1. Introduction
The nitrogen biogeochemical cycle requires reduction of atmospheric nitrogen to ammonia. About half of the 413 Tg reactive nitrogen introduced annually to the biosphere is derived through biological nitrogen fixation by the prokaryotic nitrogenase enzyme complex, with 140 Tg.yr
−1 fixation in marine environments and 58 Tg.yr
−1 through non-agricultural terrestrial fixation, and only 2% (5 Tg.yr
−1) contributed by lightning [
1]. The other half is contributed through a combination of synthetically fixed nitrogen and agricultural promotion of bacterial fixation in legumes. Biological nitrogen fixation or diazotrophy probably evolved 3.6–3.2 Ga ago to support expansion of biota in the nitrogen poor environment of that era [
2,
3]. Today it is the second most vital process for life on earth after photosynthesis [
4]. Diazotrophs are divided into two major types: (1) symbiotic nitrogen-fixing bacteria which form symbiotic relation with legumes like
Rhizobium, with actinorhizal plants such as
Frankia, and
Cyanobacteria associated with cycads, and (2) free-living nitrogen fixers belonging to genera such as
Azotobacter and
Clostridium [
5].
Irrespective of their lifestyle all diazotrophs have a nitrogenase multi-subunit enzyme complex which is the only known natural system that catalyzes the breakdown of the triple bond between two nitrogen atoms in N
2 [
6]. This oxygen-sensitive nitrogenase complex has evolved into three variants; molybdenum nitrogenase (Nif), the rarer vanadium nitrogenase (Vnf), and iron-only nitrogenase (Anf) [
7]. Vnf and Anf nitrogenase are also known as alternative nitrogenase. An oxygen insensitive nitrogenase was reported in
Streptomyces thermoautotrophicus [
8], but this has recently been refuted [
9]. Molybdenum nitrogenase encoded by the
nifH,
nifD, and
nifK genes are the most prevalent nitrogenase. Nitrogenase and its homologs have been classified into five phylogroups based on NifH and NifD proteins and their homologs by Raymond et al. [
10]. Group I contains Mo-dependent nitrogenase from aerobic and facultative bacteria and II contains the Mo-dependent nitrogenase from anaerobic bacteria. Group III contains both the alternative forms of nitrogenase. Groups IV and V contain uncharacterized nitrogenase homologs and chlorophyll/bacterio-chlorophyll biosynthesis genes, respectively. Even though Nif homologs in Groups IV and V were thought to be unable to reduce nitrogen, one report suggested that
Endomicrobium proavitum, which encodes a type IV nitrogenase, can fix nitrogen [
11].
NifH is the most sequenced and studied of the three core nitrogenase components, NifH, NifD, and NifK [
12]. Since development of the first
nifH PCR primers by Zehr and McReynolds in 1989, the number of complete
nifH sequences has skyrocketed from 1500 [
13] to more than 8000 sequences at present, available in a curated NifH database (
https://wwwzehr.pmc.ucsc.edu/nifH_Database_Public/, accessed on 2 October 2020). NifH phylogeny conducted by Zehr et al. in 2003 is one of the most comprehensive and is in close agreement with other NifH phylogenies [
14,
15]. They classified NifH into four clusters. Cluster I consists of Mo-containing
nifH and some
vnfH, Cluster II consists of
anfH and some
nifH from Archaea, and Cluster III consists of
nifH sequences from a diverse group of anaerobic bacteria. Cluster IV consists of
nifH homologs, some of which are uncharacterized
nif-like sequences and others are chlorophyllide reductase genes. According to a cross-system comparison of
nifH diversity using 16,989 publicly available
nifH sequences, Cyanobacteria and the Proteobacteria are the most common taxa containing a
nifH gene [
16]. These authors also reported that soil has the most diverse
nifH sequences compared to marine or mat habitats.
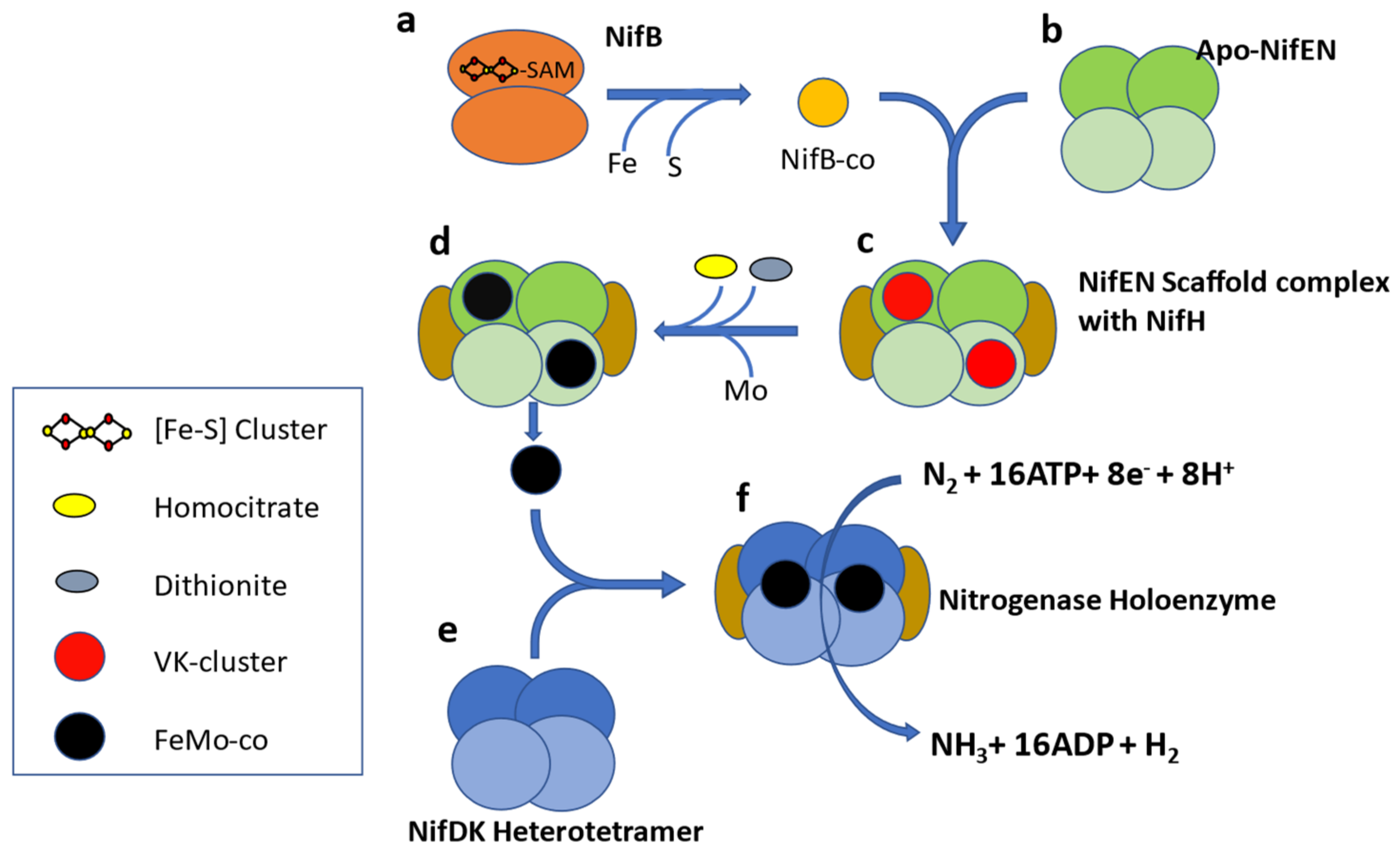
Molybdenum nitrogenase is found in all diazotrophs and is a complex enzyme with two components. Component I, or Dinitrogenase, is a heterotetramer of NifD and NifK proteins which contains an iron-molybdenum cofactor (FeMo-co) in the active site of NifD. The second component, or Dinitrogenase reductase, is a NifH homodimer [
17] (
Figure 1). In addition to these structural proteins, several ancillary proteins are required for diazotrophy, including but not limited to proteins for FeMo-co biosynthesis (NifENBXUSVYQ), nitrogenase maturation (NifZM), and
nif gene expression activation (NifA) [
18]. In some diazotrophs, NifA activity is controlled by the anti-activator NifL [
19], so the list of accessory proteins varies according to the species and physiological condition under which it fixes nitrogen. A study of 2000 complete genomes available in 2012 led to proposing NifHDKENB (
Figure 1) as the minimum criteria for computational prediction of diazotrophy [
20]. This six gene criterion has been widely used as diagnostic for diazotrophy in culture-independent studies [
21,
22,
23,
24].
