, P.A.; Ross, R.P.; Satokari, R.; Hill, C. Long-term colonisation with donor bacteriophages following successful faecal microbial transplantation. Microbiome 2018, 6, 220. [Google Scholar] [CrossRef] [Green Version]
- Zitvogel, L.; Ma, Y.; Raoult, D.; Kroemer, G.; Gajewski, T.F. The microbiome in cancer immunotherapy: Diagnostic tools and therapeutic strategies. Science 2018, 359, 1366–1370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davar, D.; Dzutsev, A.K.; McCulloch, J.A.; Rodrigues, R.R.; Chauvin, J.M.; Morrison, R.M.; Deblasio, R.N.; Menna, C.; Ding, Q.; Pagliano, O.; et al. Fecal microbiota transplant overcomes resistance to anti-PD-1 therapy in melanoma patients. Science 2021, 371, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Baruch, E.N.; Youngster, I.; Ben-Betzalel, G.; Ortenberg, R.; Lahat, A.; Katz, L.; Adler, K.; Dick-Necula, D.; Raskin, S.; Bloch, N.; et al. Fecal microbiota transplant promotes response in immunotherapy-refractory melanoma patients. Science 2021, 371, 602–609. [Google Scholar] [CrossRef]
- Pinato, D.J.; Howlett, S.; Ottaviani, D.; Urus, H.; Patel, A.; Mineo, T.; Brock, C.; Power, D.; Hatcher, O.; Falconer, A.; et al. Association of Prior Antibiotic Treatment With Survival and Response to Immune Checkpoint Inhibitor Therapy in Patients With Cancer. JAMA Oncol. 2019, 5, 1774–1778. [Google Scholar] [CrossRef] [PubMed]
- Vétizou, M.; Pitt, J.M.; Daillère, R.; Lepage, P.; Waldschmitt, N.; Flament, C.; Rusakiewicz, S.; Routy, B.; Roberti, M.P.; Duong, C.P.; et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 2015, 350, 1079–1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillère, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef] [Green Version]
- Matson, V.; Fessler, J.; Bao, R.; Chongsuwat, T.; Zha, Y.; Alegre, M.L.; Luke, J.J.; Gajewski, T.F. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science 2018, 359, 104–108. [Google Scholar] [CrossRef] [Green Version]
- Aarnoutse, R.; Ziemons, J.; Penders, J.; Rensen, S.S.; de Vos-Geelen, J.; Smidt, M.L. The Clinical Link between Human Intestinal Microbiota and Systemic Cancer Therapy. Int. J. Mol. Sci. 2019, 20, 4145. [Google Scholar] [CrossRef] [Green Version]
- Fluckiger, A.; Daillère, R.; Sassi, M.; Sixt, B.S.; Liu, P.; Loos, F.; Richard, C.; Rabu, C.; Alou, M.T.; Goubet, A.G.; et al. Cross-reactivity between tumor MHC class I-restricted antigens and an enterococcal bacteriophage. Science 2020, 369, 936–942. [Google Scholar] [CrossRef] [PubMed]
- Neil, J.A.; Cadwell, K. The Intestinal Virome and Immunity. J. Immunol. 2018, 201, 1615–1624. [Google Scholar] [CrossRef] [Green Version]
- Dzutsev, A.; Goldszmid, R.S.; Viaud, S.; Zitvogel, L.; Trinchieri, G. The role of the microbiota in inflammation, carcinogenesis, and cancer therapy. Eur. J. Immunol. 2015, 45, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Zur Hausen, H.; Bund, T.; de Villiers, E.M. Infectious Agents in Bovine Red Meat and Milk and Their Potential Role in Cancer and Other Chronic Diseases. Curr. Top. Microbiol. Immunol. 2017, 407, 83–116. [Google Scholar]
- Arroyo Mühr, L.S.; Bzhalava, Z.; Hortlund, M.; Lagheden, C.; Nordqvist Kleppe, S.; Bzhalava, D.; Hultin, E.; Dillner, J. Viruses in cancers among the immunosuppressed. Int. J. Cancer 2017, 141, 2498–2504. [Google Scholar] [CrossRef] [Green Version]
- Brunetti, C.R.; Amano, H.; Ueda, Y.; Qin, J.; Miyamura, T.; Suzuki, T.; Li, X.; Barrett, J.W.; McFadden, G. Complete genomic sequence and comparative analysis of the tumorigenic poxvirus Yaba monkey tumor virus. J. Virol. 2003, 77, 13335–13347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chehoud, C.; Dryga, A.; Hwang, Y.; Nagy-Szakal, D.; Hollister, E.B.; Luna, R.A.; Versalovic, J.; Kellermayer, R.; Bushman, F.D. Transfer of Viral Communities between Human Individuals during Fecal Microbiota Transplantation. mBio 2016, 7, e00322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunse, A.; Deng, L.; Pan, X.; Hui, Y.; Castro-Mejía, J.L.; Kot, W.; Nguyen, D.N.; Secher, J.B.; Nielsen, D.S.; Thymann, T. Fecal filtrate transplantation protects against necrotizing enterocolitis. ISME J. 2021. Epub ahead of print. [Google Scholar] [CrossRef]
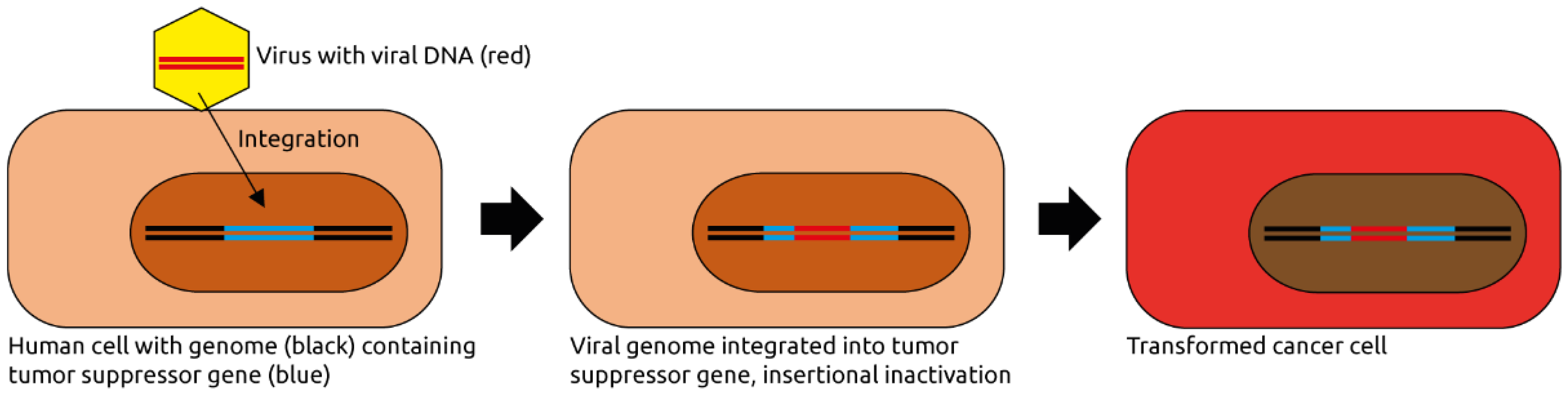
Figure 1.
