
Uncovering the Worldwide Diversity and Evolution of the Virome of the Mosquitoes Aedes aegypti and Aedes albopictus



1
School of Chemistry and Molecular Biosciences, The University of Queensland, Brisbane, QLD 4072, Australia
2
School of Biological Sciences, The University of Queensland, Brisbane, QLD 4072, Australia
*
Author to whom correspondence should be addressed.
Microorganisms 2021, 9(8), 1653; https://doi.org/10.3390/microorganisms9081653
Submission received: 19 June 2021
/
Revised: 13 July 2021
/
Accepted: 27 July 2021
/
Published: 3 August 2021
(This article belongs to the Special Issue Viral Metagenomics and Metatranscriptomics: Virus Discovery, Ecology, and Evolution)
Abstract
:Aedes aegypti, the yellow fever mosquito, and Aedes albopictus, the Asian tiger mosquito, are the most significant vectors of dengue, Zika, and Chikungunya viruses globally. Studies examining host factors that control arbovirus transmission demonstrate that insect-specific viruses (ISVs) can modulate mosquitoes’ susceptibility to arbovirus infection in both in vivo and in vitro co-infection models. While research is ongoing to implicate individual ISVs as proviral or antiviral factors, we have a limited understanding of the composition and diversity of the Aedes virome. To address this gap, we used a meta-analysis approach to uncover virome diversity by analysing ~3000 available RNA sequencing libraries representing a worldwide geographic range for both mosquitoes. We identified ten novel viruses and previously characterised viruses, including mononegaviruses, orthomyxoviruses, negeviruses, and a novel bi-segmented negev-like group. Phylogenetic analysis suggests close relatedness to mosquito viruses implying likely insect host range except for one arbovirus, the multi-segmented Jingmen tick virus (Flaviviridae) in an Italian colony of Ae. albopictus. Individual mosquito transcriptomes revealed remarkable inter-host variation of ISVs within individuals from the same colony and heterogeneity between different laboratory strains. Additionally, we identified striking virus diversity in Wolbachia infected Aedes cell lines. This study expands our understanding of the virome of these important vectors. It provides a resource for further assessing the ecology, evolution, and interaction of ISVs with their mosquito hosts and the arboviruses they transmit.
Keywords:
insect viruses; Aedes aegypti; Aedes albopictus; ISV; virome; Jingmen tick virus; Wolbachia1. Introduction
The yellow fever mosquito Aedes aegypti, and the Asian tiger mosquito Aedes albopictus, are invasive hematophagous insects distributed worldwide within the tropical and subtropical zones [1]. Both species have expanded from ancestral forest niches into peri-domestic settings facilitated by artificial containers and global shipping routes [2,3].
Laboratory studies of Ae. aegypti and Ae. albopictus have shown that both are competent vectors of numerous arthropod-borne viruses (arboviruses) such as yellow fever virus (YFV), Zika virus (ZIKV), and dengue virus (DENV) (reviewed in [4,5]). Chikungunya virus (CHIKV) (Togaviridae) is also competently vectored by both mosquitoes [6]. DENV is the most widespread arbovirus worldwide, ubiquitous in the tropics, and recently introduced into Europe [7]. Estimates of DENV infection suggest up to 390 million annual cases, of which ~100 million manifests into various disease severity levels [8]. While both Ae. aegypti and Ae. albopictus can vector DENV, reviews of vector competence between the two species indicated that DENV dissemination rates are lower in Ae. albopictus [9], which is considered a secondary or maintenance vector relative to Ae. aegypti [5]. While ZIKV and YFV are estimated to cause fewer annual cases than DENV, both viruses have been responsible for devastating outbreaks in endemic regions, placing billions of people at risk [10,11]. During the 2015–2016 Central and South American ZIKV outbreak, there were an estimated 400,000 cases of ZIKV [12]. Experimental determination of vector competence for both these species for ZIKV suggests that Ae. aegypti is a superior vector to Ae. albopictus [13]. YFV is endemic in regions of up to 47 countries in Africa and Central and South America, with approximately 200,000 annual infections resulting in at least 30,000 fatalities [14].