The most updated phylogenies of nitrogenase are from 2011 based on NifH only [
16], and 2013 using NifHDK [
25], and introduction of multiple
nifH primers has caused a surge of
nifH sequences which in turn has increased the tendency to assign diazotropy to new species without strong biochemical evidence. This has led to a significant deviation from the gold standard of biochemical evidence and caused potential incorrect assignment of phylogeny to new sequences. Hence there is an acute need for a rigorously curated
nif database based on high quality sequences obtained from the complete genomes of cultured isolates. We exploited the recent surge in the number of species with fully sequenced genomes (28,483) to conduct a phylogenetic study of diazotrophs using the six core Nif proteins H, D, K, E, N, and B from publicly available complete genomes. Nif genes were assigned to taxa using a reference phylogeny of bacterial and archaeal genomes, based on a comprehensive set of 381 marker genes [
26] to present a clear view of distribution of diazotrophic species among different prokaryotic phyla. This holistic approach also provides a curated database of six Nif proteins and identifies several genera hitherto not known to fix nitrogen but with potential to do so. The higher resolution of this phylogenetic analysis enabled us to track the evolution and horizontal gene transfer of nitrogenase in individual phyla.
2. Materials and Methods
Annotated NifH, NifD, NifK, NifE, NifN, NifB, VnfH, VnfD, VnfK, AnfH, AnfD, and AnfK protein sequences were downloaded from the InterPro database (
www.ebi.ac.uk/interpro/ accessed on 4 April 2019). Since InterPro is an integrated database built upon the signatures from several member databases like Pfam, PANTHER, and TIGRFAMs, annotation in InterPro is more reliable than individual databases. Sequences were grouped together by taxon name and only genome sequences having all the six genes (
nifHDKENB) were selected for further study. In cases where there is more than one copy of
nif gene per organism, only that
nif gene occurring in the same genomic neighborhood of the remaining minimal
nif gene complement was manually selected to yield a single set of
nifHDKENB sequences for each organism. Organisms with fused
nifEN and
nifNB were also identified based on their sequence length and included. The genomes of organisms containing the defined
nif genes were obtained from NCBI (
www.ncbi.nlm.nih.gov, accessed on 5 April 2020) using CDS IDs obtained from mapping function in uniport (
www.uniprot.org, accessed on 5 April 2020). Protein sequences are included as
Supplemental Data S1.
Biosample IDs associated with the genomes in NCBI were used to obtain the attributes describing sample type (cultured or metagenome), source of isolation, assembly status, biotic relationship (free-living or host associated), host (if applicable), env_material, and env_feature. These attributes were used to get information on the habitat and lifestyle adopted by diazotrophs. All pertinent information is included as
Supplemental Data S1 and S2.
Protein sequences were aligned individually using ClustalW multiple sequence alignment (version 2.1) in Galaxy (
https://usegalaxy.org/, accessed on 7 August 2019) with default parameters and concatenated in R using the Phylotools package (version 0.2.2). Phylogenetic trees from the concatenated sequences were constructed using PhyML 3 [
28], FastTree V2.1.10 [
29] using the JTT+CAT evolution model, and RapidNJ [
30] using the Kimura model. For both FastTree and RapidNJ default amino acid substitution model was used in the final analysis as other models also produced similar tree topologies. Branch support for PhyML 3 was calculated using a Bayesian-like transformation of aLRT (aBayes) [
31] as it was the only method computationally feasible to run at HPC facility. Phylogeny of NifHDKENB was also compared by combining individual Nif trees as well. ASTRAL-III [
32] was used to obtain a combined tree from six individual Nif trees obtained by using FastTree using the JTT+CAT evolutionary model. The ASTRAL tree was compared with concatenated trees by forming a consensus tree of three concatenated trees by using consensus clustering of phylogenetic trees obtained using Rphylip [
33] which is an R implementation of Phylip [
34]. Trees were annotated using iTOL [
35] and Fig Tree v1.4 [
36].
The 16S rRNA sequences were obtained from genomic assemblies in NCBI using the annotation key words 16S rRNA or 16S ribosomal RNA. The tree of life proposed by Zhu et al. [
26] was used to delineate distribution of diazotrophs across taxa and to estimate geological time for the evolution of diazotrophy in different phyla.
3. Results
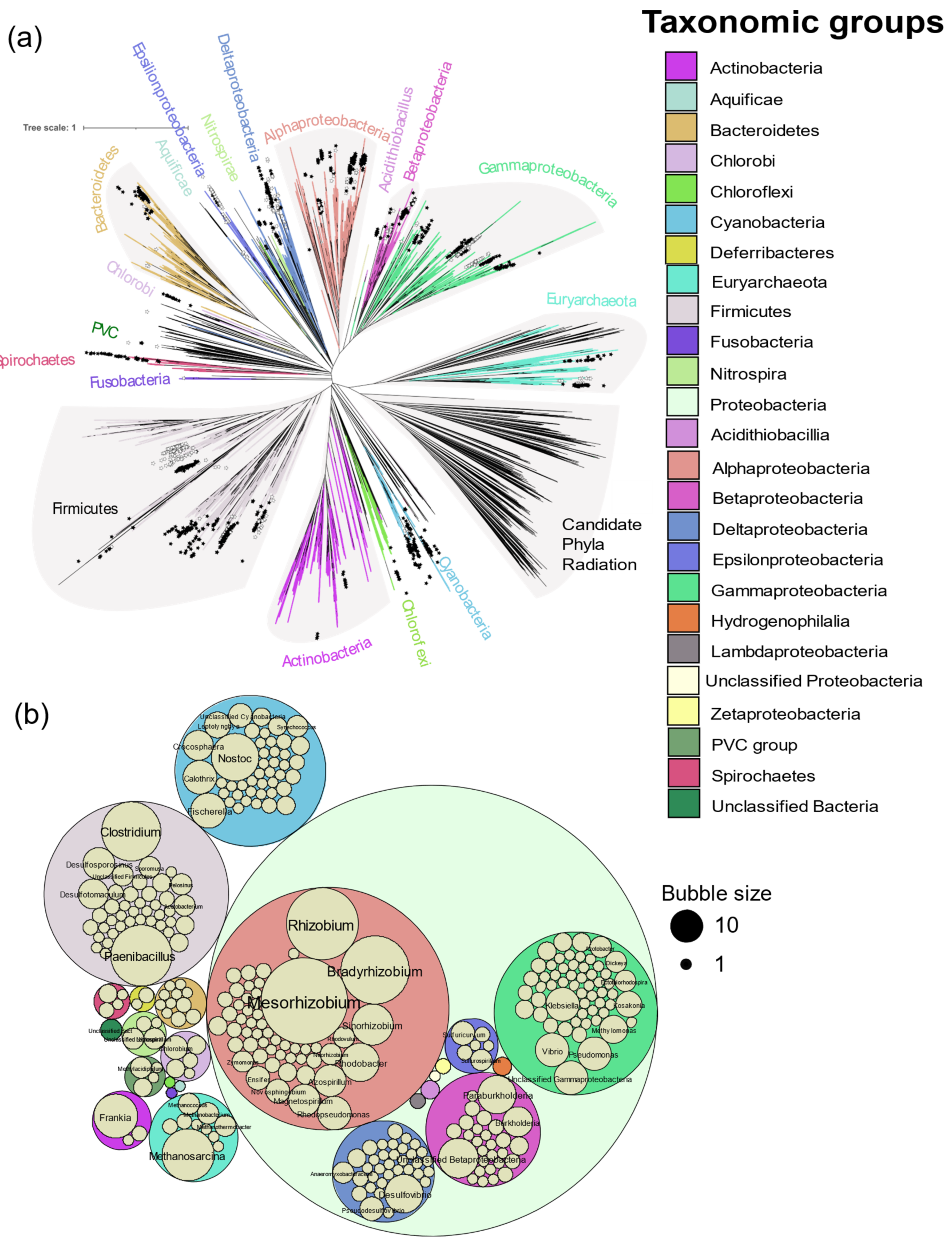
In total, 963 genomes or genomes from metagenomes containing all six
nif genes were obtained, falling into 325 genera from Actinobacteria, Aquificae, Bacteroidetes, Chlorobi, Chloroflexi, Cyanobacteria, Deferribacteres, Euryarchaeota, Firmicutes, Fusobacteria, Nitrospira, Proteobacteria, PVC group, and Spirochaetes (
Figure 2). Of the 24,168 genomes available in NCBI (5 April 2020),
nif genes were found in limited proportion, 0.2 to 41.1% per phylum, showing the wide but patchy distribution of diazotropy in both bacterial and archaeal phyla (
Figure 2 and
Figure 3). Most organisms were from Proteobacteria, followed by Firmicutes and Cyanobacteria. Some smaller phyla like Aquificae, Chloroflexi, and Fusobacteria had only one representative each.
Mesorhizobium with 66 strains was the most sequenced diazotroph followed by
Rhizobium (44), and
Bradyrhizobium (41) (
Figure 3).