Eukaryotic virus-induced carcinogenesis. A virus infects a human cell and integrates its genome into the host cell genome. In this example, the viral genome integrates into and thereby inactivates a tumor suppressor gene, contributing to oncogenic transformation. The viral genome may also express viral oncogenes, or it activates a near-by host proto-oncogene (not shown). Figure adapted from Marônek et al. [7].
Figure 1.
Eukaryotic virus-induced carcinogenesis. A virus infects a human cell and integrates its genome into the host cell genome. In this example, the viral genome integrates into and thereby inactivates a tumor suppressor gene, contributing to oncogenic transformation. The viral genome may also express viral oncogenes, or it activates a near-by host proto-oncogene (not shown). Figure adapted from Marônek et al. [7].

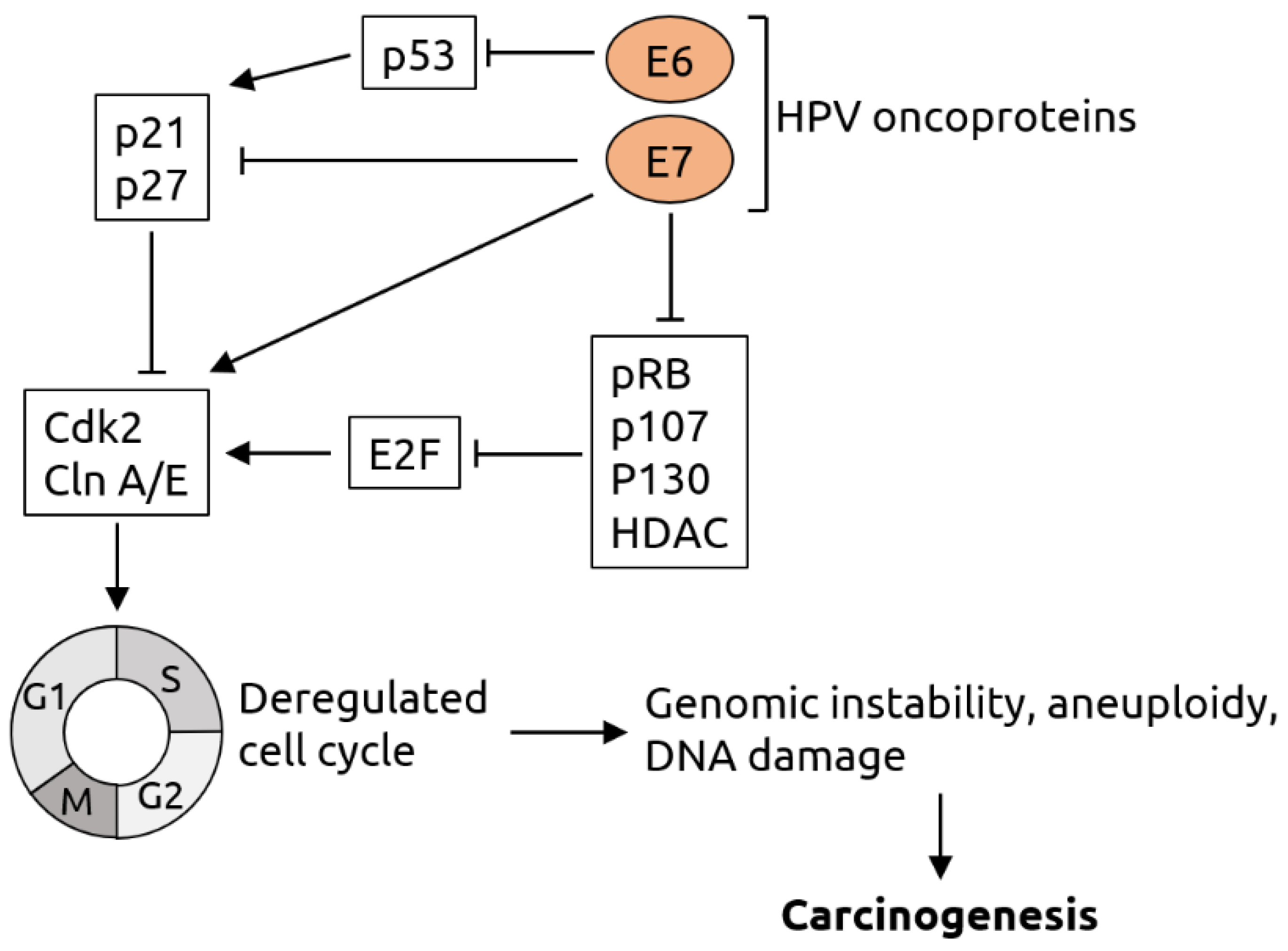
Figure 2.
Molecular mechanisms of HPV-induced carcinogenesis. The viral oncoprotein E6 inhibits tumor suppressor p53 and thereby indirectly suppresses the p21 and p27 proteins, which are negative regulators of cyclin-dependent kinase 2 (Cdk2) and cyclin (Cln) A/E. Cdk2 and Cln A/E are important factors for cell cycle regulation. The viral oncoprotein E7 inhibits p21 and p27 and directly activates Cdk2 and Cln A/E. In addition, E7 inhibits retinoblastoma protein pRB and related pocket proteins p107 and p130 as well as specific histone deacetylases (HDAC). pRB, p107, p130 and HDAC are inhibitors of E2F transcription factors, which activate Cdk2 and Cln A/E. Thus, E6 and E7 act synergistically in cell cycle deregulation, which results in genomic instability, aneuploidy and DNA damage and, consequently, carcinogenesis. Figure adapted from Lehoux et al. [70].
Figure 2.
Molecular mechanisms of HPV-induced carcinogenesis. The viral oncoprotein E6 inhibits tumor suppressor p53 and thereby indirectly suppresses the p21 and p27 proteins, which are negative regulators of cyclin-dependent kinase 2 (Cdk2) and cyclin (Cln) A/E. Cdk2 and Cln A/E are important factors for cell cycle regulation. The viral oncoprotein E7 inhibits p21 and p27 and directly activates Cdk2 and Cln A/E. In addition, E7 inhibits retinoblastoma protein pRB and related pocket proteins p107 and p130 as well as specific histone deacetylases (HDAC). pRB, p107, p130 and HDAC are inhibitors of E2F transcription factors, which activate Cdk2 and Cln A/E. Thus, E6 and E7 act synergistically in cell cycle deregulation, which results in genomic instability, aneuploidy and DNA damage and, consequently, carcinogenesis. Figure adapted from Lehoux et al. [70].

