
Challenges in Determining the Role of Microbiome Evolution in Barrett’s Esophagus and Progression to Esophageal Adenocarcinoma
 ,
, 
Abstract
:1. Introduction
2. Epidemiology of Barrett’s Esophagus and EAC
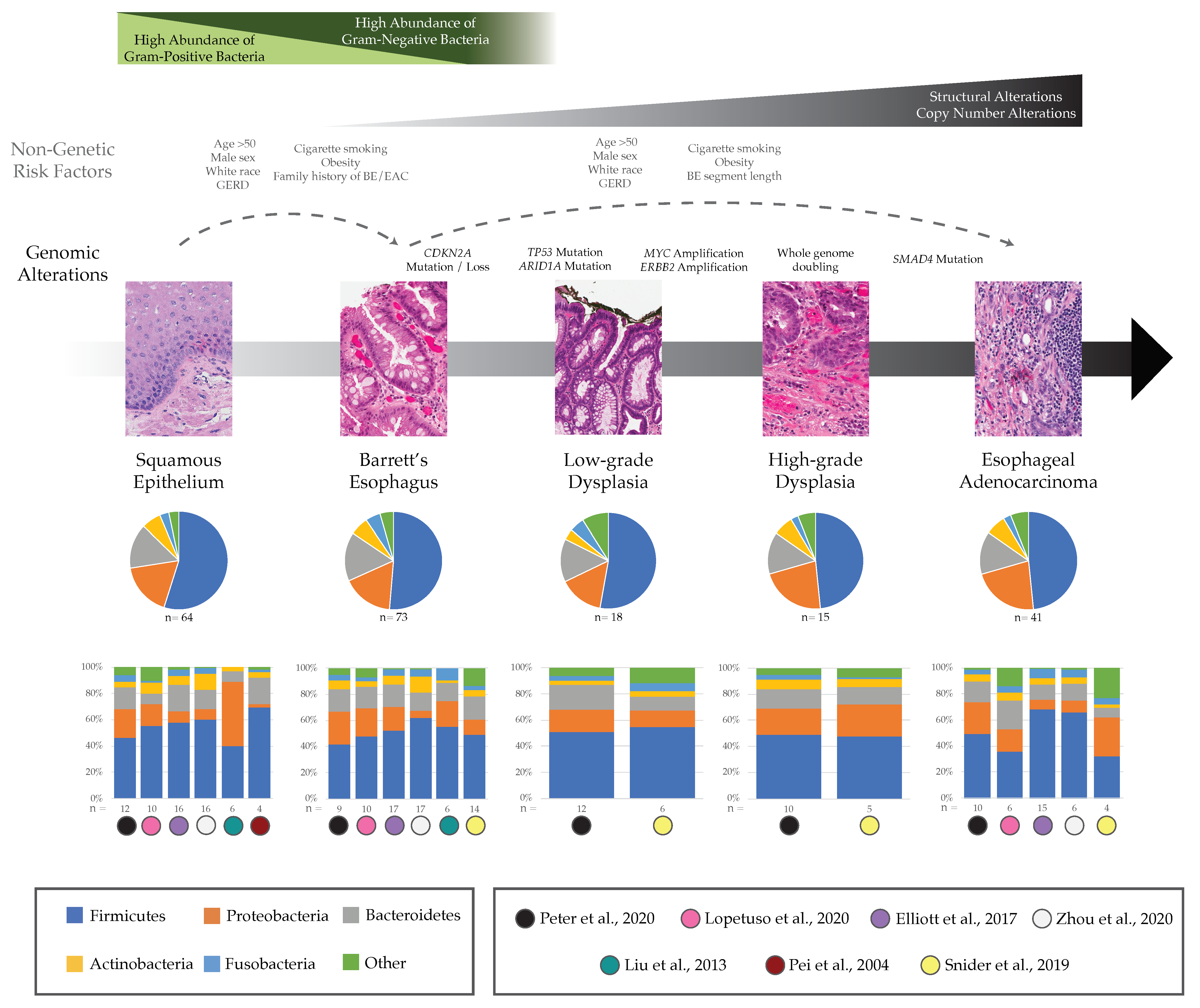
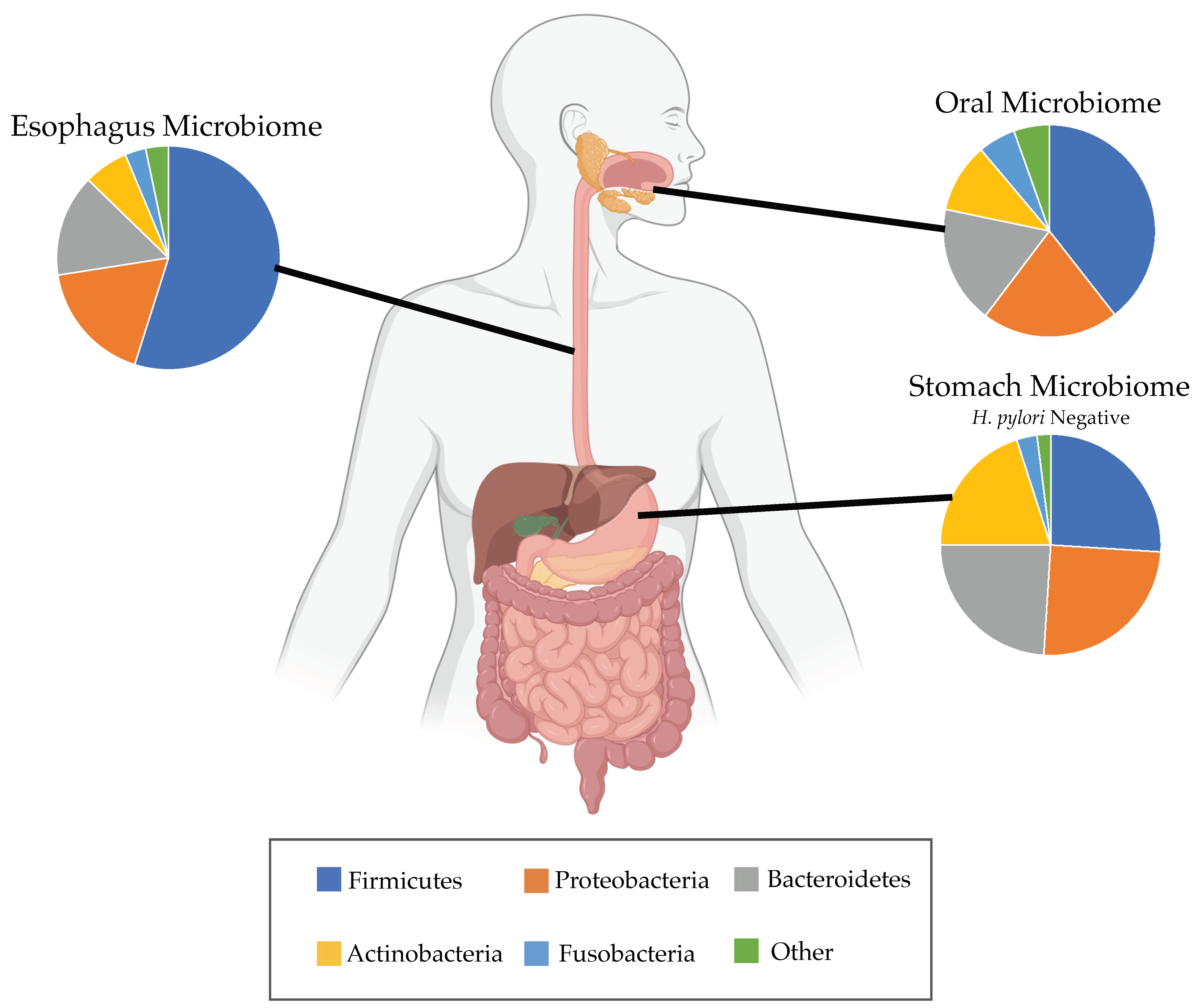
3. Potential Role of Esophageal Microbiome in BE and EAC
3.1. Barrett’s Esophagus
3.2. Esophageal Adenocarcinoma
3.3. Proton Pump Inhibitors
3.4. Helicobacter pylori
4. Current Challenges
4.1. How Do Microbial Communities Influence Precancer and Cancer Genomes, Specifically during the Early Growth of EAC Malignancies?
4.2. What Evolutionary Forces Shape Microbial Communities throughout the Body, and Specifically in BE and EAC?
4.3. How Can We Increase the Number of Longitudinal Clinical Datasets That Capture Evolutionary Dynamic Changes in the Esophageal Microbiome?
5. Future Directions
5.1. Open Access to Large Microbial Datasets Integrated with Host Genomics
5.2. Improve Detection of Microbes at the Species Level
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kong, C.Y.; Kroep, S.; Curtius, K.; Hazelton, W.D.; Jeon, J.; Meza, R.; Heberle, C.R.; Miller, M.C.; Choi, S.E.; Lansdorp-Vogelaar, I.; et al. Exploring the Recent Trend in Esophageal Adenocarcinoma Incidence and Mortality Using Comparative Simulation Modeling. Cancer Epidemiol. Biomark. Prev. 2014, 23, 997–1006. [Google Scholar] [CrossRef] [Green Version]
- SEER*Explorer: An Interactive Website for SEER Cancer Statistics [Internet]. Surveillance Research Program, National Cancer Institute. Available online: https://seer.cancer.gov/explorer/ (accessed on 15 April 2021).
- Vaughan, T.L.; Fitzgerald, R.C. Precision Prevention of Oesophageal Adenocarcinoma. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Curtius, K.; Rubenstein, J.H.; Chak, A.; Inadomi, J.M. Computational Modelling Suggests That Barrett’s Oesophagus May Be the Precursor of All Oesophageal Adenocarcinomas. Gut 2021, 70, 1435–1440. [Google Scholar] [CrossRef]
- Nowicki-Osuch, K.; Zhuang, L.; Jammula, S.; Bleaney, C.W.; Mahbubani, K.T.; Devonshire, G.; Katz-Summercorn, A.; Eling, N.; Wilbrey-Clark, A.; Madissoon, E.; et al. Molecular Phenotyping Reveals the Identity of Barrett’s Esophagus and Its Malignant Transition. Science 2021, 373, 760–767. [Google Scholar] [CrossRef]
- Bhat, S.; Coleman, H.G.; Yousef, F.; Johnston, B.T.; McManus, D.T.; Gavin, A.T.; Murray, L.J. Risk of Malignant Progression in Barrett’s Esophagus Patients: Results from a Large Population-Based Study. J. Natl. Cancer Inst. 2011, 103, 1049–1057. [Google Scholar] [CrossRef] [Green Version]
- Thrift, A.P. Barrett’s Esophagus and Esophageal Adenocarcinoma: How Common Are They Really? Dig. Dis. Sci. 2018, 63, 1988–1996. [Google Scholar] [CrossRef]
- Zagari, R.M.; Fuccio, L.; Wallander, M.-A.; Johansson, S.; Fiocca, R.; Casanova, S.; Farahmand, B.Y.; Winchester, C.C.; Roda, E.; Bazzoli, F. Gastro-Oesophageal Reflux Symptoms, Oesophagitis and Barrett’s Oesophagus in the General Population: The Loiano-Monghidoro Study. Gut 2008, 57, 1354–1359. [Google Scholar] [CrossRef] [PubMed]
- Ronkainen, J.; Aro, P.; Storskrubb, T.; Johansson, S.-E.; Lind, T.; Bolling-Sternevald, E.; Vieth, M.; Stolte, M.; Talley, N.J.; Agréus, L. Prevalence of Barrett’s Esophagus in the General Population: An Endoscopic Study. Gastroenterology 2005, 129, 1825–1831. [Google Scholar] [CrossRef] [PubMed]
- Herrera Elizondo, J.L.; Monreal Robles, R.; García Compean, D.; González Moreno, E.I.; Borjas Almaguer, O.D.; Maldonado Garza, H.J.; González González, J.A. Prevalence of Barrett’s Esophagus: An Observational Study from a Gastroenterology Clinic. Rev. Gastroenterol. Méx. Engl. Ed. 2017, 82, 296–300. [Google Scholar] [CrossRef]