Most of the genomes (867) were from cultured isolates, followed by metagenomic assembly (93), and single cell genomes (3). Diazotrophic organisms were from a wide range of habitats (
Figure 2). The largest proportion of diazotrophs were isolated from root nodules (180), followed by terrestrial soil (109), fresh water (99), and biogas digesters (68). More than half (562) of the organisms were free living followed by a symbiotic lifestyle. Most of the symbiotic bacteria were rhizobia (165), fixing nitrogen in root nodules of leguminous plants. Other modes of symbiosis observed were root nodules in actinorhizal plants by
Frankia sp. (15), cyanobacterial endosymbionts of diatoms (3), syntrophic coculture (
Syntrophobotulus glycolicus,
Chlorobium ferrooxidans), phototrophic consortium (
Chlorobium chlorochromatii), Proteobacteria from gill tissue of bivalves (5), Spirochaetes from termite gut (3),
Nostoc in coralloid roots (3), moss carpet (5), and Azolla (1). Thirty-six strains were isolated from thermophilic sites and eight strains isolated from psychrophilic sites (
Supplementary Data S2).
3.1. Biochemical Evidence of Diazotrophy
Incorporation of
15N
2 and the acetylene reduction assay have been used to biochemically confirm diazotrophy. With the growing ease of genome sequencing, isolates can be tentatively reported to fix nitrogen based on occurrence of a
nif operon in their genome. Of the 325 genera containing
nifHDKENB genes, 156 were without biochemical evidence of diazotrophy and most of them were represented by single strains (
Supplemental Data S3).
Figure 3 shows the distribution of prokaryotic genera with and without biochemical evidence in a tree of life proposed by Zhu et al. [
26]. No biochemical evidence was found in the phyla Aquificae and Fusobacteria, both of which are represented here by a single strain. Most of the isolates in the delta and epsilon subdivisions of the Proteobacteria do not have biochemical evidence of N
2 fixation to date.
3.2. Phylogeny of Nitrogenase
A total of 6 genes were obtained from all 963 organisms, but 29 assemblies from Cyanobacteria and Nitrospira had fused
nifE and
nifN, and 44 assemblies from Clostridia which had fused
nifN and
nifB. In addition to the Mo-Fe nitrogenase, 31 assemblies had vanadium containing V-Fe nitrogenase and 44 assemblies had iron only containing Fe-Fe nitrogenase. Species of
Azotobacter,
Clostridium,
Methanosarcina,
Paenibacillus, and
Rhodopseudomonas were found to harbor all three forms of nitrogenase. Not all alternative nitrogenases were found to carry their own cofactor synthesis genes (
nifENB), hence phylogenetic analysis with six genes was limited to the Mo-Fe nitrogenase only. Phylogenetic analysis of Mo-Fe nitrogenase was done using multiple approaches. Reconstruction of species trees from six individual Nif protein trees (
Supplementary Figures S1–S6) using ASTRAL-III gave similar patterns of clustering to the tree constructed using concatenated sequences. Trees constructed with multiple algorithms (PhyML, FASTTREE, and Rapid NJ) from concatenated sequences also agree on the major clusters (
Supplementary Figures S7–S9).
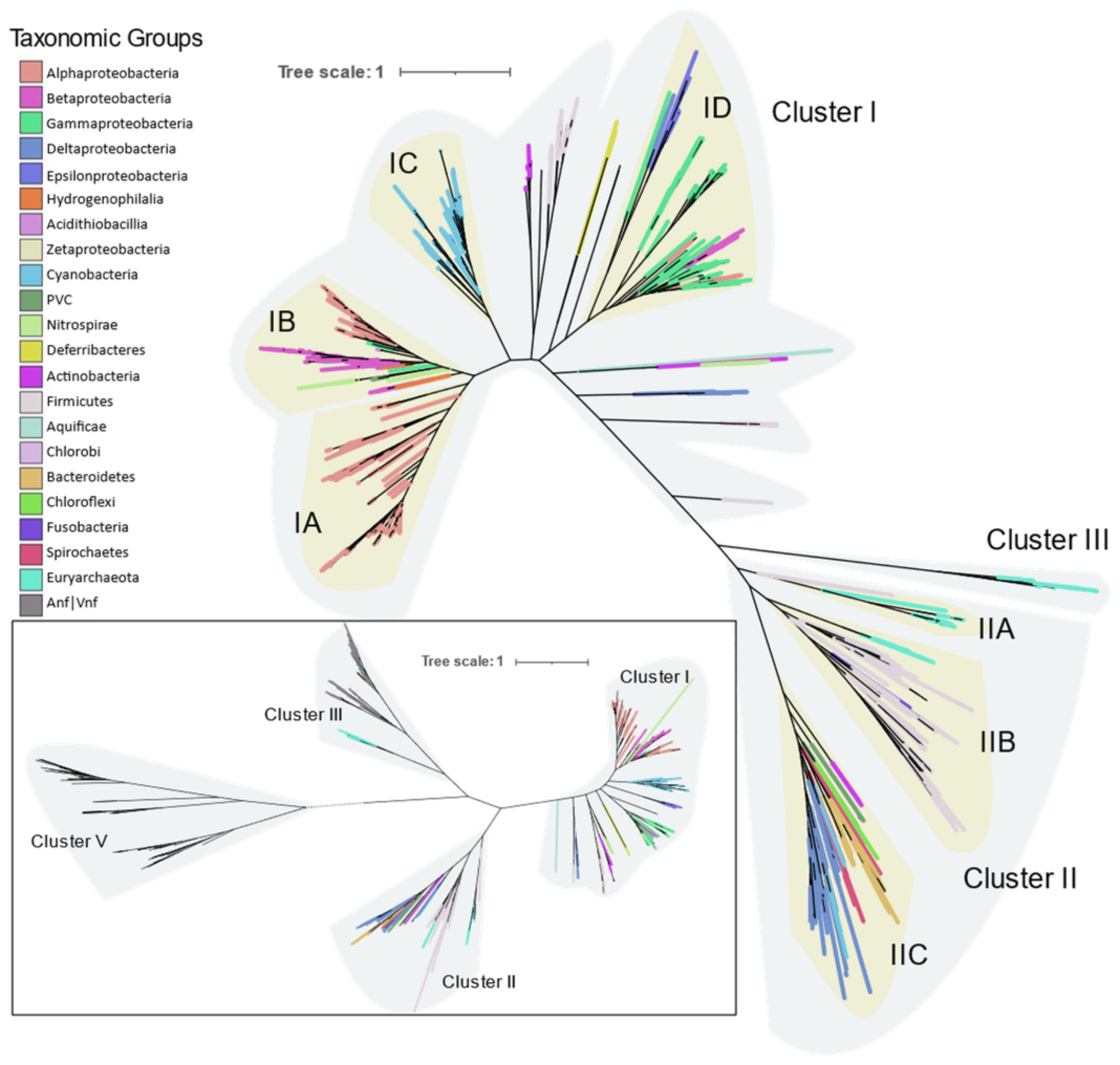
Phylogeny of the Mo-Fe nitrogenase using concatenated NifHDKENB obtained by FASTTREE is given in
Figure 4 and the clusters are labeled according to Raymond et al. [
10]. Cluster I contains aerobic and facultative bacteria from Proteobacteria, Cyanobacteria, Firmicutes, Actinobacteria, and other smaller phyla Aquificae, Deferribacteres, and Nitrospira. Cluster II consists of anaerobic bacteria (Firmicutes, Bacteroidetes, Chlorobi, Chloroflexi, Fusobacteria) and Archaea (Euryarchaeota). Cluster III consists of Mo-Fe nitrogenase from the Methanomada clade of Euryarchaeota and alternative nitrogenase as evident from the NifHDK tree (inset in
Figure 4). Cluster III in the
nifHDKENB tree appears small compared to
nifHDK because alternative nitrogenases were not included as cofactor biosynthesis proteins were not universally present in alternative nitrogenase operons.
3.3. Distribution and Phylogeny of Nitrogenase in Various Phyla
3.3.1. Proteobacteria
Proteobacteria is the largest bacterial phylum and consists of bacteria with diverse morphology and physiology, yet are united by 16S rRNA phylogeny. It had the largest number of representatives in this study as well (574). The nitrogenases of Proteobacteria occur entirely in Cluster I, with the exception of the Deltaproteobacteria whose nitrogenases cluster together with other anaerobic bacteria in Cluster II.