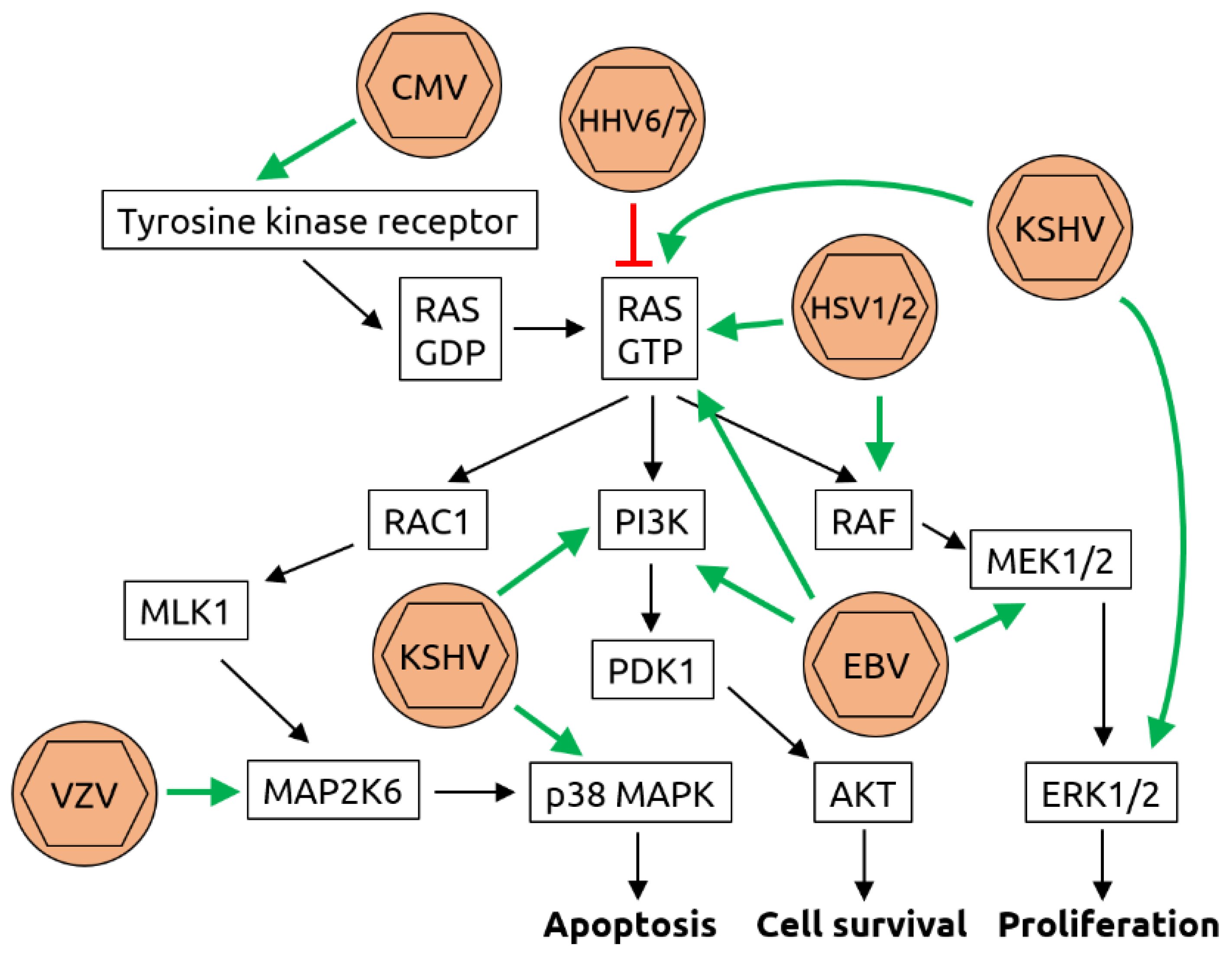
Figure 3.
Molecular mechanisms of herpesvirus-induced carcinogenesis. Shown is a graphical representation of the key mediators of the Ras/Raf/MEK/ERK pathway. The known interactions between herpesviruses and proteins of this pathway are indicated by green arrows (activation) or red T-bars (inhibition). Cytomegalovirus (CMV) has been shown to bind to and activate tyrosine kinase receptors such as epidermal growth factor receptor (EGFR). While human herpesviruses (HHV) 6 and 7 inhibit rat sarcoma (RAS) bound to guanosine triphosphate (RAS-GTP), herpes simplex viruses 1 and 2 (HSV1/2), Kaposi sarcoma-associated herpesvirus (KSHV) and Epstein–Barr virus (EBV) all activate RAS-GTP. In addition, KSHV activates extracellular signal-regulated kinases 1/2 (ERK1/2), phosphoinositide 3-kinase (PI3K) and p38 mitogen-activated protein kinase (MAPK). EBV also activates PI3K as well as meiosis-specific kinases MEK1/2 and HSV1/2 activate rat fibrosarcoma (RAF). Although an association of varicella zoster virus (VZV, also known as HHV3) with cancer is not established, it has been shown to be capable of transforming cells in vitro and to activate dual specificity mitogen-activated protein kinase 6 (MAP2K6). Through these manipulations of the pathway, the processes of apoptosis, cell survival and proliferation are deregulated, contributing to carcinogenesis. Figure adapted from Filippakis et al. [75], a review in which details can be found.
Figure 3.
Molecular mechanisms of herpesvirus-induced carcinogenesis. Shown is a graphical representation of the key mediators of the Ras/Raf/MEK/ERK pathway. The known interactions between herpesviruses and proteins of this pathway are indicated by green arrows (activation) or red T-bars (inhibition). Cytomegalovirus (CMV) has been shown to bind to and activate tyrosine kinase receptors such as epidermal growth factor receptor (EGFR). While human herpesviruses (HHV) 6 and 7 inhibit rat sarcoma (RAS) bound to guanosine triphosphate (RAS-GTP), herpes simplex viruses 1 and 2 (HSV1/2), Kaposi sarcoma-associated herpesvirus (KSHV) and Epstein–Barr virus (EBV) all activate RAS-GTP. In addition, KSHV activates extracellular signal-regulated kinases 1/2 (ERK1/2), phosphoinositide 3-kinase (PI3K) and p38 mitogen-activated protein kinase (MAPK). EBV also activates PI3K as well as meiosis-specific kinases MEK1/2 and HSV1/2 activate rat fibrosarcoma (RAF). Although an association of varicella zoster virus (VZV, also known as HHV3) with cancer is not established, it has been shown to be capable of transforming cells in vitro and to activate dual specificity mitogen-activated protein kinase 6 (MAP2K6). Through these manipulations of the pathway, the processes of apoptosis, cell survival and proliferation are deregulated, contributing to carcinogenesis. Figure adapted from Filippakis et al. [75], a review in which details can be found.