- Rubenstein, J.H.; Mattek, N.; Eisen, G. Age- and Gender-Specific Yield of Barrett’s Esophagus by Endoscopy Indication. Gastrointest. Endosc. 2010, 71, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masclee, G.M.C.; Coloma, P.M.; de Wilde, M.; Kuipers, E.J.; Sturkenboom, M.C.J.M. The Incidence of Barrett’s Oesophagus and Oesophageal Adenocarcinoma in the United Kingdom and the Netherlands Is Levelling Off. Aliment. Pharmacol. Ther. 2014, 39, 1321–1330. [Google Scholar] [CrossRef] [PubMed]
- Corley, D.A.; Kubo, A.; Levin, T.R.; Block, G.; Habel, L.; Rumore, G.; Quesenberry, C.; Buffler, P. Race, Ethnicity, Sex and Temporal Differences in Barrett’s Oesophagus Diagnosis: A Large Community-Based Study, 1994–2006. Gut 2009, 58, 182–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snider, E.J.; Freedberg, D.E.; Abrams, J.A. Potential Role of the Microbiome in Barrett’s Esophagus and Esophageal Adenocarcinoma. Dig. Dis. Sci. 2016, 61, 2217–2225. [Google Scholar] [CrossRef] [Green Version]
- Bhat, S.K.; McManus, D.T.; Coleman, H.G.; Johnston, B.T.; Cardwell, C.R.; McMenamin, Ú.; Bannon, F.; Hicks, B.; Kennedy, G.; Gavin, A.T.; et al. Oesophageal Adenocarcinoma and Prior Diagnosis of Barrett’s Oesophagus: A Population-Based Study. Gut 2015, 64, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Shaheen, N.J.; Falk, G.W.; Iyer, P.G.; Gerson, L.B. ACG Clinical Guideline: Diagnosis and Management of Barrett’s Esophagus. Am. J. Gastroenterol. 2016, 111, 30–50. [Google Scholar] [CrossRef]
- Fitzgerald, R.C.; di Pietro, M.; O’Donovan, M.; Maroni, R.; Muldrew, B.; Debiram-Beecham, I.; Gehrung, M.; Offman, J.; Tripathi, M.; Smith, S.G.; et al. Cytosponge-Trefoil Factor 3 versus Usual Care to Identify Barrett’s Oesophagus in a Primary Care Setting: A Multicentre, Pragmatic, Randomised Controlled Trial. Lancet 2020, 396, 333–344. [Google Scholar] [CrossRef]
- Snider, E.J.; Compres, G.; Freedberg, D.E.; Giddins, M.J.; Khiabanian, H.; Lightdale, C.J.; Nobel, Y.R.; Toussaint, N.C.; Uhlemann, A.-C.; Abrams, J.A. Barrett’s Esophagus Is Associated with a Distinct Oral Microbiome. Clin. Transl. Gastroenterol. 2018, 9, 135. [Google Scholar] [CrossRef]
- Shah, P.M.; Gerdes, H. Endoscopic Options for Early Stage Esophageal Cancer. J. Gastrointest. Oncol. 2015, 6, 20–30. [Google Scholar] [CrossRef]
- Wani, S.; Qumseya, B.; Sultan, S.; Agrawal, D.; Chandrasekhara, V.; Harnke, B.; Kothari, S.; McCarter, M.; Shaukat, A.; Wang, A.; et al. Endoscopic Eradication Therapy for Patients with Barrett’s Esophagus–Associated Dysplasia and Intramucosal Cancer. Gastrointest. Endosc. 2018, 87, 907–931.e9. [Google Scholar] [CrossRef] [Green Version]
- Shaheen, N.J.; Sharma, P.; Overholt, B.F.; Wolfsen, H.C.; Sampliner, R.E.; Wang, K.K.; Galanko, J.A.; Bronner, M.P.; Goldblum, J.R.; Bennett, A.E.; et al. Radiofrequency Ablation in Barrett’s Esophagus with Dysplasia. N. Engl. J. Med. 2009, 360, 2277–2288. [Google Scholar] [CrossRef] [Green Version]
- Kroep, S.; Lansdorp-Vogelaar, I.; Rubenstein, J.H.; de Koning, H.J.; Meester, R.; Inadomi, J.M.; van Ballegooijen, M. An Accurate Cancer Incidence in Barrett’s Esophagus: A Best Estimate Using Published Data and Modeling. Gastroenterology 2015, 149, 577–585.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacy, B.E.; Spiegel, B. Introduction to the Gut Microbiome Special Issue. Off. J. Am. Coll. Gastroenterol. 2019, 114, 1013. [Google Scholar] [CrossRef]
- Perillo, F.; Amoroso, C.; Strati, F.; Giuffrè, M.R.; Díaz-Basabe, A.; Lattanzi, G.; Facciotti, F. Gut Microbiota Manipulation as a Tool for Colorectal Cancer Management: Recent Advances in Its Use for Therapeutic Purposes. Int. J. Mol. Sci. 2020, 21, 5389. [Google Scholar] [CrossRef] [PubMed]