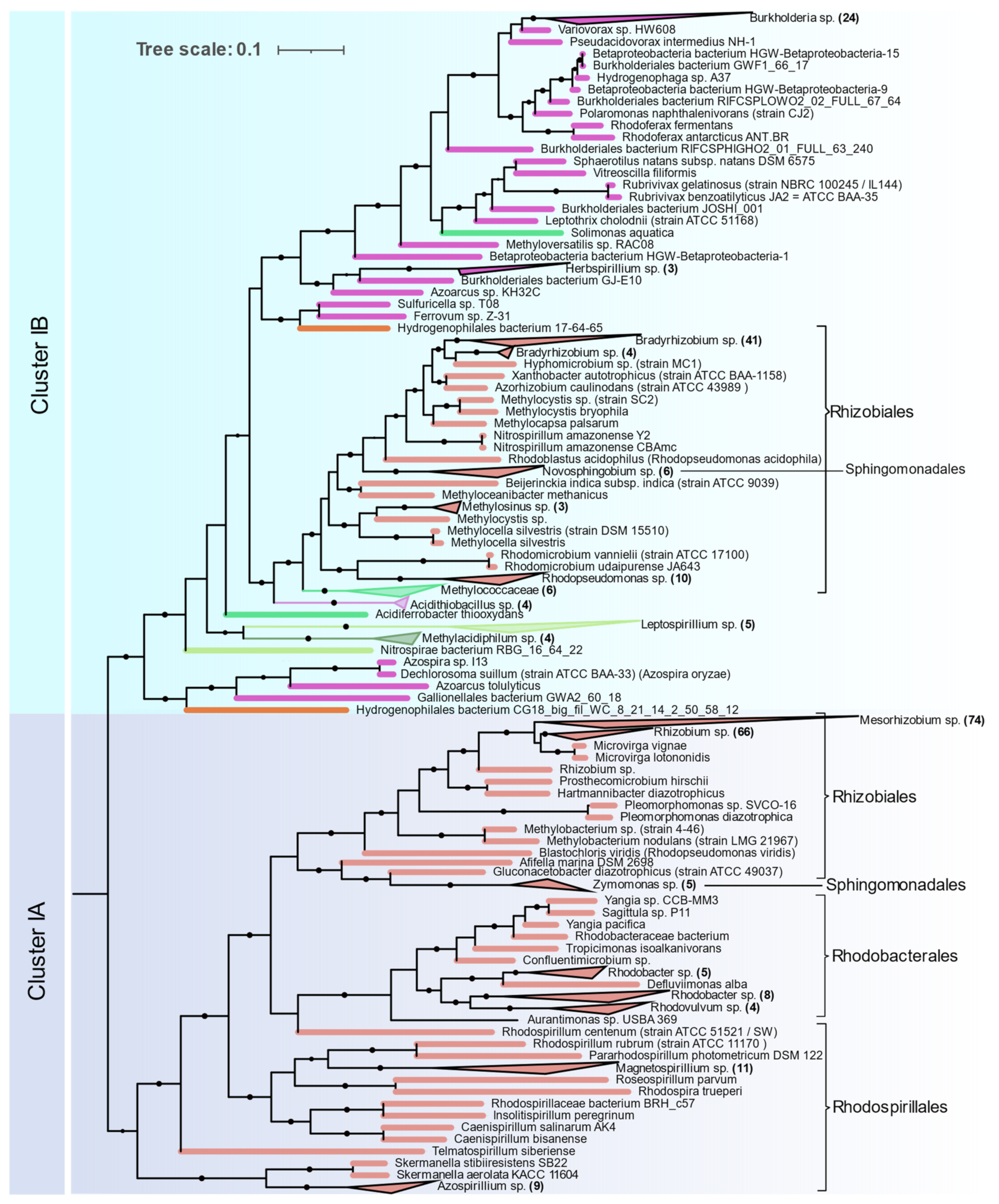
Alphaproteobacteria are distributed in Clusters IA and IB (
Figure 5). The orders Magnetococcales, Rhodospirillales, Rhodobacterales, Sphingomonadales, and Rhizobiales were found to contain nif genes. Cluster IA is coherent with 16S rRNA phylogeny of Alphaproteobacteria where primitive Rhodospirillales, Rhodobacterales, and Sphingomonadales branch out earlier than Rhizobiales. Rhizobiales are distributed between Cluster IA and IB. Phyllobacteriaceae, Rhizobiaceae, and Rhodobiaceae form a monophyletic cluster in Cluster IA whereas Bradyrhizobiaceae, Xanthobacteraceae, and Beijerinckiaceae form a monophyletic cluster in IB along with Betaproteobacteria and other acidophilic methanotrophic bacteria. Some Alphaproteobacteria (
Magnetococcus,
Magnetovibrio,
Martelella, and
Cohaesibacter) occur in Cluster ID along with Gammaproteobacteria.
Nitrogenases from Betaproteobacteria are present in Cluster IB and are represented by the orders Ferrovales, Neisseriales, Nitrosomonadales, and Burkholderiales. The Rhodocyclales are present in Cluster ID. The Burkholderiales were the most abundant diazotrophic Betaproteobacteria in this study.
Gammaproteobacterial nitrogenases form a phylogenetically coherent cluster in ID. Enterobacterales and Pseudomonadales were the most common diazotrophic Gammaproteobacterial. Methanotrophic Methylococcales form a deep branch in the Rhizobiales cluster in IB.
Nitrogenases from
Deltaproteobacteria occur in a monophyletic group in Cluster IIC except for the orders
Desulfuromonadales and
Myxococcales which occur at the base of cluster I. Most of the
Deltaproteobacteria genomes in Cluster IIC are from
Desulfovibrionales and
Desulfobacterales and for several of these isolates, no biochemical evidence could be found (
Supplement File S3). Distribution of nitrogenase in Deltaproteobacteria in Cluster IIC is also coherent with 16S rRNA phylogeny. Similarly,
Desulfuromonadales (strictly anaerobic) and
Myxococcales (aerobic/facultative anaerobic) that occur in Cluster I were found to be phylogenetically distinct from the rest of the Deltaproteobacteria in a recent phylogenetic study [
26].
Epsilonproteobacteria are the next most abundant Proteobacteria represented by 15 isolates, all of which occur in a monophyletic cluster in ID along with Gammaproteobacteria.
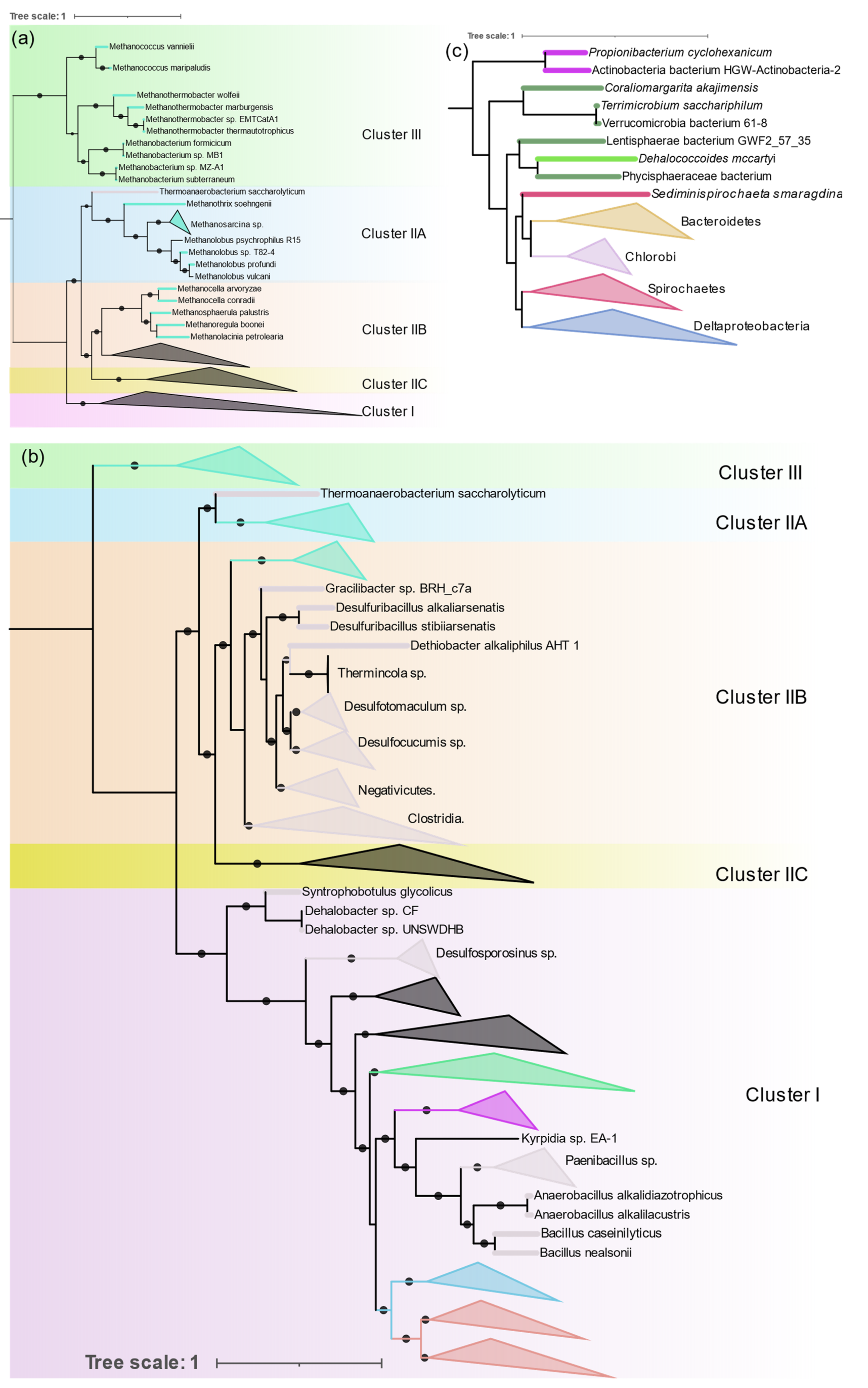
3.3.2. Archaea
Diazotrophic Archaea were only found in methanogenic Euryarchaeota, but they are distributed in both Clusters II and III (
Figure 6a).