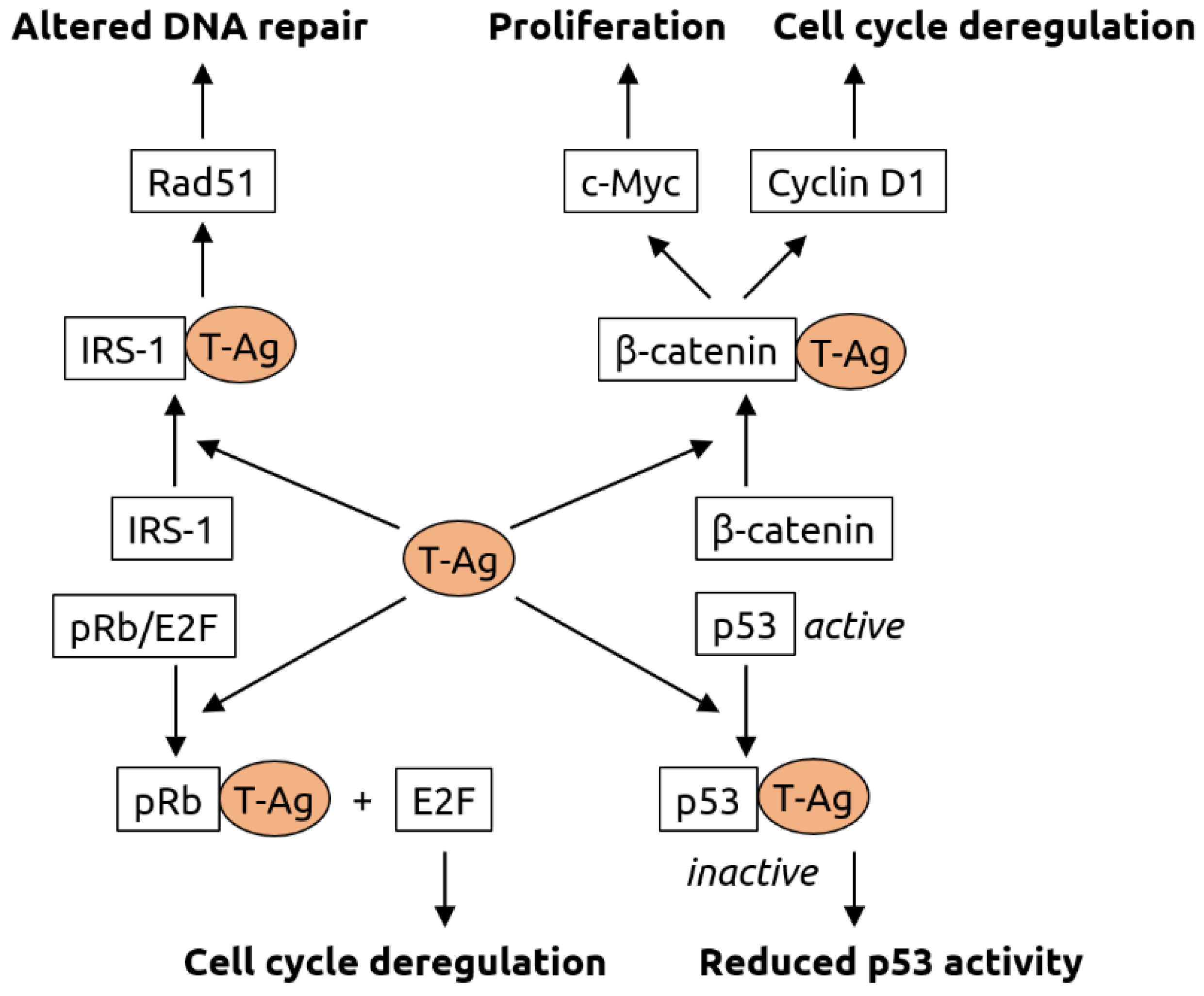
Figure 4.
Molecular mechanisms of carcinogenesis induced by the T-antigen (T-Ag) of polyomaviruses. T-Ag has been shown to bind to insulin receptor substrate 1 (IRS-1), causing its translocation into the nucleus, where it is likely involved Rad51 trafficking. Rad51 is known to be involved in DNA repair. T-Ag also associates with β-catenin, leading to its translocation into the nucleus. There, β-catenin activates the c-Myc proto-oncogene (involved in cell proliferation) and cyclin D1 (involved in cell cycle regulation). Interaction of T-Ag with retinoblastoma protein pRb leads to the release of transcription factor E2F that is involved in cell cycle regulation. T-Ag also inhibits tumor suppressor p53. Figure adapted from White and Khalili [80], a review in which details can be found.
Figure 4.
Molecular mechanisms of carcinogenesis induced by the T-antigen (T-Ag) of polyomaviruses. T-Ag has been shown to bind to insulin receptor substrate 1 (IRS-1), causing its translocation into the nucleus, where it is likely involved Rad51 trafficking. Rad51 is known to be involved in DNA repair. T-Ag also associates with β-catenin, leading to its translocation into the nucleus. There, β-catenin activates the c-Myc proto-oncogene (involved in cell proliferation) and cyclin D1 (involved in cell cycle regulation). Interaction of T-Ag with retinoblastoma protein pRb leads to the release of transcription factor E2F that is involved in cell cycle regulation. T-Ag also inhibits tumor suppressor p53. Figure adapted from White and Khalili [80], a review in which details can be found.

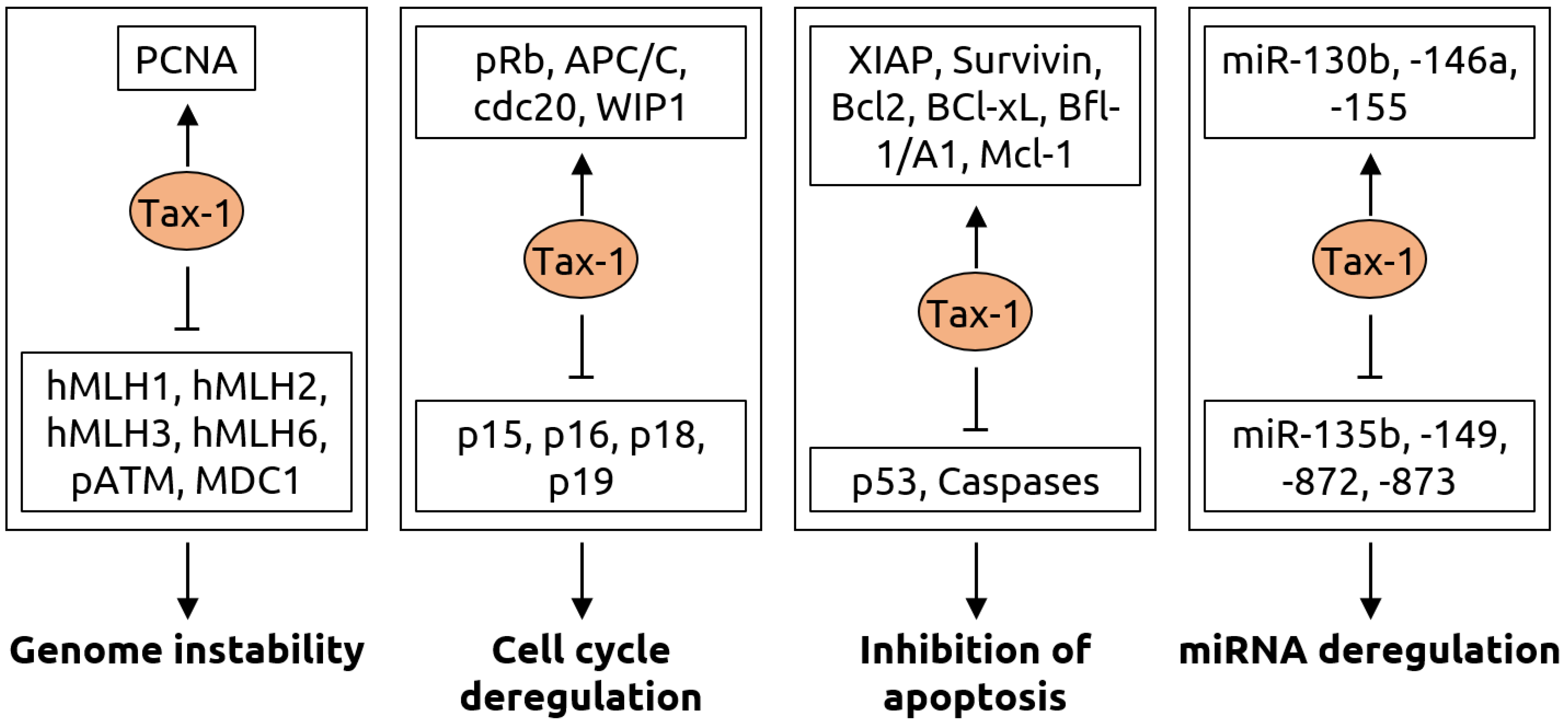
Figure 5.