- Zaharuddin, L.; Mokhtar, N.M.; Muhammad Nawawi, K.N.; Raja Ali, R.A. A Randomized Double-Blind Placebo-Controlled Trial of Probiotics in Post-Surgical Colorectal Cancer. BMC Gastroenterol. 2019, 19, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khanna, S.; Vazquez-Baeza, Y.; González, A.; Weiss, S.; Schmidt, B.; Muñiz-Pedrogo, D.A.; Rainey, J.F.; Kammer, P.; Nelson, H.; Sadowsky, M.; et al. Changes in Microbial Ecology after Fecal Microbiota Transplantation for Recurrent C. Difficile Infection Affected by Underlying Inflammatory Bowel Disease. Microbiome 2017, 5, 55. [Google Scholar] [CrossRef] [PubMed]
- Charlson, E.S.; Bittinger, K.; Chen, J.; Diamond, J.M.; Li, H.; Collman, R.G.; Bushman, F.D. Assessing Bacterial Populations in the Lung by Replicate Analysis of Samples from the Upper and Lower Respiratory Tracts. PLoS ONE 2012, 7, e42786. [Google Scholar] [CrossRef] [Green Version]
- Annavajhala, M.K.; May, M.; Compres, G.; Freedberg, D.E.; Graham, R.; Stump, S.; Que, J.; Korem, T.; Uhlemann, A.-C.; Abrams, J.A. Relationship of the Esophageal Microbiome and Tissue Gene Expression and Links to the Oral Microbiome: A Randomized Clinical Trial. Clin. Transl. Gastroenterol. 2020, 11, e00235. [Google Scholar] [CrossRef]
- Hasan, A.; Hasan, L.K.; Schnabl, B.; Greytak, M.; Yadlapati, R. Microbiome of the Aerodigestive Tract in Health and Esophageal Disease. Dig. Dis. Sci. 2021, 66, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Alemán, J.O.; Eusebi, L.H.; Ricciardiello, L.; Patidar, K.; Sanyal, A.J.; Holt, P.R. Mechanisms of Obesity-Induced Gastrointestinal Neoplasia. Gastroenterology 2014, 146, 357–373. [Google Scholar] [CrossRef] [Green Version]
- Lv, J.; Guo, L.; Liu, J.-J.; Zhao, H.-P.; Zhang, J.; Wang, J.-H. Alteration of the Esophageal Microbiota in Barrett’s Esophagus and Esophageal Adenocarcinoma. World J. Gastroenterol. 2019, 25, 2149–2161. [Google Scholar] [CrossRef]
- Gall, A.; Fero, J.; McCoy, C.; Claywell, B.C.; Sanchez, C.A.; Blount, P.L.; Li, X.; Vaughan, T.L.; Matsen, F.A.; Reid, B.J.; et al. Bacterial Composition of the Human Upper Gastrointestinal Tract Microbiome Is Dynamic and Associated with Genomic Instability in a Barrett’s Esophagus Cohort. PLoS ONE 2015, 10, e0129055. [Google Scholar] [CrossRef]
- Kramer, J.R.; Fischbach, L.A.; Richardson, P.; Alsarraj, A.; Fitzgerald, S.; Shaib, Y.; Abraham, N.S.; Velez, M.; Cole, R.; Anand, B.; et al. Waist-to-Hip Ratio, but Not Body Mass Index, Is Associated with an Increased Risk of Barrett’s Esophagus in White Men. Clin. Gastroenterol. Hepatol. 2013, 11, 373–381.e1. [Google Scholar] [CrossRef] [Green Version]
- Peter, S.; Pendergraft, A.; VanDerPol, W.; Wilcox, C.M.; Kyanam Kabir Baig, K.R.; Morrow, C.; Izard, J.; Mannon, P.J. Mucosa-Associated Microbiota in Barrett’s Esophagus, Dysplasia, and Esophageal Adenocarcinoma Differ Similarly Compared with Healthy Controls. Clin. Transl. Gastroenterol. 2020, 11, e00199. [Google Scholar] [CrossRef]
- Lopetuso, L.R.; Severgnini, M.; Pecere, S.; Ponziani, F.R.; Boskoski, I.; Larghi, A.; Quaranta, G.; Masucci, L.; Ianiro, G.; Camboni, T.; et al. Esophageal Microbiome Signature in Patients with Barrett’s Esophagus and Esophageal Adenocarcinoma. PLoS ONE 2020, 15, e0231789. [Google Scholar] [CrossRef]