Methanosarcina is the most sequenced diazotrophic archaea (24 out of 44 genomes). Its nitrogenase forms a single monophyletic cluster in IIA along with other
Methanosarcinales (
Methanothrix and
Methanolobus).
Methanomicrobiales (
Methanosphaerula,
Methanolacina, and
Methanoregula), and
Methanocellales (
Methanocella) form a monophyletic group in ClusterIIB together with
Firmicutes. Nitrogenases from the group “Methanomada” (
Methanococcales (
Methanococcus), and
Methanobacteriales (
Methanobacterium,
Methanothermobacter)) are different from any archaeal nitrogenase and form a deep branch in Cluster III along with the alternative nitrogenases. Nif phylogeny strictly follows the 16S rRNA phylogeny, indicating the vertical transfer of
nif genes within Archaea.
3.3.3. Firmicutes
Genomes of Firmicutes (145) from Bacilli (41), Clostridia (90), Negativicutes (13), and Tissierellia (1) were found to contain all six
nif genes.
Clostridium and
Paenibacillus are the most prevalent diazotrophs in Firmicutes.
Nif phylogeny shows three distinct evolutionary patterns of nitrogenase in Firmicutes (
Figure 6b). Although distribution of Firmicute
nif appears to be patchy and interspersed by other taxa, within each patch 16S rRNA phylogeny is highly conserved. All Bacilli (except
Desulfuribacillus) are present as a monophyletic cluster in Cluster I together with aerobic high G + C gram positive Actinobacteria. Paenibacillaceae (
Paenibacillus and
Fontibacillus), and Bacillaceae (
Anaerobacillus and
Bacillus) form their own distinct cluster and
Kyrpidia branches out early, which is in close harmony with the 16S rRNA phylogeny of the Bacilli. Only two
Bacillus (
B. nealsonii and
B. caseinilyticus) were found to contain complete
nif operons.
In Cluster IIB Firmicutes occur as a monophyletic cluster which comprises Clostridia, Negativicutes, and Tissierellia. Peptococcaceae (
Desulfosporosinus,
Dehalobacter,
Syntrophobotulus, and
Desulfitobacterium) occur at the base of Cluster I. The division of Peptococcaceae into two phylogroups is also evident in 16S rRNA phylogeny of Peptococcaceae [
37].
Thermoanaerobacterium which branches deep in 16S rRNA phylogeny also has very distinct nif genes that branch out earlier than other Firmicutes and occur in IIA.
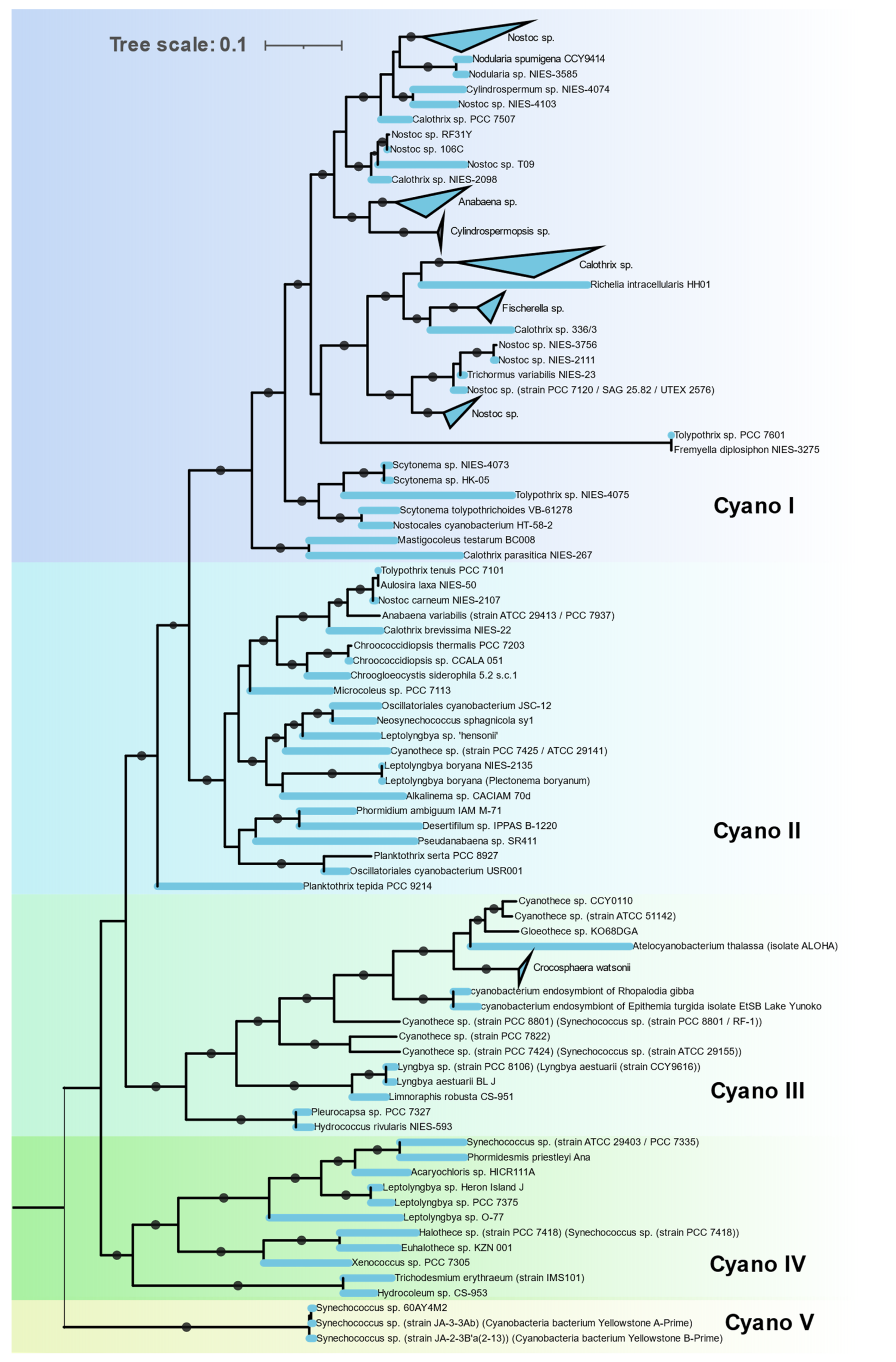
3.3.4. Cyanobacteria
The Nif proteins of all 115 cyanobacterial genomes clustered together in a single cluster (Cluster IC) with no representatives of non-cyanobacterial taxa (
Figure 7). This indicates a monophyletic origin of cyanobacterial
nif genes. The
nif phylogeny agrees with the 16S rRNA phylogeny of these cyanobacterial isolates, indicating all the cyanobacteria derived their
nif operon from a common cyanobacterial ancestor. Cyanobacteria that possess
nif genes were found to occur in various habitats from hot springs (
Synechococus sp. Strain JA-2-3B’a-2-13), antartic endolith (
Chroococcidopsis sp.), marine (
Trichodesmium erythraeum,
Crocosphaera) to fresh water (Oscillatoria). Within the cyanobacterial cluster these isolates form five distinct clades.
Clade I is homogenous in terms of 16S rRNA phylogeny, containing all the Nostocales. These are filamentous cyanobacteria producing specialized, terminally differentiated cells called heterocysts for nitrogen fixation. Since photosynthesis and nitrogen fixation are spatially separated from each other, these cyanobacteria are capable of fixing nitrogen in aerobic condition [
38]. Major representatives in this clade are
Nostoc,
Fischerella,
Calothrix,
Nodularia, and
Anabaena.
Clade II is polyphyletic, containing Synechococcales, Oscillatoriales, Chroococcidiopsidales, and a single representative of Chroococcales,
Chroogloeocystis where no biochemical evidence has been reported. Chroococcidiopsidales are unicellular, free-living cyanobacteria shown to fix nitrogen in anaerobic condition only [
39]. Other cyanobacteria in this clade are all filamentous but lack heterocysts and fix nitrogen in anaerobic or microaerophilic conditions only [
40].
Clade III consists of unicellular Pleurocapsales and Chroococcales along with single filamentous genus
Lyngbya. All Chroococcales of Cluster III form a single cluster and contain some of the most important marine diazotrophs (
Crocosphaera/UCYN B,
Atelocyanobacterium/UCYN A,
Gloethece,
Rippkaea, and
Zehria). Chroococcales use temporal separation of photosynthesis and diazotrophy by fixing nitrogen in the dark cycle only [
41].
Clade IV is also polyphyletic, consisting of Synechococcales, Pleurocapsales, Oscillatoriales, and Chroococcales.
Clade V contains three unique cyanobacteria characterized by deep branching from the rest of the cyanobacteria in both 16S rRNA and NifHDKENB. These are unicellular, thermophilic cyanobacteria isolated from hot springs in Yellowstone National Park [
42]. Although all three strains have all six
nif genes, diazotrophic growth has not been reported yet.