Molecular mechanisms of carcinogenesis induced by the Tax-1 protein of HTLV-1. Genome instability is caused by Tax-1 through activation of proliferating cell nuclear antigen (PCNA) and suppression of hMLH mismatch repair proteins, ataxia-telangiectasia mutated (ATM) phosphorylation (pATM) and mediator of DNA damage checkpoint 1 (MDC1). Cell cycle deregulation occurs via activation of retinoblastoma protein pRb, anaphase-promoting complex/cyclosome (APC/C), its binding partner cdc20 and wild-type p53-induced phosphatase 1 (WIP1) as well as inhibition of p15, p16, p18 and p19, which are inhibitors of cyclin-dependent kinase 4 (CDK4). Tax-1 leads to evasion of apoptosis by activating various anti-apoptotic proteins including X-linked inhibitor of apoptosis (XIAP), Survivin, the B-cell lymphoma (Bcl) family proteins Bcl-2 and Bcl-xL, Bcl-2-related protein Bfl-1/A and myeloid cell factor-1 (Mcl-1) as well as by suppressing tumor suppressor p53 and Caspase-3, -7, -8 and-9. Furthermore, Tax-1 alters numerous microRNAs resulting in a fine-tuning of gene expression for oncogenic transformation, with an upregulation of miR-130b, -146a, -155 and downregulation of miR-135b, -149, -872 and-873. Figure adapted from Mohanty and Harjah [81], a review in which details can be found.
Figure 5.
Molecular mechanisms of carcinogenesis induced by the Tax-1 protein of HTLV-1. Genome instability is caused by Tax-1 through activation of proliferating cell nuclear antigen (PCNA) and suppression of hMLH mismatch repair proteins, ataxia-telangiectasia mutated (ATM) phosphorylation (pATM) and mediator of DNA damage checkpoint 1 (MDC1). Cell cycle deregulation occurs via activation of retinoblastoma protein pRb, anaphase-promoting complex/cyclosome (APC/C), its binding partner cdc20 and wild-type p53-induced phosphatase 1 (WIP1) as well as inhibition of p15, p16, p18 and p19, which are inhibitors of cyclin-dependent kinase 4 (CDK4). Tax-1 leads to evasion of apoptosis by activating various anti-apoptotic proteins including X-linked inhibitor of apoptosis (XIAP), Survivin, the B-cell lymphoma (Bcl) family proteins Bcl-2 and Bcl-xL, Bcl-2-related protein Bfl-1/A and myeloid cell factor-1 (Mcl-1) as well as by suppressing tumor suppressor p53 and Caspase-3, -7, -8 and-9. Furthermore, Tax-1 alters numerous microRNAs resulting in a fine-tuning of gene expression for oncogenic transformation, with an upregulation of miR-130b, -146a, -155 and downregulation of miR-135b, -149, -872 and-873. Figure adapted from Mohanty and Harjah [81], a review in which details can be found.

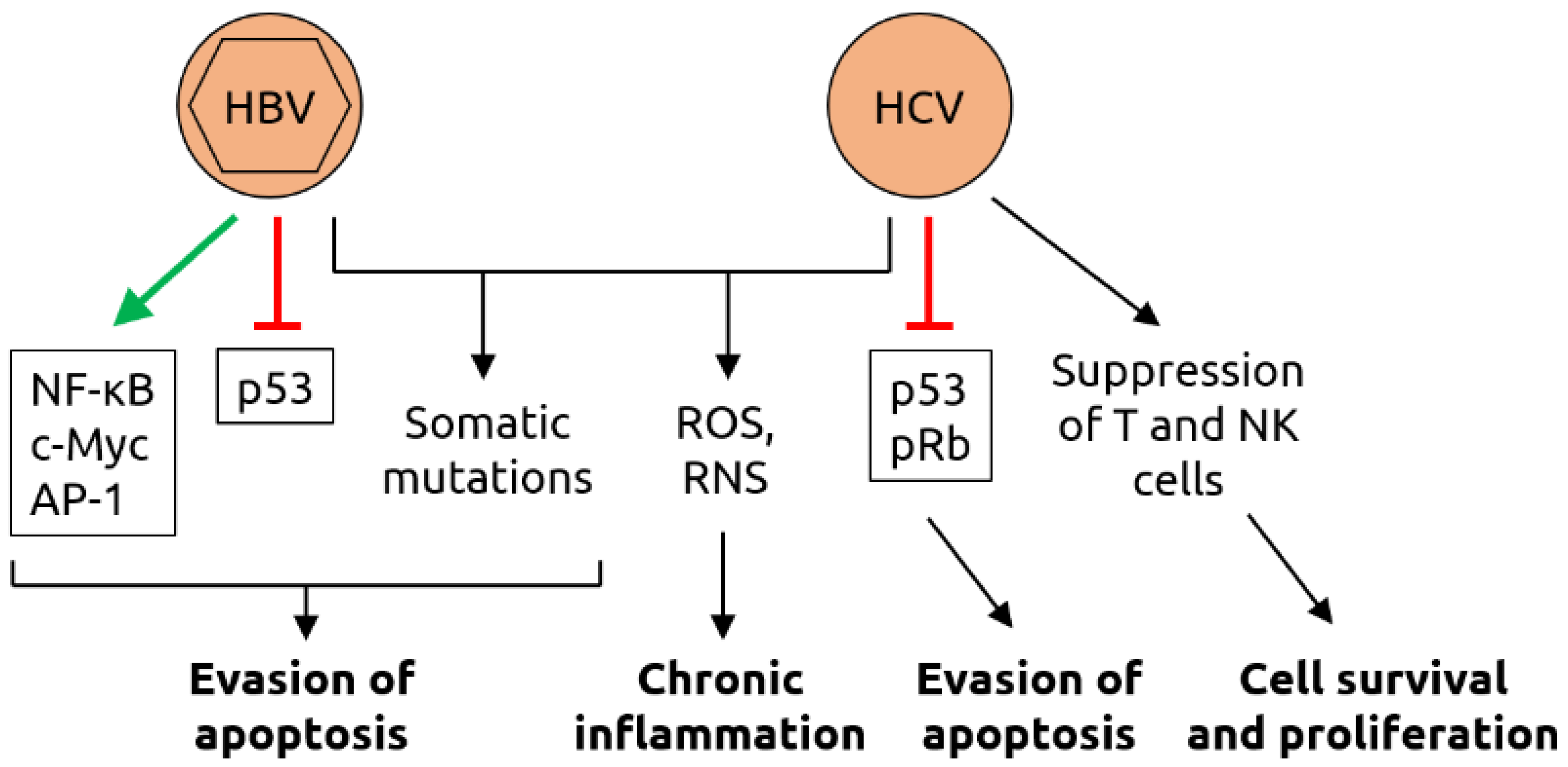
Figure 6.