- Liu, N.; Ando, T.; Ishiguro, K.; Maeda, O.; Watanabe, O.; Funasaka, K.; Nakamura, M.; Miyahara, R.; Ohmiya, N.; Goto, H. Characterization of Bacterial Biota in the Distal Esophagus of Japanese Patients with Reflux Esophagitis and Barrett’s Esophagus. BMC Infect. Dis. 2013, 13, 130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snider, E.J.; Compres, G.; Freedberg, D.E.; Khiabanian, H.; Nobel, Y.R.; Stump, S.; Uhlemann, A.-C.; Lightdale, C.J.; Abrams, J.A. Alterations to the Esophageal Microbiome Associated with Progression from Barrett’s Esophagus to Esophageal Adenocarcinoma. Cancer Epidemiol. Biomark. Prev. 2019, 28, 1687–1693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Shrestha, P.; Qiu, Z.; Harman, D.G.; Teoh, W.-C.; Al-Sohaily, S.; Liem, H.; Turner, I.; Ho, V. Distinct Microbiota Dysbiosis in Patients with Non-Erosive Reflux Disease and Esophageal Adenocarcinoma. J. Clin. Med. 2020, 9, 2162. [Google Scholar] [CrossRef] [PubMed]
- Elliott, D.R.F.; Walker, A.W.; O’Donovan, M.; Parkhill, J.; Fitzgerald, R.C. A Non-Endoscopic Device to Sample the Oesophageal Microbiota: A Case-Control Study. Lancet Gastroenterol. Hepatol. 2017, 2, 32–42. [Google Scholar] [CrossRef] [Green Version]
- Qumseya, B.; Sultan, S.; Bain, P.; Jamil, L.; Jacobson, B.; Anandasabapathy, S.; Agrawal, D.; Buxbaum, J.L.; Fishman, D.S.; Gurudu, S.R.; et al. ASGE Guideline on Screening and Surveillance of Barrett’s Esophagus. Gastrointest. Endosc. 2019, 90, 335–359.e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qumseya, B.J.; Bukannan, A.; Gendy, S.; Ahemd, Y.; Sultan, S.; Bain, P.; Gross, S.A.; Iyer, P.; Wani, S. Systematic Review and Meta-Analysis of Prevalence and Risk Factors for Barrett’s Esophagus. Gastrointest. Endosc. 2019, 90, 707–717.e1. [Google Scholar] [CrossRef] [PubMed]
- Prasad, G.A.; Bansal, A.; Sharma, P.; Wang, K.K. Predictors of Progression in Barrett’s Esophagus: Current Knowledge and Future Directions. Am. J. Gastroenterol. 2010, 105, 1490–1502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solanky, D.; Krishnamoorthi, R.; Crews, N.; Johnson, M.; Wang, K.; Wolfsen, H.; Fleischer, D.; Ramirez, F.C.; Katzka, D.; Buttar, N.; et al. Barrett Esophagus Length, Nodularity, and Low-Grade Dysplasia Are Predictive of Progression to Esophageal Adenocarcinoma. J. Clin. Gastroenterol. 2019, 53, 361–365. [Google Scholar] [CrossRef]
- Li, X.; Galipeau, P.C.; Paulson, T.G.; Sanchez, C.A.; Arnaudo, J.; Liu, K.; Sather, C.L.; Kostadinov, R.L.; Odze, R.D.; Kuhner, M.K.; et al. Temporal and Spatial Evolution of Somatic Chromosomal Alterations: A Case-Cohort Study of Barrett’s Esophagus. Cancer Prev. Res. 2014, 7, 114–127. [Google Scholar] [CrossRef] [Green Version]
- Killcoyne, S.; Gregson, E.; Wedge, D.C.; Woodcock, D.J.; Eldridge, M.D.; de la Rue, R.; Miremadi, A.; Abbas, S.; Blasko, A.; Kosmidou, C.; et al. Genomic Copy Number Predicts Esophageal Cancer Years before Transformation. Nat. Med. 2020, 26, 1726–1732. [Google Scholar] [CrossRef] [PubMed]
- Stachler, M.D.; Camarda, N.D.; Deitrick, C.; Kim, A.; Agoston, A.T.; Odze, R.D.; Hornick, J.L.; Nag, A.; Thorner, A.R.; Ducar, M.; et al. Detection of Mutations in Barrett’s Esophagus Before Progression to High-Grade Dysplasia or Adenocarcinoma. Gastroenterology 2018, 155, 156–167. [Google Scholar] [CrossRef] [Green Version]
- Ross-Innes, C.S.; Becq, J.; Warren, A.; Cheetham, R.K.; Northen, H.; O’Donovan, M.; Malhotra, S.; di Pietro, M.; Ivakhno, S.; He, M.; et al. Whole-Genome Sequencing Provides New Insights into the Clonal Architecture of Barrett’s Esophagus and Esophageal Adenocarcinoma. Nat. Genet. 2015, 47, 1038–1046. [Google Scholar] [CrossRef]
- Stachler, M.D.; Taylor-Weiner, A.; Peng, S.; McKenna, A.; Agoston, A.T.; Odze, R.D.; Davison, J.M.; Nason, K.S.; Loda, M.; Leshchiner, I.; et al. Paired Exome Analysis of Barrett’s Esophagus and Adenocarcinoma. Nat. Genet. 2015, 47, 1047–1055. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; Maden, S.K.; Stachler, M.; Kaz, A.M.; Ayers, J.; Guo, Y.; Carter, K.T.; Willbanks, A.; Heinzerling, T.J.; O’Leary, R.M.; et al. Subtypes of Barrett’s Oesophagus and Oesophageal Adenocarcinoma Based on Genome-Wide Methylation Analysis. Gut 2019, 68, 389–399. [Google Scholar] [CrossRef]