3.3.5. Actinobacteria
There were 21 Actinobacterial genomes containing all 6
nif genes, 18 were
Frankia, 1
Propionibacterium, and 2 unclassified actinobacteria. All the
Frankia make a monophyletic cluster in Cluster I (
Figure 6b) along with other gram-positive bacteria. Nevertheless, anaerobic
Propionibacterium and one of the unclassified actinobacteria cluster together with
nif from
Verrucomicrobium in Cluster IIC (
Figure 6). The remaining unclassified actinobacterial
nif align
nif from Nitrospira. All
nif containing Actinobacteria cluster together by 16S rRNA indicating polyphyletic evolution of nitrogenase in Actinobacteria.
3.3.6. Bacteroidetes and Chlorobi
These phylogenetically related phyla occurred as a monophyletic group in Cluster IIC (
Figure 6c) with a separate branch for each phylum. Only 11 genera (
Bacteroides,
Dysgonomonas,
Paludibacter,
Azobacteroides,
Labilibaculum,
Alkalitalea,
Geofilum,
Saccharicrinis,
Draconibacterium,
Lutibacter, and
Arcticibacter) in Bacteroidetes were found to have all
nif genes, of which only five have been shown to fix nitrogen. Chlorobi is a small phylum of green sulfur bacteria and all the sequenced members of this phylum have
nif genes except
Ignavibacteria. The evolution of nitrogenase in these two phyla is in close alignment with their 16S RNA phylogeny.
3.3.7. Spirochaetes
All the Spirochaete nitrogenases form a monophyletic cluster in IIB (
Figure 6c). All the diazotrophic Spirochaetes belonged to three genera (
Spirochaeta thermophila,
Treponema azotonutricum,
Treponema primitia, and
Sediminispirochaeta smaragdinae). All of them are strictly anaerobic.
3.3.8. Other Anaerobes
Verrucomicrobia, Chloroflexi, Planctomycetes, and Lentisphaerae form a single cluster in IIB (
Figure 6c). Verrucomicrobia, Planctomycetes, and Lentisphaerae are phylogenetically related and group together as the PCV clade. One methanotrophic genus of Verrucomicrobia, Methylacidiphilum forms a separate group in Cluster IB with Nitrospira and other acidophilic Gammaproteobacteria. The Planctomycetes and Lentisphaerae representative were obtained from metagenomic sequencing. The only Chloroflexi found to harbor nitrogenase was Dehalococcoides mccartyi (D. ethenogenes) and the Fusobacteria were also represented by a single observation, Ilyobacter polytropus, both of which occur in Cluster IIB.
3.3.9. Nitrospira, Deferribacterales, and Aquificacea
These are small phyla represented by 10, 3, and 1 strain, respectively. Nitrospira and Aquificae form a single cluster at the base of Cluster I. Acidophilic Leptospirillium sp. of the Nitrospira occur at the base of Cluster IB. Deferribacterales form a single deep branch in Cluster ID.
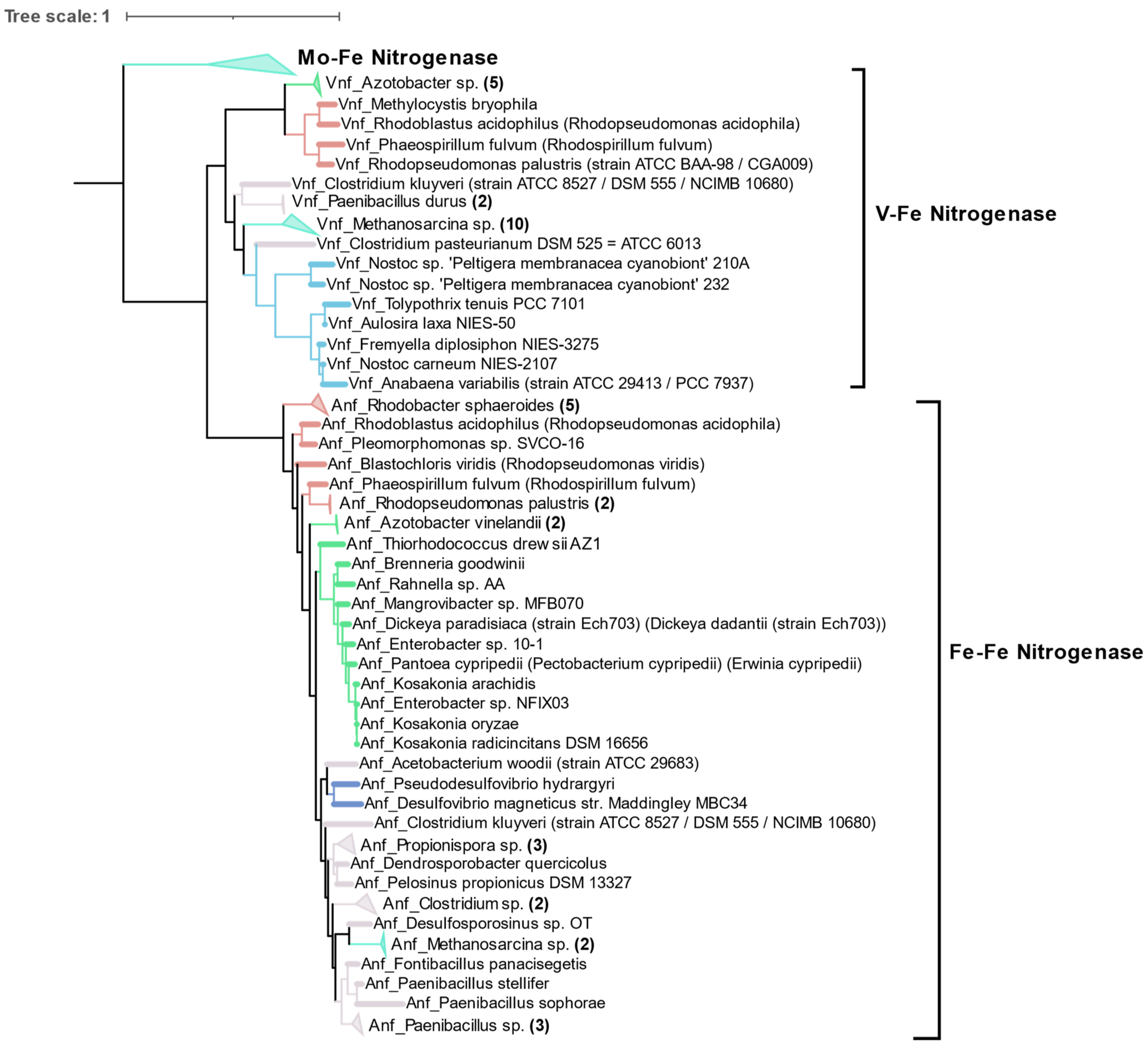
3.4. Alternative Nitrogenases
Compared to the diversity of Mo-Fe nitrogenases, alternative forms of nitrogenase are limited to very few taxonomic groups as shown by phylogeny of concatenated HDK proteins. Vanadium containing nitrogenase (Vnf) is found only in some Alphaproteobacteria, Gammaproteobacteria, Firmicutes, Archaea (
Methanosarcina sp.), and Cyanobacteria. Similarly, Fe only containing nitrogenase (Anf) is found in some Alphaproteobacteria, Gammaproteobacteria, Deltaproteobacteria, Archaea (
Methanosarcina sp.), and Firmicutes (
Figure 8). Phylogenetically, Vnfs and Anfs form their own distinct clades, but are very similar to Mo-Fe nitrogenases from the Methanomada clade of Euryarchaeota. Anfs and Vnfs are very similar to each other, suggesting their very recent evolution from Mo-Fe nitrogenase (inset in
Figure 4).