Molecular mechanisms of carcinogenesis induced by HBV and HCV. HBV infection leads to the activation of various transcription factors, including nuclear factor of activated B cells (NF-κB), c-Myc and activator protein 1 (AP-1), and the suppression of tumor suppressor p53 which, together with virus-induced somatic mutations, contributing to evasion of apoptosis. As HBV, HCV infection induces somatic mutations and increases the levels of reactive oxygen species (ROS) and reactive nitrogen species (RNS); ROS and RNS contribute to chronic inflammation. HCV also suppresses p53 and retinoblastoma protein pRb, contributing to evasion of apoptosis. Suppression of T cells and natural killer (NK) cells by HCV support cell survival and proliferation. Figure adapted from Karpiński [84], a review in which details can be found.
Figure 6.
Molecular mechanisms of carcinogenesis induced by HBV and HCV. HBV infection leads to the activation of various transcription factors, including nuclear factor of activated B cells (NF-κB), c-Myc and activator protein 1 (AP-1), and the suppression of tumor suppressor p53 which, together with virus-induced somatic mutations, contributing to evasion of apoptosis. As HBV, HCV infection induces somatic mutations and increases the levels of reactive oxygen species (ROS) and reactive nitrogen species (RNS); ROS and RNS contribute to chronic inflammation. HCV also suppresses p53 and retinoblastoma protein pRb, contributing to evasion of apoptosis. Suppression of T cells and natural killer (NK) cells by HCV support cell survival and proliferation. Figure adapted from Karpiński [84], a review in which details can be found.

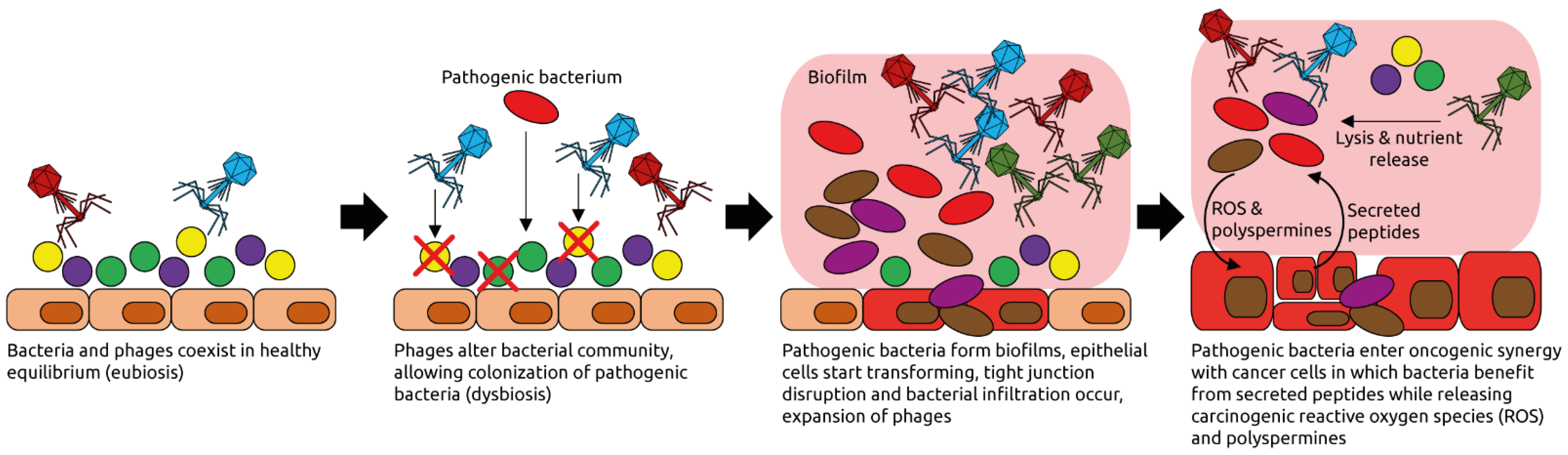
Figure 7.
Examples of phage-induced carcinogenesis. Phages coexist with their host bacteria in a well-balanced equilibrium, for example in the intestinal tract. Some phages might get activated pathologically and alter the bacterial community structure through lysis. Pathogenic bacteria can then thrive and form biofilms. Phages populations expand, typical for dysbiotic, intestinal microbiota and inflammation [95]. Phages lyse commensal bacteria, releasing nutrients that are used by the pathogenic ones. Reactive oxygen species (ROS) and polyspermines produced in the biofilms contribute to DNA damage of host cells, and thereby oncogenic transformation [94]. Peptides released by the transformed cells are metabolized by the pathogenic bacteria. Figure adapted from Hannigan et al. [92].
Figure 7.
Examples of phage-induced carcinogenesis. Phages coexist with their host bacteria in a well-balanced equilibrium, for example in the intestinal tract. Some phages might get activated pathologically and alter the bacterial community structure through lysis. Pathogenic bacteria can then thrive and form biofilms. Phages populations expand, typical for dysbiotic, intestinal microbiota and inflammation [95]. Phages lyse commensal bacteria, releasing nutrients that are used by the pathogenic ones. Reactive oxygen species (ROS) and polyspermines produced in the biofilms contribute to DNA damage of host cells, and thereby oncogenic transformation [94]. Peptides released by the transformed cells are metabolized by the pathogenic bacteria. Figure adapted from Hannigan et al. [92].