- Luebeck, E.G.; Curtius, K.; Hazelton, W.D.; Maden, S.; Yu, M.; Thota, P.N.; Patil, D.T.; Chak, A.; Willis, J.E.; Grady, W.M. Identification of a Key Role of Widespread Epigenetic Drift in Barrett’s Esophagus and Esophageal Adenocarcinoma. Clin. Epigenet. 2017, 9, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregson, E.M.; Bornschein, J.; Fitzgerald, R.C. Genetic Progression of Barrett’s Oesophagus to Oesophageal Adenocarcinoma. Br. J. Cancer 2016, 115, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Lu, X.; Nossa, C.W.; Francois, F.; Peek, R.M.; Pei, Z. Inflammation and Intestinal Metaplasia of the Distal Esophagus Are Associated with Alterations in the Microbiome. Gastroenterology 2009, 137, 588–597. [Google Scholar] [CrossRef] [Green Version]
- Pei, Z.; Bini, E.J.; Yang, L.; Zhou, M.; Francois, F.; Blaser, M.J. Bacterial Biota in the Human Distal Esophagus. Proc. Natl. Acad. Sci. USA 2004, 101, 4250–4255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morton, J.T.; Marotz, C.; Washburne, A.; Silverman, J.; Zaramela, L.S.; Edlund, A.; Zengler, K.; Knight, R. Establishing Microbial Composition Measurement Standards with Reference Frames. Nat. Commun. 2019, 10, 2719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blackett, K.L.; Siddhi, S.S.; Cleary, S.; Steed, H.; Miller, M.H.; Macfarlane, S.; Macfarlane, G.T.; Dillon, J.F. Oesophageal Bacterial Biofilm Changes in Gastro-Oesophageal Reflux Disease, Barrett’s and Oesophageal Carcinoma: Association or Causality? Aliment. Pharmacol. Ther. 2013, 37, 1084–1092. [Google Scholar] [CrossRef] [PubMed]
- Garrett, W.S.; Gallini, C.A.; Yatsunenko, T.; Michaud, M.; DuBois, A.; Delaney, M.L.; Punit, S.; Karlsson, M.; Bry, L.; Glickman, J.N.; et al. Enterobacteriaceae Act in Concert with the Gut Microbiota to Induce Spontaneous and Maternally Transmitted Colitis. Cell Host Microbe 2010, 8, 292–300. [Google Scholar] [CrossRef] [Green Version]
- Derrien, M.; Belzer, C.; de Vos, W.M. Akkermansia Muciniphila and Its Role in Regulating Host Functions. Microb. Pathog. 2017, 106, 171–181. [Google Scholar] [CrossRef] [Green Version]
- McDonald, D.; Hyde, E.; Debelius, J.W.; Morton, J.T.; Gonzalez, A.; Ackermann, G.; Aksenov, A.A.; Behsaz, B.; Brennan, C.; Chen, Y.; et al. American Gut: An Open Platform for Citizen Science Microbiome Research. mSystems 2018, 3, e00031-18. [Google Scholar] [CrossRef] [Green Version]
- Kusters, J.G.; van Vliet, A.H.M.; Kuipers, E.J. Pathogenesis of Helicobacter Pylori Infection. Clin. Microbiol. Rev. 2006, 19, 449–490. [Google Scholar] [CrossRef] [Green Version]
- Alfarouk, K.O.; Bashir, A.H.H.; Aljarbou, A.N.; Ramadan, A.M.; Muddathir, A.K.; AlHoufie, S.T.S.; Hifny, A.; Elhassan, G.O.; Ibrahim, M.E.; Alqahtani, S.S.; et al. The Possible Role of Helicobacter Pylori in Gastric Cancer and Its Management. Front. Oncol. 2019, 9, 75. [Google Scholar] [CrossRef]
- Islami, F.; Kamangar, F. Helicobacter Pylori and Esophageal Cancer Risk—A Meta-Analysis. Cancer Prev. Res. 2008, 1, 329–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, S.C.; McKinley, M.; Gupta, S.; Peek, R.M.; Martinez, M.E.; Gomez, S.L. Population-Based Analysis of Differences in Gastric Cancer Incidence Among Races and Ethnicities in Individuals Age 50 Years and Older. Gastroenterology 2020, 159, 1705–1714.e2. [Google Scholar] [CrossRef] [PubMed]