Vector competence encompasses all the intrinsic and extrinsic host factors and the interplay between these factors (reviewed in [15]). Identifying host factors that modulate vector competence to arboviruses in mosquitoes is an attractive area of research. Exploiting these mechanisms offers the potential to curb the burden of arbovirus disease [16]. Extrinsic or environmental factors such as host-associated microbiota can profoundly impact the vector competence of mosquitoes (reviewed in [17]). Due to arbovirus surveillance programs, several viruses isolated from Ae. aegypti and Ae. albopictus have been discovered, which do not productively infect vertebrate cells and only replicate in mosquito or insect cells. These insect-specific viruses (ISVs) are unlikely to exist in a vector-borne transmission cycle and are maintained through horizontal or vertical transmission. The presence of ISVs can suppress or enhance the subsequent replication of arboviruses. Experimentally ISV-infected Ae. aegypti cells modulate the ability of cells to produce DENV-2 [18,19]. Many in vivo and in vitro studies in Ae. aegypti and Ae. albopictus have shown that ISV-arbovirus interference can be from the same virus family as the arbovirus [20,21,22,23] or completely different virus groups [24,25,26]. The mechanism(s) of this interference phenomenon remains the subject of ongoing research.
Virus discovery generally takes either a culture-dependent or culture-independent approach. In culture-independent methods, samples are subjected to high-throughput total RNA sequencing (RNA-Seq), allowing extensive resolution of the ecology and incidence of viruses. These techniques have been used extensively across eukaryotic systems [27,28,29,30]. Library preparation for metatranscriptomics studies typically involves the clearance or depletion of ribosomal RNA and is not enriched for poly-A mRNA transcripts, a library preparation step typically used for differential gene expression analysis. While the poly-A enrichment step does bias the population of viruses identified, with sufficient depth, many non-poly-A enriched viruses can also be assembled and identified from these data.
In addition to total-RNA sequencing, small RNA (16–32 nt) sequencing is also especially useful for virus discovery; as during virus infection and replication, the host endoribonuclease III enzyme Dicer-2 processes dsRNA produced by viruses during replication into short interfering RNAs (siRNAs) between 20–22 nt length with a strong bias for 21 nt [31,32,33]. Collectively, this pathway is referred to as the RNA interference (RNAi) response and can promote tolerance to virus infection. Furthermore, active RNAi response targeting both strands of a virus genome suggests strong evidence for ongoing replication of viruses, and de novo assembly using these sRNAs has allowed for the recovery of virus contigs [34,35].
As we slowly appreciate the role the virome can play in vector competence, we sought to uncover the incidence, diversity, and evolution of these viruses in Ae. aegypti and Ae. albopictus from previously published RNA-Seq data. We included cell lines, laboratory strains, and wild-caught mosquito pools in our analyses. The outcomes provide a comprehensive list of ISVs present in these mosquitoes and their diversity between and within mosquito populations.
2. Materials and Methods
2.1. Collation of Metadata from Ae. aegypti and Ae. albopictus Virus Publications
To identify publications utilising high-throughput RNA-Seq of Ae. aegypti and Ae. albopictus, we searched Web of Science (http://www.webofknowledge.com; accessed on 20 April 2020) and PubMed (https://www.ncbi.nlm.nih.gov/pubmed; accessed on 20 April 2020) using the following search terms: (“Aedes”[Title] OR “Aedes”[All Fields]) AND (RNA-Seq[Title] OR transcriptomic[Title] OR transcriptome[Title] OR sequencing[Title]). We cross-referenced these publications with raw data deposited in Sequence Read Archive (SRA) hosted by NCBI. We excluded libraries and publications where metadata was not usable, conflicted with methods described within the publication or if uploaders of data were uncontactable. Original catch locations of colonies latitude and longitude positions were approximated from metadata and plotted using ggplot2 (H. Wickham, 2016) and the package “maps” (v 3.3.0, https://CRAN.R-project.org/package=maps; accessed on 20 April 2020) in the RStudio environment (Version 0.99.491). Fastq files from associated SRA accession numbers were downloaded using a Perl script written by Michael Gerth (Oxford Brookes University) and available from GitHub (https://github.com/gerthmicha/perlscripts/blob/master/sra_download.pl; accessed on 20 April 2020).