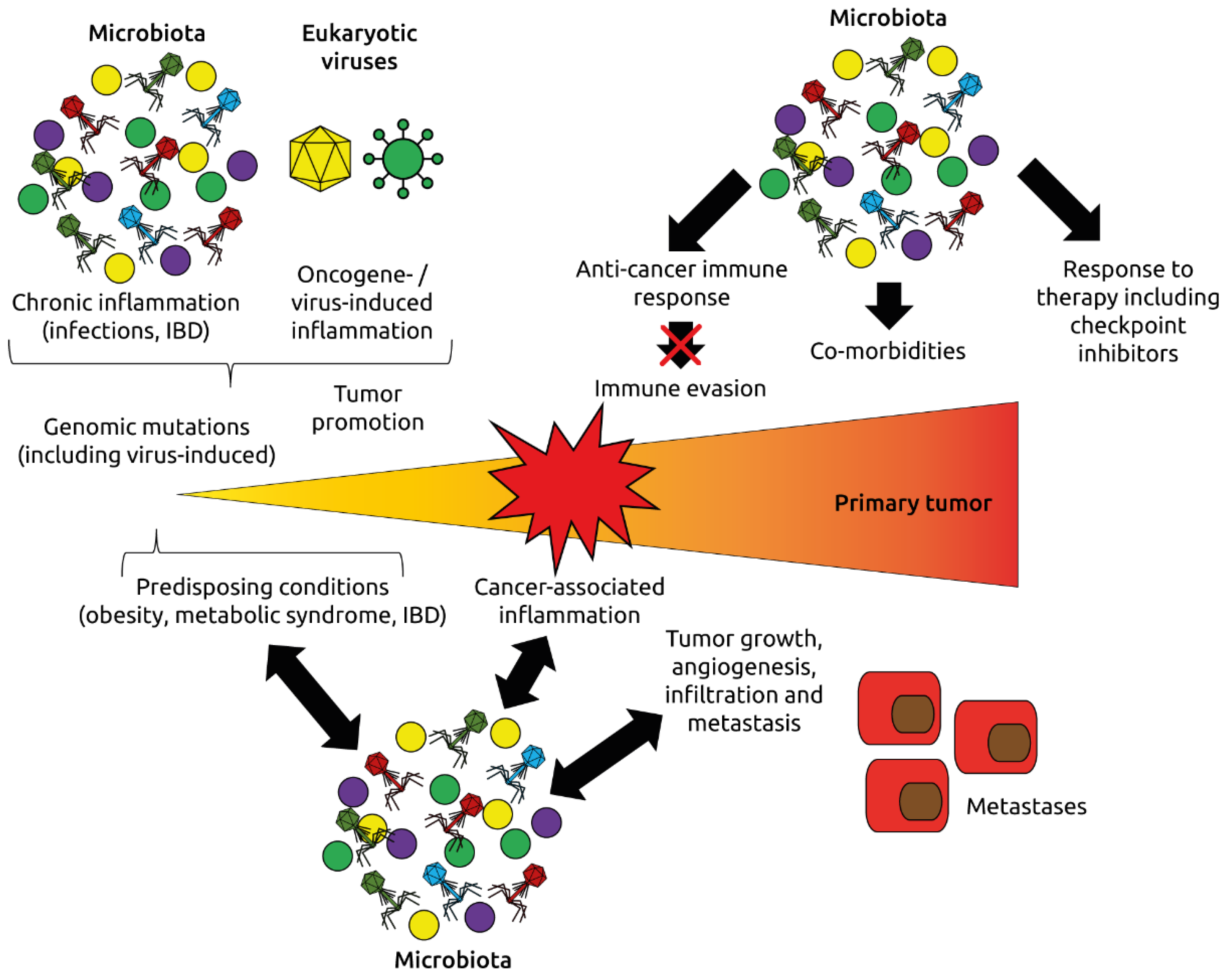
Figure 8.
Roles of the microbiota and virome (eukaryotic viruses and phages) in oncogenesis and response to immunotherapy. The microbiota and virome affect inflammation, one of the hallmarks of cancer, the development of cancer-promoting conditions such as obesity, metabolic syndrome and inflammatory bowel disease (IBD), and modulate immune mechanisms regulating cancer initiation and progression. Adapted from Dzutsev et al. [115].
Figure 8.
Roles of the microbiota and virome (eukaryotic viruses and phages) in oncogenesis and response to immunotherapy. The microbiota and virome affect inflammation, one of the hallmarks of cancer, the development of cancer-promoting conditions such as obesity, metabolic syndrome and inflammatory bowel disease (IBD), and modulate immune mechanisms regulating cancer initiation and progression. Adapted from Dzutsev et al. [115].


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Eukaryotic viruses linked to cancer. Abbreviations: ATLL, adult T-cell leukemia/lymphoma; BKV; BK polyomavirus; CMV, cytomegalovirus; CTCL, cutaneous T-cell lymphoma; DLBCL, diffuse large B-cell lymphoma; EBV, Epstein–Barr virus; HBoV; human bocavirus; HBV, hepatitis B virus; HCC, hepatocellular carcinoma; HCV, hepatitis C virus; HHV, human herpesvirus; HIV, human immunodeficiency virus; HPV, human papillomavirus; HSV, herpes simplex virus; HTLV-1, human T-lymphotropic virus type 1; JCV, JC polyomavirus; KSHV, Kaposi sarcoma-associated herpesvirus; MCV, Merkel cell polyomavirus; NHL, non-Hodgkin lymphoma; PTCL, peripheral T-cell lymphoma; SCC, squamous cell carcinoma; TTV, torque teno virus.
Table 1.
Eukaryotic viruses linked to cancer. Abbreviations: ATLL, adult T-cell leukemia/lymphoma; BKV; BK polyomavirus; CMV, cytomegalovirus; CTCL, cutaneous T-cell lymphoma; DLBCL, diffuse large B-cell lymphoma; EBV, Epstein–Barr virus; HBoV; human bocavirus; HBV, hepatitis B virus; HCC, hepatocellular carcinoma; HCV, hepatitis C virus; HHV, human herpesvirus; HIV, human immunodeficiency virus; HPV, human papillomavirus; HSV, herpes simplex virus; HTLV-1, human T-lymphotropic virus type 1; JCV, JC polyomavirus; KSHV, Kaposi sarcoma-associated herpesvirus; MCV, Merkel cell polyomavirus; NHL, non-Hodgkin lymphoma; PTCL, peripheral T-cell lymphoma; SCC, squamous cell carcinoma; TTV, torque teno virus.