- Thrift, A.P. Global Burden and Epidemiology of Barrett Oesophagus and Oesophageal Cancer. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 432–443. [Google Scholar] [CrossRef]
- Nguyen, T.; Ramsey, D.; Graham, D.; Shaib, Y.; Shiota, S.; Velez, M.; Cole, R.; Anand, B.; Vela, M.; El-Serag, H.B. The Prevalence of Helicobacter Pylori Remains High in African American and Hispanic Veterans. Helicobacter 2015, 20, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Anandasabapathy, S.; Jhamb, J.; Davila, M.; Wei, C.; Morris, J.; Bresalier, R. Clinical and Endoscopic Factors Predict Higher Pathologic Grades of Barrett Dysplasia. Cancer 2007, 109, 668–674. [Google Scholar] [CrossRef]
- Pero, R.; Angrisano, T.; Brancaccio, M.; Falanga, A.; Lombardi, L.; Natale, F.; Laneri, S.; Lombardo, B.; Galdiero, S.; Scudiero, O. Beta-Defensins and Analogs in Helicobacter Pylori Infections: MRNA Expression Levels, DNA Methylation, and Antibacterial Activity. PLoS ONE 2019, 14, e0222295. [Google Scholar] [CrossRef] [Green Version]
- Angrisano, T.; Pero, R.; Brancaccio, M.; Coretti, L.; Florio, E.; Pezone, A.; Calabrò, V.; Falco, G.; Keller, S.; Lembo, F.; et al. Cyclical DNA Methylation and Histone Changes Are Induced by LPS to Activate COX-2 in Human Intestinal Epithelial Cells. PLoS ONE 2016, 11, e0156671. [Google Scholar] [CrossRef] [PubMed]
- Pero, R.; Brancaccio, M.; Laneri, S.; De Biasi, M.-G.; Lombardo, B.; Scudiero, O. A Novel View of Human Helicobacter Pylori Infections: Interplay between Microbiota and Beta-Defensins. Biomolecules 2019, 9, 237. [Google Scholar] [CrossRef] [Green Version]
- Poore, G.D.; Kopylova, E.; Zhu, Q.; Carpenter, C.; Fraraccio, S.; Wandro, S.; Kosciolek, T.; Janssen, S.; Metcalf, J.; Song, S.J.; et al. Microbiome Analyses of Blood and Tissues Suggest Cancer Diagnostic Approach. Nature 2020, 579, 567–574. [Google Scholar] [CrossRef]
- Manzoor, S.S.; Doedens, A.; Burns, M.B. The Promise and Challenge of Cancer Microbiome Research. Genome Biol. 2020, 21, 131. [Google Scholar] [CrossRef]
- Sender, R.; Fuchs, S.; Milo, R. Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLoS Biol. 2016, 14, e1002533. [Google Scholar] [CrossRef] [Green Version]
- Bertocchi, A.; Carloni, S.; Ravenda, P.S.; Bertalot, G.; Spadoni, I.; Lo Cascio, A.; Gandini, S.; Lizier, M.; Braga, D.; Asnicar, F.; et al. Gut Vascular Barrier Impairment Leads to Intestinal Bacteria Dissemination and Colorectal Cancer Metastasis to Liver. Cancer Cell 2021, 39, 708–724.e11. [Google Scholar] [CrossRef] [PubMed]
- Münch, N.S.; Fang, H.-Y.; Ingermann, J.; Maurer, H.C.; Anand, A.; Kellner, V.; Sahm, V.; Wiethaler, M.; Baumeister, T.; Wein, F.; et al. High-Fat Diet Accelerates Carcinogenesis in a Mouse Model of Barrett’s Esophagus via Interleukin 8 and Alterations to the Gut Microbiome. Gastroenterology 2019, 157, 492–506.e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freeman, M.; Offman, J.; Walter, F.M.; Sasieni, P.; Smith, S.G. Acceptability of the Cytosponge Procedure for Detecting Barrett’s Oesophagus: A Qualitative Study. BMJ Open 2017, 7, e013901. [Google Scholar] [CrossRef]
- Sanghi, V.; Thota, P.N. Barrett’s Esophagus: Novel Strategies for Screening and Surveillance. Ther. Adv. Chronic Dis. 2019, 10, 2040622319837851. [Google Scholar] [CrossRef] [PubMed]
- Jovel, J.; Patterson, J.; Wang, W.; Hotte, N.; O’Keefe, S.; Mitchel, T.; Perry, T.; Kao, D.; Mason, A.L.; Madsen, K.L.; et al. Characterization of the Gut Microbiome Using 16S or Shotgun Metagenomics. Front. Microbiol. 2016, 7, 459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laudadio, I.; Fulci, V.; Palone, F.; Stronati, L.; Cucchiara, S.; Carissimi, C. Quantitative Assessment of Shotgun Metagenomics and 16S RDNA Amplicon Sequencing in the Study of Human Gut Microbiome. OMICS J. Integr. Biol. 2018, 22, 248–254. [Google Scholar] [CrossRef]