2.2. Virus Discovery Pipeline
For virus discovery, we split computational tasks evenly across the Galaxy workbench (version 18.09) and CLC Genomics workbench (12.0.3). RNA-Seq fastq files were downloaded, and FastQC (Version 0.11.8, https://www.bioinformatics.babraham.ac.uk/projects/fastqc/; accessed on 20 April 2020) was used to identify adapters. Adapters and low-quality regions of sequencing were trimmed using the CLC in-house trimming tool (quality = 0.05, ambiguous nucleotides = 2) or Trimmomatic (galaxy version: 0.36.4) [36] under the following conditions (Sliding window = 4, average quality = 20). De novo assembly was then undertaken using CLC Genomics Workbench under default conditions or using the Trinity Galaxy wrapper (version 2.4.0.2) [37]. To identify novel viruses, we created a local BLAST database from virus protein sequences deposited in the non-redundant NCBI database (NCBI:txid10239). We excluded several large dsDNA virus families to reduce computational requirements, owing to a high proportion of false-positive BLASTx hits with host and phage proteins. The resulting file (n = 581,356) was collapsed for similarity using the CD-hit tool (Version 4.7) [38] under default conditions (n = 62,840). Assembled contigs were then queried against the representative virus protein database using BLASTx (Expect: 10, Word size: 3, Matrix BLOSUM45, Gap cost Existence 15, Extension 2) [39]. To incriminate viruses as bona fide, we considered several criteria: completeness of coding regions, evidence of sub-genomic transcription, and conserved virus protein domains. For multi-segmented viruses, all segments had to co-appear in multiple libraries or within sequencing libraries from individual mosquitoes; a reciprocal BLASTn analysis of independently assembled virus strains from different libraries allowed to identify chimeric virus assemblies. Putative virus strains were re-mapped to RNA-Seq data to inspect for sufficient coverage and possible misassembly.
As endogenous viral elements (EVEs) are abundant in mosquito nuclear genomes and transcriptionally active [40,41], we excluded the possibility that virus contigs are derived from these EVEs by identifying retrotransposable elements or also partial fragments of totally unrelated viruses, a feature of viral EVE clusters [40]. As PIWI-RNAs (piRNAs) against a single orientation of a viral genome with no virus-derived siRNA (vsiRNA) response in mosquitoes is a classic demonstration of endogenisation of a virus, this was also used to exclude “viruses” that are potentially endogenised. Finally, we screened all the resultant viruses with the recent assemblies of the Ae. aegypti (AaegL5) and Ae. albopictus Foshan (AaloF1) genomes through BLASTn analysis.
2.3. Virus Genome Annotation
Assembled contigs were subjected to BLASTx analysis using our curated database to identify contigs bearing high identity to previously described viruses. Hits bearing an expected value (E-value) < 10−5 were manually inspected for chimeric contigs and then annotated. We predicted the open reading frames of these viruses using the NCBI Open Reading Frame Finder (https://www.ncbi.nlm.nih.gov/orffinder/; accessed on 20 April 2020) with a minimum ORF length of 150 aa and using standard genetic code. Translated ORFs were then analyzed for putative domains with the NCBI Conserved Domain Search (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi; accessed on 20 April 2020) and cross-referenced with the Pfam protein database (v 33.1) search (https://pfam.xfam.org; accessed on 20 April 2020). Predicted ORFs with no known similarity to any proteins based on BLASTp analysis were also analyzed using PSI-BLAST to identify highly divergent homologs [42]. Predicted transmembrane domain helices and signal peptides were identified using the TMHMM Server v. 2.0 (http://www.cbs.dtu.dk/services/TMHMM/; accessed on 20 April 2020) and using the SignalP 3.0 Server (http://www.cbs.dtu.dk/services/SignalP-3.0/; accessed on 20 April 2020), respectively. Nuclear Localisation Signals were predicted using the cNLS Mapper under default conditions [43].
2.4. Virus Coverage Statistics and Virus Incidence Heat Maps
To obtain coverage statistics and to validate potential misassemblies, clean fastq reads were mapped using BWA-MEM [44] and visually inspected using the Integrative Genomics Viewer (IGV v2.6.2) [45]. Coverage was calculated from the resultant Binary Alignment (BAM) files using bedtools [46] based on 5′ positions and graphed using GraphPad Prism (v9.0.0). For the representation of the incidence of viral RNA composition between RNA-Seq libraries, adapter, and quality trimmed fastq libraries were mapped against a representative virus database using the CLC Genomics Workbench mapping program (coverage = 0.9, identity = 0.9). Total mapped reads are presented as heat maps using the pheatmap package (v 1.0.12) (https://cran.r-project.org/web/packages/pheatmap/; accessed on 20 April 2020) in the RStudio environment (Version 0.99.491).