| Virus Family | Virus | Cancer Type | Observations |
|---|---|---|---|
| Papillomaviridae | HPV-16 | Anal | HPV, especially HPV-16, is a possible risk factor for anal and rectal cancer [8,9] and a significant prognostic marker, especially for locally advanced disease [10] |
| HPV | Bladder | HPV (different serotypes) may be linked to bladder cancer in a small number of cases [11] | |
| HPV-16, -18 | Cervical | Association between infection with high-risk HPV serotype (mainly HPV-16 and-18) and development of cervical cancer [11,12,13] | |
| HPV-18 | Colorectal | HPV, especially HPV-18, is a possible risk factor for colorectal cancer [14,15], however, another study found no association [16] | |
| HPV-16, -18, -26, -57 | Esophageal | HPV-16 is a risk factor for esophageal carcinoma [17,18]; HPV infection (mainly HPV-16, -18, -26 and-57) is common in esophageal carcinoma [19] | |
| HPV-16 | Head and neck (SCC) | HPV infection, especially HPV-16, is associated with head and neck cancer [11,20,21] and better long-term outcome [22] | |
| HPV-6 | Oral | Association of HPV-6 with oral cancer [23] | |
| HPV-16 | Prostate | Association of HPV-16 with prostate cancer [24] | |
| HPV-16, -18, -58 | Renal | Association of HPV-16, -18 and-58 with renal cell carcinoma [25] | |
| HPV-5, -8 | Skin and mucosal | Papillomavirus DNA frequently detected in skin-and mucosa-associated cancers [26]; HPV-5 and-8 are associated with epidermodysplasia verruciformis associated with a high risk of skin cancer [27,28] | |
| HPV-16 | Vulvar | Association between HPV, especially HPV-16, and vulvar squamous cell carcinoma [29] | |
| Herpesviridae | CMV (HHV5) | Colorectal | CMV DNA is more abundant cancer tissues compared to healthy tissues [30]; CMV-positive tumors in non-elderly patients are associated with increased disease-free survival rate [31]; specific genetic polymorphisms of CMV are linked to different clinical outcomes [32] |
| EBV (HHV4) | Colorectal | Possible association of EBV with colorectal carcinoma [33], however, no association found in another study [16] | |
| EBV (HHV4) | Esophageal | EBV is associated with esophageal cancer [34] | |
| EBV (HHV4) | Gastric | Possible involvement of EBV in gastric cancer and precursor lesions [35]; patients with EBV-positive gastric cancer had a better response to chemotherapy and better survival [36] | |
| EBV (HHV4) | Hepatic | EBV infections detected in HCC tissues [37] | |
| EBV (HHV4) | Lymphoma (Burkitt) | EBV infections contribute to Burkitt lymphoma [38] | |
| EBV (HHV4) | Lymphoma (DLBCL) | EBV RNA detected in B-cell lymphoma samples [39] | |
| EBV (HHV4) | Lymphoma (PTCL) | EBV expression associated with some subtypes of peripheral T-cell lymphomas [40] | |
| EBV (HHV4) | Oral | Higher proportion of EBV-positive oral squamous cell carcinoma in industrialized countries [41] | |
| EBV (HHV4) | Skin and mucosal | EBV DNA frequently detected in skin and mucosal cancers [26] | |
| HHV6 | Lymphoma (DLBCL) | HHV6 RNA detected in B-cell lymphoma samples [39] | |
| HHV6 | Malignant melanoma | HHV6 DNA frequently detected in malignant melanoma [26] | |
| HHV7 | Bladder | HHV7 DNA frequently detected in bladder cancer [26] | |
| HHV7 | Lymphoma (CTCL) | HHV7 DNA frequently detected in cutaneous T-cell lymphoma (Mycosis fungoides) [26] | |
| HHV7 | Oral | HHV7 DNA frequently detected in oral cavity cancer [26] | |
| HSV (HHV1/2) | Oral | Higher proportion of HSV-positive oral squamous cell carcinoma in industrialized countries [41] | |
| KSHV (HHV8) | Kaposi sarcoma | In HIV-infected individuals, KSHV infection is associated with Kaposi sarcoma [42] | |
| Polyomaviridae | BKV | Bladder | Possible association of BKV with bladder cancer [26] |
| BKV | Colorectal | Possible association of BKV with colorectal cancer [43,44], however, other studies found no association [16,45] | |
| JCV | Colorectal | JCV is associated with colorectal cancer [45,46] and may be involved in carcinogenesis [47], specifically in chromosomal instability [48]; JCV T-antigen is expressed in early-stage colorectal cancer [49], however, another study found no association [16] | |
| MCV | Merkel cell carcinoma | MCV is the major causative factor for Merkel cell carcinoma [50,51] | |
| Retroviridae | HIV | Anal | HIV-positive people have increased risk for anal cancer [52,53] and worse overall colostomy-free survival rates [54] |
| HIV | Cervical | Cervical cancer is more prevalent in HIV-positive individuals, likely because of increased susceptibility to HPV infection [55,56] | |
| HIV | Kaposi sarcoma | Kaposi sarcoma is more prevalent in HIV-positive individuals, likely because of increased susceptibility to KSHV infection [55,56] | |
| HIV | Lymphoma (NHL) | Aggressive B cell non-Hodgkin lymphoma is more prevalent in HIV-positive individuals, likely because of increased susceptibility to EBV infection [55,56] | |
| HTLV-1 | Lymphoma (ATLL) | HTLV-1 induces adult T-cell leukemia/lymphoma in 5% of infected individuals [57] through random integration into the host genome [58] | |
| Others | HBV | Bile duct | HBV is a risk factor for bile duct cancer [59] |
| HBV | Colorectal | Chronic HBV infection is a risk factor for colorectal cancer [60] | |
| HBV | Hepatic | Liver cancer is associated with HBV [11] | |
| HBV | Pancreatic | Chronic HBV infection [61] or past exposure [62] are risk factors for pancreatic cancer | |
| HCV | Bile duct | HCV is a risk factor for bile duct cancer [59] | |
| HCV | Hepatic | Liver cancer is associated with HCV [11] | |
| TTV | Hepatic | TTV is a risk factor for hepatocellular carcinoma [63] | |
| HBoV | Colorectal | Some colorectal cancers are associated with HBoV [64] | |
| HBoV | Lung | Some lung cancers are associated with HBoV [64] | |
| HBoV | Tonsillar | Association of HBoV with tonsil squamous cell carcinoma [65] | |
| Orthobunyaviruses | Colorectal | High abundance of orthobunyaviruses in colorectal cancer [66] | |
| Parvoviruses | Skin | Parvovirus DNA frequently detected in skin-associated cancers [26] | |
| Anelloviruses | Mucosal | Anellovirus DNA frequently detected in mucosal cancers [26] | |
| Anelloviruses | Leukemias | Anellovirus DNA frequently detected in leukemias [26] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Broecker, F.; Moelling, K. The Roles of the Virome in Cancer. Microorganisms 2021, 9, 2538. https://doi.org/10.3390/microorganisms9122538
AMA Style
Broecker F, Moelling K. The Roles of the Virome in Cancer. Microorganisms. 2021; 9(12):2538. https://doi.org/10.3390/microorganisms9122538
Chicago/Turabian StyleBroecker, Felix, and Karin Moelling. 2021. "The Roles of the Virome in Cancer" Microorganisms 9, no. 12: 2538. https://doi.org/10.3390/microorganisms9122538
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.