- Durazzi, F.; Sala, C.; Castellani, G.; Manfreda, G.; Remondini, D.; De Cesare, A. Comparison between 16S RRNA and Shotgun Sequencing Data for the Taxonomic Characterization of the Gut Microbiota. Sci. Rep. 2021, 11, 3030. [Google Scholar] [CrossRef]
- Davenport, E.R.; Sanders, J.G.; Song, S.J.; Amato, K.R.; Clark, A.G.; Knight, R. The Human Microbiome in Evolution. BMC Biol. 2017, 15, 127. [Google Scholar] [CrossRef]
- Rosenberg, E.; Sharon, G.; Zilber-Rosenberg, I. The Hologenome Theory of Evolution Contains Lamarckian Aspects within a Darwinian Framework. Environ. Microbiol. 2009, 11, 2959–2962. [Google Scholar] [CrossRef]
- Messer, J.S.; Liechty, E.R.; Vogel, O.A.; Chang, E.B. Evolutionary and Ecological Forces That Shape the Bacterial Communities of the Human Gut. Mucosal Immunol. 2017, 10, 567–579. [Google Scholar] [CrossRef] [Green Version]
- Stewart, C.J.; Ajami, N.J.; O’Brien, J.L.; Hutchinson, D.S.; Smith, D.P.; Wong, M.C.; Ross, M.C.; Lloyd, R.E.; Doddapaneni, H.; Metcalf, G.A.; et al. Temporal Development of the Gut Microbiome in Early Childhood from the TEDDY Study. Nature 2018, 562, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Sieber, M.; Pita, L.; Weiland-Bräuer, N.; Dirksen, P.; Wang, J.; Mortzfeld, B.; Franzenburg, S.; Schmitz, R.A.; Baines, J.F.; Fraune, S.; et al. Neutrality in the Metaorganism. PLOS Biol. 2019, 17, e3000298. [Google Scholar] [CrossRef] [Green Version]
- Bansept, F.; Obeng, N.; Schulenburg, H.; Traulsen, A. Modeling Host-Associating Microbes under Selection. ISME J. 2021. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Ma, Z. Testing the Neutral Theory of Biodiversity with Human Microbiome Datasets. Sci. Rep. 2016, 6, 31448. [Google Scholar] [CrossRef]
- Kim, H.-J.; Kim, H.; Kim, J.J.; Myeong, N.R.; Kim, T.; Park, T.; Kim, E.; Choi, J.; Lee, J.; An, S.; et al. Fragile Skin Microbiomes in Megacities Are Assembled by a Predominantly Niche-Based Process. Sci. Adv. 2018, 4, e1701581. [Google Scholar] [CrossRef] [Green Version]
- Venkataraman, A.; Bassis, C.M.; Beck, J.M.; Young, V.B.; Curtis, J.L.; Huffnagle, G.B.; Schmidt, T.M. Application of a Neutral Community Model to Assess Structuring of the Human Lung Microbiome. mBio 2015, 6, e02284-14. [Google Scholar] [CrossRef] [Green Version]
- Deng, F.; Li, Y.; Zhao, J. The Gut Microbiome of Healthy Long-Living People. Aging 2019, 11, 289–290. [Google Scholar] [CrossRef]
- Dewhirst, F.E.; Chen, T.; Izard, J.; Paster, B.J.; Tanner, A.C.R.; Yu, W.-H.; Lakshmanan, A.; Wade, W.G. The Human Oral Microbiome. J. Bacteriol. 2010, 192, 5002–5017. [Google Scholar] [CrossRef] [Green Version]
- May, M.; Abrams, J.A. Emerging Insights into the Esophageal Microbiome. Curr. Treat. Options Gastroenterol. 2018, 16, 72–85. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Zhang, Y.; Zhao, Q.; Yan, Y.; Yang, T.; Xia, Y.; Chen, H. Patients with Reflux Esophagitis Possess a Possible Different Oral Microbiota Compared with Healthy Controls. Front. Pharmacol. 2020, 11, 1000. [Google Scholar] [CrossRef]
- Gonzalez, A.; Navas-Molina, J.A.; Kosciolek, T.; McDonald, D.; Vázquez-Baeza, Y.; Ackermann, G.; DeReus, J.; Janssen, S.; Swafford, A.D.; Orchanian, S.B.; et al. Qiita: Rapid, Web-Enabled Microbiome Meta-Analysis. Nat. Methods 2018, 15, 796–798. [Google Scholar] [CrossRef]
- Proctor, L.M.; Creasy, H.H.; Fettweis, J.M.; Lloyd-Price, J.; Mahurkar, A.; Zhou, W.; Buck, G.A.; Snyder, M.P.; Strauss, J.F.; Weinstock, G.M.; et al. The Integrative Human Microbiome Project. Nature 2019, 569, 641–648. [Google Scholar] [CrossRef] [Green Version]