2.5. Analysis of Virus-Derived Small RNAs in Aedes
For examination of the viral RNAi response, we used a previously described pipeline [47]. Briefly, adapter and quality trimmed fastq files (>18 nt) from small RNA datasets were mapped to virus genomes using Bowtie2 (Galaxy Version 2.3.4.3) [48] and coverage calculated as above. Extracted sequence signatures of 27 nt virus-derived piRNAs (vpiRNAs) were generated using Weblogo3 from mapped sense and reverse-complemented antisense reads. Sequence overlaps of vpiRNAs were determined using the small RNA signatures tool version 3.1.0 hosted on the Mississippi Galaxy server (https://mississippi.snv.jussieu.fr; accessed on 20 April 2020) with 24–29 nt viral mapped reads as input.
2.6. Phylogenetic Analysis of Aedes Viruses
For the phylogenetic placement of novel viruses within respective families, we used predicted RNA-dependent RNA polymerase (RdRp) proteins. Closely related viruses from BLASTp analysis of the NCBI non-redundant protein database were aligned using MUSCLE [49]. Ambiguously aligned regions were removed from the alignment using TrimAl (v. 1.3) under the automated 1 method [50]. For phylogenetic inferences of viruses, maximum likelihood trees were produced using IQ-TREE (v1.6.10) [51] using the Le-Gascuel (LG) model [52] with discrete gamma model with 4 rate categories (+G4) and 50,000 Ultrafast bootstraps [53]. FigTree version 1.4 (A. Rambaut; https://github.com/rambaut/figtree/releases; accessed on 20 April 2020) was used to visualise consensus trees.
3. Results and Discussion
3.1. Many Novel Viruses Are Associated with Aedes aegypti and Aedes albopictus Colonies and Cell Lines
To discover and profile viruses infecting Ae. aegypti and Ae. albopictus, we downloaded high-throughput RNA-Seq libraries deposited in the Sequence Read Archive (SRA) database (Figure 1A). These samples represent tissues from various laboratory colonies lab-adapted from various geographic locations or wild-caught outright (Figure 1B, Supplementary Tables S1 and S2) and cell line RNA-Seq data produced from both species. In total, we identified up to 10 novel viruses (Table 1). BLASTp analysis of predicted novel virus sequences based on the RdRp genes suggests viruses presented here are closely related to those known to infect mosquitoes or other insect species (Table 1).
3.2. Novel Negative-Sense RNA Viruses Infecting Ae. aegypti and Ae. albopictus
We identified three novel negative-strand mononegaviruses (Supplementary File S1, Figures S1–S3 for details). The first, Formosus virus, a Rhabdovirus from a lab colony of Ae. aegypti formosus was established from wild-caught samples from Bundibugyo, Uganda [54]. Formosus virus shared close pairwise amino acid similarity to the Culex rhabdo-like virus assembled from a metagenomic analysis from Culex mosquitoes in California, USA [55] (Figure 2B). Phylogenetic placement of Formosus virus from alignments of the L protein suggests Formosus virus groups within Rhabdoviridae.
All members of Mononegavirales have a negative-stranded RNA genome encapsidated within the capsid and the RNA polymerase complex [56]. The RNA polymerase complex sequentially transcribes discrete mRNAs from the genome with mRNA from each gene, which are then capped and polyadenylated. Importantly, all three viruses had sub-genomic mapping profiles typical of mononegaviruses from poly-A enriched libraries (Supplementary File S1: Figures S1–S3)
We identified a multi-segmented negative-sense RNA virus from the Orthomyxoviridae family (Figure 3) infecting Ae. albopictus samples, which we name Aedes orthomyxo-like virus 2 (AOMV-2). This virus is very lowly abundant in Foshan Ae. albopictus RNA-Seq data [69,70] but abundant in RNA-Seq data from a lab colony established from the Torres Strait in Australia [72,123]. Phylogenetic placement of this virus based on alignments of the RdRp polymerase basic 1 subunit (PB1) places AOMV-2 within a well-supported mosquito clade of unassigned orthomyxoviruses (Figure 3B). Orthomyxoviruses have a nuclear replication strategy, and we predicted several monopartite and bipartite nuclear localisation signals on multiple proteins from AOMV-2, showing a close association with the host nucleus (Supplementary File S1: Table S7).
3.3. Novel Positive-Sense RNA Negev-like Viruses Infecting Ae. aegypti and Ae. albopictus
Negeviruses are positive-sense RNA viruses with genome sizes between 9–10 kb encoding three ORFs that infect multiple mosquito species and sandflies. All negeviruses currently reported have been unable to replicate in vertebrate cell cultures [124]. We identified one novel coding complete negev-like virus (Figure 4A; details in Supplementary File S1) in Ae. aegypti originating from two colonies collected in Rabai, Kenya (K2, K4), and therefore, we named the virus Rabai virus [54]. Phylogenetic analyses of the Rabai virus based on the highly conserved VMet, HEL, and RdRp domains suggests a close relatedness to other negev-like viruses assembled from metagenomic studies of mosquitoes (Figure 4B) but does not belong to the monophyletic Nelorpivirus and Sandewavirus taxons as previously reported [125].