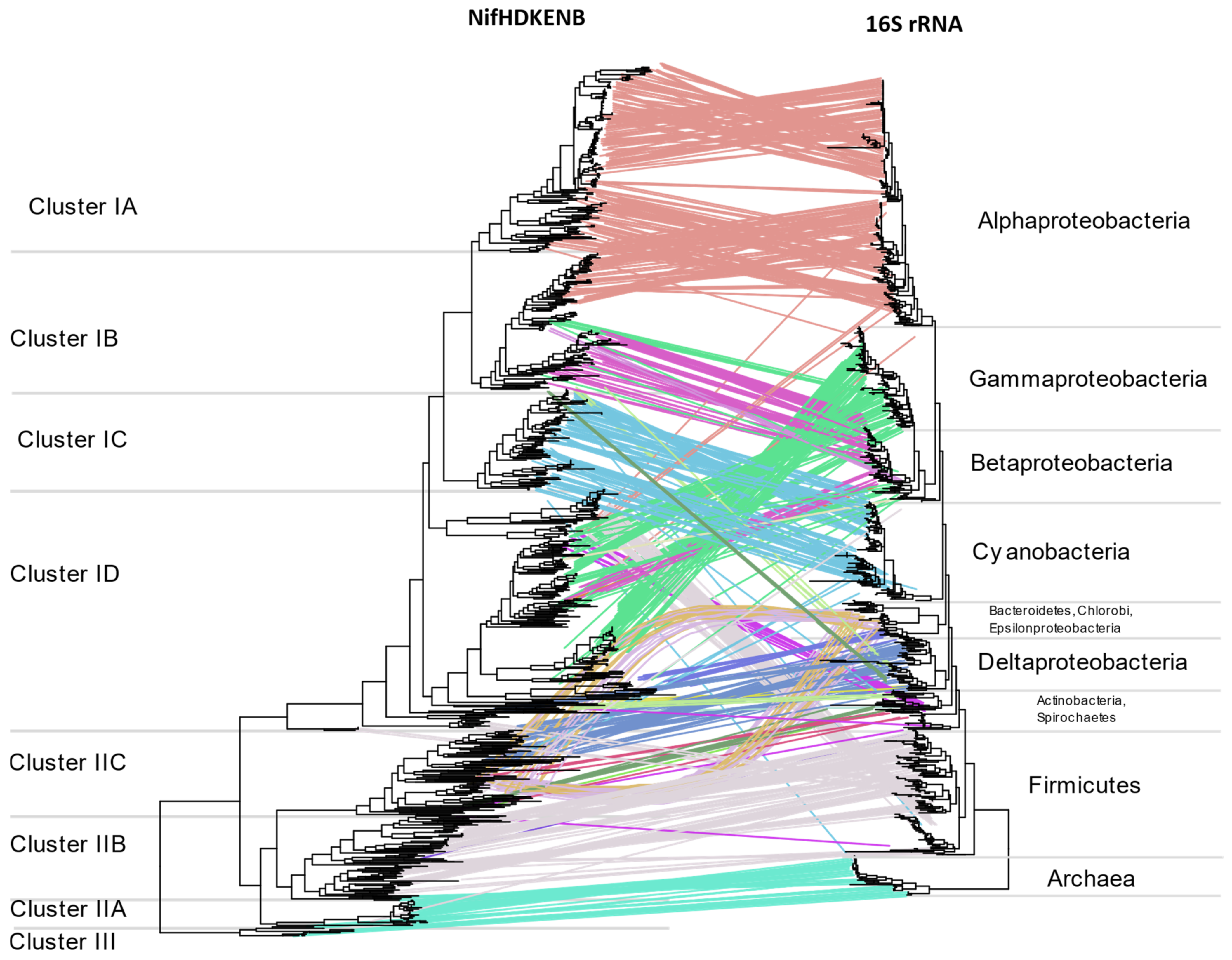
3.5. Horizontal Gene Transfer (HGT)
Multiple instances of HGT are evident when comparing
nif phylogeny with16S rRNA phylogeny (
Figure 9) or the tree of life reported by Zhu et al. [
26] (
Supplementary Figure S10). The two most evident instances of HGT are within anaerobic niches and acidic niches where methylotrophs are concentrated. Regardless of their 16S rRNA phylogeny, the NifHDKENB sequences of strict anaerobes belonging to diverse phyla were similar, aligning together in Cluster II. This included NifHDKENB sequences from strict anaerobes like Clostridia, Bacteroidetes, Chlorobi, PVC, Chloroflexi, Fusobacteria, Deltaproteobacteria, and Spirochaetes that cluster together in Cluster II with Euryarchaea. Within Cluster II, Clostridia and other anaerobes form two clear clusters, indicating two major instances of HGT. Details are apparent when zooming in to the
Supplementary Tree File.
Cluster IB represents another instance of HGT where acidophilic and methanotrophic bacteria cluster together irrespective of their phylogeny. At the root of this cluster are
Methylacidiphilum sp. from PVC,
Leptospirillium sp. from Nitrospira,
Acidithiobacillus from Proteobacteria, and species of Methylococcaceae from Gammaproteobacteria, all of which might have obtained their
nif genes from ancient Alphaproteobacteria. In addition to these, there are also several instances where one or more genus appears to cluster with phylogenetically unrelated bacteria (
Figure 9,
Supplementary Tree File) indicating promiscuous lateral transfer during the early divergence of bacteria.
4. Discussion
Using the minimal structural (NifHDK) and assembly (NifENB) protein components of the nitrogenase enzyme as a marker of diazotrophy, we found diazotrophs in the Proteobacteria, Firmicutes, Cyanobacteria, Bacteroidetes, Chlorobi, Nitrospira, PVC group, Spirochaetes, Deferribacteres, Aquificae, Fusobacteria, Chloroflexi, and Euryarchaeota. In addition to these phyla, diazotrophy has been reported in Chrysiogenales [
20], Acidobacteria [
43] and Elusimicrobia [
11], and
Azoarcus-
Aromatoleum groups [
44] but these were not included in this study as at least one gene was absent or misannotated according to the KEGG database. Although the Candidate Phyla Radiation group (CPR) accounts for over 15% of bacterial diversity [
45], no evidence of diazotrophy was found in the genomes available for this prokaryotic group. CPR bacteria have reduced genome size and lack basic pathways like the citric acid cycle and respiratory chains [
46], indicating the primitive nature of these bacteria. Among non-CPR bacteria, Proteobacteria accounted for more than half of the diazotrophic species and some smaller phyla are represented by only a single species. While distribution of diazotrophs appears widespread across the non-CPR bacteria, only 3.9% of the bacterial whole genomes harbor the six core
nif genes (
Figure 2a). With the consumption of 16 ATPs per dinitrogen reduced, the nitrogenase system is very costly for a bacterium; hence it is energy efficient for species living in nitrogen-rich environments to rescind the nitrogenase enzyme. In fact, multiple studies have shown a decrease in diversity of diazotrophs when treated with nitrogen fertilizer [
47,
48]. Phyla represented by photosynthetic species like Cyanobacteria (22.2%) and Chlorobi (41.2%), that are primarily found in nitrogen deficient aquatic habitats, have significantly higher representation of diazotrophic members. This indicates that the maintenance of nitrogenase systems is heavily dependent on the scarcity of nitrogen in the niche, and coupling of the two most important biological pathways, photosynthesis and nitrogen fixation, is a more efficient method in fixing both C and N.
Traditionally, phylogeny of nitrogenase has been based on either
nifH or
nifD but use of one or both of these was reportedly prone to a false positive rate of diazotrophy [
20]. By using all six genes, we found 84% of the sequenced isolates to have biochemical evidence, and the remaining 16% are recently isolated strains (after 2010) (
Supplementary Figure S11). None of the information available for these strains included mention of experimental testing for diazotrophy, so these could all be new potential diazotrophs not yet characterized. Conversely, the literature abounds with reports of bacterial genera able to fix nitrogen but without genetic or biochemical evidence, for example the genus
Bacillus. Most
Bacillus found to have
nif genes, such as
B. polymyxa and
B. macerans, have been shown to have multiple plant beneficial properties along with nitrogen fixation, and have been reclassified as
Paenibacillus [
49].
B. nealsonii and
B. caseinilyticus, the only
Bacillus that possesses all core six
nif genes, are extremophiles and lack biochemical evidence for nitrogen fixation. This is in contrast to multiple claims of diazotrophic
Bacillus isolates, supported only by culture evidence for growth on nitrogen free media [
50,
51,
52,
53]. However, none of these claims have been supported by genetic evidence. In addition to the archaeal diazotophs proposed by Leigh [
54], potential diazotrophic species were observed in the orders
Methanocellales and
Methanomicrobiales as well. Although Actinobacteria is a large phylum where diazotrophy has been reported in multiple genera [
55], only
Frankia and
Propionibacterium were found to have all six
nif genes in this study.