- Yachida, S.; Mizutani, S.; Shiroma, H.; Shiba, S.; Nakajima, T.; Sakamoto, T.; Watanabe, H.; Masuda, K.; Nishimoto, Y.; Kubo, M.; et al. Metagenomic and Metabolomic Analyses Reveal Distinct Stage-Specific Phenotypes of the Gut Microbiota in Colorectal Cancer. Nat. Med. 2019, 25, 968–976. [Google Scholar] [CrossRef]
- Buas, M.F.; Gu, H.; Djukovic, D.; Zhu, J.; Onstad, L.; Reid, B.J.; Raftery, D.; Vaughan, T.L. Candidate Serum Metabolite Biomarkers for Differentiating Gastroesophageal Reflux Disease, Barrett’s Esophagus, and High-Grade Dysplasia/Esophageal Adenocarcinoma. Metab. Off. J. Metab. Soc. 2017, 13, 23. [Google Scholar] [CrossRef] [Green Version]
- Proctor, L. Priorities for the next 10 Years of Human Microbiome Research. Nature 2019, 569, 623–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martiny, J.B.H.; Whiteson, K.L.; Bohannan, B.J.M.; David, L.A.; Hynson, N.A.; McFall-Ngai, M.; Rawls, J.F.; Schmidt, T.M.; Abdo, Z.; Blaser, M.J.; et al. The Emergence of Microbiome Centres. Nat. Microbiol. 2020, 5, 2–3. [Google Scholar] [CrossRef] [PubMed]
- Brumfield, K.D.; Huq, A.; Colwell, R.R.; Olds, J.L.; Leddy, M.B. Microbial Resolution of Whole Genome Shotgun and 16S Amplicon Metagenomic Sequencing Using Publicly Available NEON Data. PLoS ONE 2020, 15, e0228899. [Google Scholar] [CrossRef]
- Hillmann, B.; Al-Ghalith, G.A.; Shields-Cutler, R.R.; Zhu, Q.; Knight, R.; Knights, D. SHOGUN: A Modular, Accurate and Scalable Framework for Microbiome Quantification. Bioinformatics 2020, 36, 4088–4090. [Google Scholar] [CrossRef] [PubMed]
- Salosensaari, A.; Laitinen, V.; Havulinna, A.S.; Meric, G.; Cheng, S.; Perola, M.; Valsta, L.; Alfthan, G.; Inouye, M.; Watrous, J.D.; et al. Taxonomic Signatures of Cause-Specific Mortality Risk in Human Gut Microbiome. Nat. Commun. 2021, 12, 2671. [Google Scholar] [CrossRef]
- Hillmann, B.; Al-Ghalith, G.A.; Shields-Cutler, R.R.; Zhu, Q.; Gohl, D.M.; Beckman, K.B.; Knight, R.; Knights, D. Evaluating the Information Content of Shallow Shotgun Metagenomics. mSystems 2018, 3, e00069-18. [Google Scholar] [CrossRef] [Green Version]
- Sinha, R.; Abnet, C.C.; White, O.; Knight, R.; Huttenhower, C. The Microbiome Quality Control Project: Baseline Study Design and Future Directions. Genome Biol. 2015, 16, 276. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Benefits | Pitfalls | Publications | |
|---|---|---|---|
| Sampling Methods | |||
| Esophageal Biopsies |
|
| [37,39] |
| Esophageal Brushings |
|
| [32,37] |
| Saliva/Oral swabs |
|
| [18] |
| Cytosponge samples |
|
| [39,74,75] |
| Sequencing Methods | |||
| 16S rRNA Sequencing |
|
| [76,77,78] |
| Whole Genome Shotgun Sequencing |
|
| [76,77,78] |
| Whole Genome Sequencing (Human DNA library prep) |
|
| [69] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guccione, C.; Yadlapati, R.; Shah, S.; Knight, R.; Curtius, K. Challenges in Determining the Role of Microbiome Evolution in Barrett’s Esophagus and Progression to Esophageal Adenocarcinoma. Microorganisms 2021, 9, 2003. https://doi.org/10.3390/microorganisms9102003
Guccione C, Yadlapati R, Shah S, Knight R, Curtius K. Challenges in Determining the Role of Microbiome Evolution in Barrett’s Esophagus and Progression to Esophageal Adenocarcinoma. Microorganisms. 2021; 9(10):2003. https://doi.org/10.3390/microorganisms9102003
Chicago/Turabian StyleGuccione, Caitlin, Rena Yadlapati, Shailja Shah, Rob Knight, and Kit Curtius. 2021. "Challenges in Determining the Role of Microbiome Evolution in Barrett’s Esophagus and Progression to Esophageal Adenocarcinoma" Microorganisms 9, no. 10: 2003. https://doi.org/10.3390/microorganisms9102003