3.4. Binegeviruses: A Novel Negev-Related Taxon with Bi-Segmented Genomes in Aedes Mosquitoes
In addition to the monopartite negev-virus genomes, we discovered two novel bi-segmented viruses distantly related to the negev-like viruses in both Ae. aegypti and Ae. albopictus (Figure 4A). The larger segment of these “binegeviruses” is ~7600 nt, encodes for two ORFs with the prototypical negevirus viral methyltransferase (VMet) and helicase (HEL) domains (Supplementary File S1: Figures S5 and S6). The second smaller segment is ~4600 nt and contains two ORFs with an RdRp. TBLASTn analysis of the predicted proteins of both segments suggests that these viruses are most closely related to the 7413 nt ssRNA virus-like sequence 6 (Genbank ID: KX148585) and the 4642 nt ssRNA virus-like sequence 5 (Genbank ID: KX148584) identified from metagenomics analysis of Anopheles mosquitoes [126]. Phylogenetic inferences based on alignments of the VMet, HEL, and RdRp domains from both viruses, tentatively named Aedes binegev-like virus 1 (AeBNV-1) and Aedes binegev-like virus 2 (AeBNV-2), suggest the formation of a phylogenetically divergent and well-supported grouping distinct from other negev-viruses (Figure 4B) and grouping with a partial RdRp from Blackford virus identified from Drosophila tristis pools [127]. Both segments of AeBNV-1 and AeBNV-2 co-appear in all mosquito pools positive for these viruses in numerous American Ae. aegypti datasets and individual Ae. albopictus mosquitoes from Longgang District, Shenzhen, China [68], supporting the likelihood of these viruses belonging to the same group. For this reason, we believe it is likely that the ssRNA virus-like sequence 5/6 previously reported belongs to the same virus as both co-appeared in pools of mosquitoes from Senegal and Liberia [126]. Bi-segmentation and tri-segmentation of viruses from the Virgaviridae family are well established [128]. The RdRp_2 domain on a smaller segment is a classic feature of the plant infecting Hordeivirus genus with the barley stripe virus as the best-known member [128].
3.5. Tombus-Noda Viruses in Aedes
Previously, fragments of a potential “Mosquito nodavirus” were assembled from small RNA-Seq data from one Liverpool colony of Ae. aegypti [34,35]. We were able to complete the genome of this virus by assembling multiple RNA-Seq libraries from numerous Liverpool Ae. aegypti datasets and find that this “Mosquito nodavirus” is not segmented like classical nodaviruses but instead has a classical monopartite 4 kb genome (Figure 5A). The virus is more closely related to the single genome Tombus-Noda arthropod lineage (Figure 5B) [73]. For this reason, we chose to re-name this virus Liverpool tombus-like virus, as it appears to infect Liverpool colonies of Ae. aegypti exclusively, with one exception being one Ae. albopictus colony co-housed with the Ae. aegypti Liverpool strain [129]. Additionally, we identified a divergent strain of a similar tombus-noda virus from the Foshan Ae. albopictus colony, which we named Foshan tombus-like virus, with similar genome features and phylogenetic position (Figure 5B). We identified a common novel multi-segmented tombus-like virus, which is a ubiquitous infectious agent in Ae. albopictus mosquitoes, termed tiger mosquito bi-segmented tombus-like virus (TMTLV). The genome orientation and structure of TMTLV is very similar to Diaphorina citri-associated C virus (DcACV) from the Asian citrus psyllid (Diaphorina citri) to which TMTLV is phylogenetically related (Figure 5B) [130], with segment one on TMTLV and DcACV encoding the RdRp_3 domain. The second segment encodes a putative DiSB-ORF2_chro domain, a putative virion glycoprotein of insect viruses (Supplementary File S1: Figure S10) [131]. This virus is ubiquitous in American, Asian, and European Ae. albopictus mosquitoes (Table 1). TMTLV is abundantly targeted by the RNAi response in the Foshan colonies and Ae. albopictus strain at the University of Pavia [93,94], and recently lab-adapted strains from Vietnam and Japan [59,86].