The six gene criterion yielded nitrogenases aligning exclusively with Clusters I, II and III in the Raymond classification [
10]. While several genomes contained
nifH aligning with Clusters IV or V, none had all six genes, with at least one of the
NifENB gene homologs either missing or not annotated in the KEGG database. As
nifH of Clusters IV and V do not encode functional nitrogenases, this result strengthens the case for the use of six genes for probing potential diazotrophs. One notable exception is
Endomicrobium proavitum reported by Zheng et al. [
11] which was experimentally shown to fix nitrogen but harbors nitrogenase homologous to Group IV nitrogenase. However,
Endomicrobium genomes available in the KEGG database lack
NifEN genes. Many operons containing alternative nitrogenases
vnfHDK or
anfHDK did not have their own set of
ENB genes. This is perhaps indicative that some bacteria encoding Group IV—NifHDK can express an active nitrogenase, even if they do not encode the NifEN assembly proteins. The exact mechanism for this is yet to be determined but suggests that there may be some exceptions to the six gene criteria established by Dos Santos et al. in 2012. Phylogenetically, the six
nif genes have very similar distribution, suggesting that the
nif operon evolved as a unit. Our analysis of the six genes points to the influence of habitat on the evolution of the nitrogenase enzyme. Smaller phyla with species adapted to a particular niche were found clustered together in a single cluster. Taxonomically unrelated species occurring in a particular niche were found to have similar
nif genes, suggesting the role of lateral gene transfer in evolution of the nitrogenase enzyme. As an example, two distinct physiological groups of Firmicutes fell in two distinct NifHDKENB clusters. Anaerobic clostridia and related taxa aligned with archaea occurring in Cluster IIB, while the aerobic firmicutes occurred in Cluster I together with the Actinobacteria. On the other hand, Cyanobacteria, which comprise photosynthetic and mostly aquatic bacteria, occurred in a monophyletic clade which is in close agreement with its phylogeny supported by the study of Esteves-Ferreira et al. [
56]. Species of larger phyla like Proteobacteria and Firmicutes which are adapted to a wide range of physiological conditions were found to have
nif genes distributed across multiple clusters in the phylogenetic tree (
Figure 4). Proteobacteria form phylogenetically coherent clusters except for
Bradyrhizobiaceae and related families of Alphaproteobacteria (
Figure 5), and Betaproteobacteria cluster together with acidophilic, methanotrophic bacteria from diverse taxonomical clades also observed by Khadem et al. [
57] with methylotrophic Verrucomicrobium,
Methylacidiphilum. Subcluster ID mainly consists of Gammaproteobacteria with some exceptions of Alphaproteobacteria like
Martellela,
Cohesibacter,
Magnetovibrio, and Betaproteobacteria like
Aquaspirillium,
Sideroxydans, and some
Rhodocyclales. Similarly, nitrogenase in Archaea is confined within the methanogenic Euryarchaeota. The Stenosarchaea group of Euryachaeota comprises most of the Archaeal diazotrophs, all of which occur in Cluster II together with other anaerobic bacteria. Smaller phyla like Bacteroidetes, Chlorobi, Spirochaetes, and the PVC group, which, in spite of the wide range of habitats, all clustered together in subcluster IIC, correlating with their anaerobic respiratory pathways.
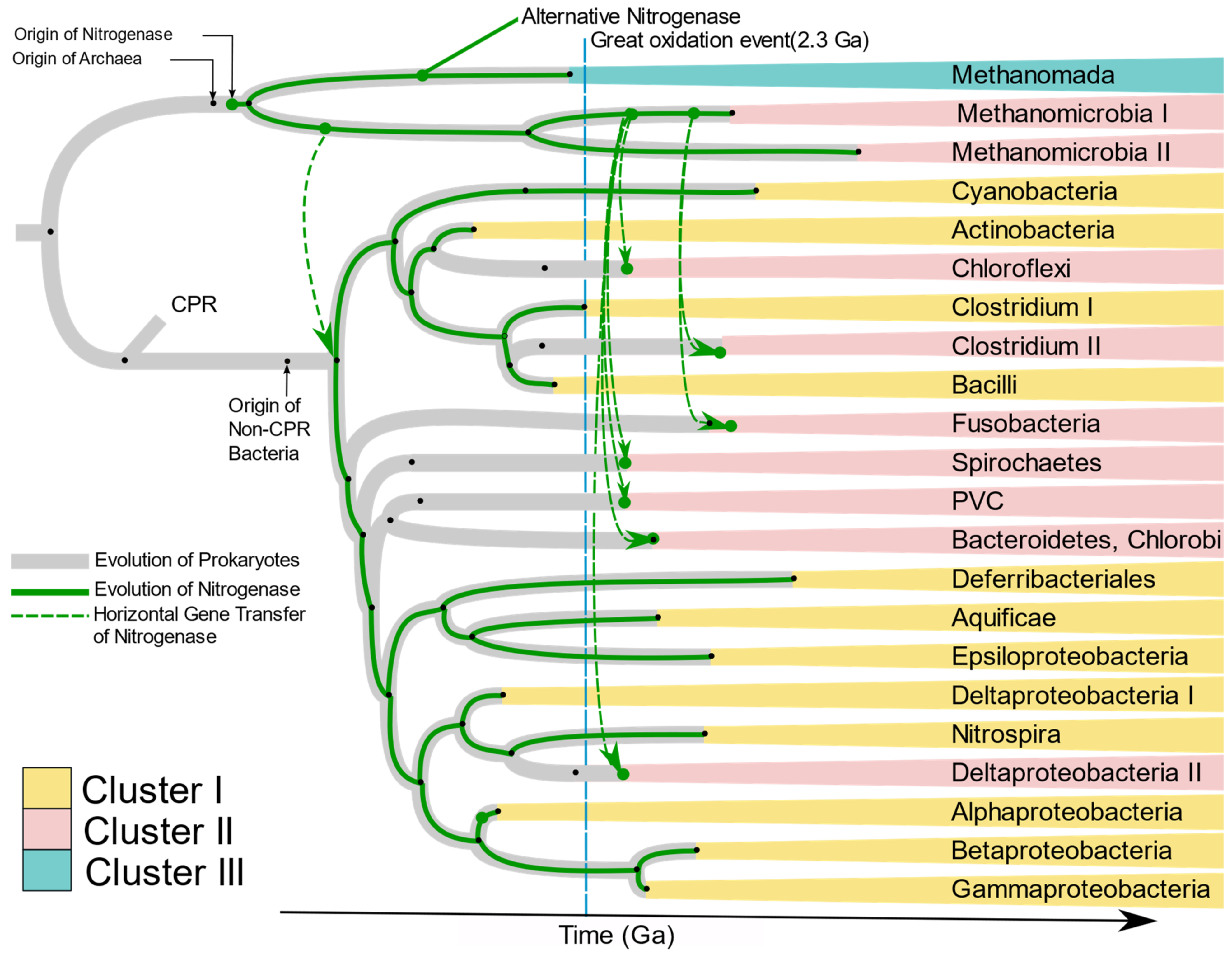
Raymond et al. [
10] proposed two plausible hypotheses of the evolution of nitrogenase. The first is that the nitrogen-fixing LUCA harbored nitrogenase, and present-day diversity was attained by loss of
nif genes in large numbers of taxonomic groups including Eukaryotes and non-methanogenic archaea. The second hypothesis has nitrogenase originate in the methanogenic archaea, with transfer of
nif genes to bacteria by HGT. Our results strongly support the latter hypothesis. The absence of
nif genes in Candidate Phylum Radiation (CPR), Eukarya, and non-methanogenic Archaea, and branching of bacterial
nif from archaea strongly suggests that nitrogenase originated first in primitive methanogenic Archaea. This hypothesis has been supported by Boyd and Peters [
25]. The present phylogenetic analysis of
nifHDKENB genes and the chronogram of prokaryotes proposed by Zhu et al. [
26] (
Supplementary Figure S10) enabled us to propose the evolution of nitrogenase in each phylum harboring diazotrophic species (
Figure 10). Kasting and Walker [
58] proposed that a nitrogen crisis occurred at ~3.5 Ga, overlapping with the origin of methanogenic Euryarchaeota (
Supplementary Figure S10); hence nitrogenase must have evolved during that time in ancient methanogenic Euryarchaeota in response to the shortage of combined nitrogen. Ancient nitrogenase was vertically transferred to two groups of Euryarchaeota, Methanomada, and Stenosarchaea. An alternative nitrogenase originated from ancient Methanomada and forms Cluster III. The first
nif in bacteria was laterally transferred to ancient bacteria from the ancient Methanomicrobia between 3.5 and 3.18 Ga, after which it was vertically transferred to all bacteria (
Figure 10). The present scattered distribution of nitrogenase across multiple phyla likely originated through loss of nitrogenase by organisms adapted to live in nitrogen-rich environments, followed by multiple lateral transfers within anaerobic niches. The great oxidation event in 2.3 Ga. must have turned many bacterial environments aerobic, creating isolated anaerobic niches. This would have brought diverse anaerobes into close proximity in the available anaerobic niches. Co-occurrence of unrelated taxa in close proximity would increase exchange of genes, thus favoring HGT among anaerobes. This is supported by the similarity of
nif genes of anaerobes like Bacteroidetes, Chlorobi, Spirochaetes, Deltaproteobacteria, Clostridium, and PVC to Methanomicrobia. Based on the branching pattern in Cluster II, two distinct HGT’s must have occurred to achieve the present distribution of
nif genes among anaerobic bacteria (
Figure 10), where Clostridium II and Fusobacteria obtained the genes much later than other anaerobes. Cluster I on the other hand is dominated by aerobic and facultative anaerobic diazotrophs, most of which belong to Alphaproteobacteria, Cyanobacteria, and Firmicutes. Although the majority of Alphaproteobacteria have lost the
nif genes, they are preserved in the species which evolved to be in symbiosis with plants (
Rhizobiales) and free-living species which primarily occur in nitrogen deficient habitats like water (
Rodospirillales and
Rhodobacterales). Occurrence of Aquificae and Nitrospirae at the root of Cluster I and clustering of Cyanobacteria, Actinobacteria, and aerobic firmicutes in the
nif tree strongly suggests nitrogenase in these phyla must have evolved by vertical transfer from ancient non-CPR bacteria.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}