3.6. Double-Stranded RNA Viruses Infecting Ae. aegypti and Ae. albopictus
Partitivirus-like sequences belonging to a putative partitivirus named Aedes partiti-like virus 1 (APLV-1) were distributed worldwide in Ae. aegypti colonies (Table 1, Figure 6A). Phylogenetic placement of the RdRp segment of APLV-1 indicated a close relationship of APLV-1 and mosquito and dipteran partitiviruses (Figure 6B). Members of the Partitiviridae family possess two genome segments, dsRNA1 (RdRp) and dsRNA2 (Virus Coat), each containing one large ORF between 1.4–2.4 kb. Each genome segment is separately encapsidated. Some partitiviruses have additional (satellite or defective) dsRNA elements. We identified two segments of a putative partiti-like virus in three Rabai, Kenya (K2, K4, K14) colonies [54]. Another colony from this study, the K27 colony, did not have any reads mapping to APLV-1 but was crossed with the APLV-1 positive colony K14. We identified both segments in all the libraries in each of the four progeny libraries (GP1, GP2, HP1, HP2). This suggests that APLV-1, like the recently identified partitivirus Verdadero virus in Ae. aegypti mosquitoes may exhibit efficient vertical transmission. Using the massively high-throughput nature of our metanalysis, we could incriminate the Chaq-like virus (Genbank ID: MT742176.1), previously identified in Verdadero virus-infected Ae. aegypti samples, as a likely satellite or additional segment of Verdadero virus. Chaq-like virus (Genbank ID: MT742176.1) sequences co-appeared in every library along with the two segments of Verdadero virus from multiple independent sequencing efforts (11/11 BioProject accessions) and importantly co-appear in up to eight individual mosquitoes from individual midgut libraries produced by Raquin et al., 2017 [119], although in low abundance. We identified a virus belonging to the Reoviridae family that is phylogenetically related to the Orbivirus genus (Figure 6C,D), which we have tentatively named Aedes orbi-like virus. Phylogenetic analysis of the VP1 segment of the virus suggests a close relationship to orbiviruses assembled from a pool of Ochlerotatus mosquitoes (Figure 6D) [132]. In total, we were able to identify seven segments designated VP1-7 as per orbivirus convention (Figure 6C).
While four of the AOLV segments had detectable homologs from other insect reo-like viruses, three out of the seven incriminated segments of AOLV had no detectable homology with any known virus from this group or RNA viruses (Supplementary File S1; Figure S8 & Table S8), suggesting a highly divergent genome within this virus taxon. Identification of these additional segments of AOLV was aided by co-appearance in numerous individual transcriptomes from a field adapted colony from Cairns, Australia [108]. In addition to co-appearance in individual mosquito transcriptomes, all seven segments are targeted by the RNAi response in lab colonies from Far North QLD, Australia [105].
3.7. DNA Viruses of Aedes Mosquitoes
Mosquito densoviruses (MDVs) are single-stranded DNA viruses of the Parvoviridae family known to infect both Ae. aegypti and Ae. albopictus (reviewed in [133]). MDVs in Ae. aegypti and Ae. albopictus belong to the type species Dipteran brevidensovirus 1 and 2 of the monosense Brevidensovirus genus [134]. We identified several datasets of Ae. albopictus colonies and cell lines infected with MDVs and were able to recover the complete coding genome (NS1/2 and VP genes) of these putative MDV strains. Phylogenetic placement of the NS1/2 and VP genes of these contigs (Figure 7) suggested these MDVs are most closely related to MDVs previously identified from other mosquitoes. Based on species demarcation criteria of the Parvoviridae (<80% amino acid identity from the replicase protein), they are not sufficiently divergent to be separate species of MDV.
After the initial assembly of a putative MDV from the Ae. albopictus U4.4 cells sequenced as part of the Arthropod Cell Line RNA-Seq initiative, Broad Institute, we examined the incidence of this MDV sequence in U4.4 cells and could not identify reads mapping to this MDV from any other U4.4 datasets (Supplementary Table S5). Given this densovirus is most closely related to Anopheles gambiae densovirus (AgDV), which has been previously isolated from the Anopheles gambiae cell line Sua5B [135], we presume that it is likely sequencing contamination from Sua5B cells. Further examination of all Aedes cells from this initiative demonstrates that the virus variant is abundant in all Aedes cell lines but absent in other U4.4 datasets (Supplementary Table S5). Additionally, we identified one Ae. albopictus C6/36 sRNA dataset that produces abundant vpiRNAs against another more distantly related MDV, Culex pipiens densovirus (CppDNV). We were even able to partially assemble the virus genome from this data [136]. Examination of the incidence of CppDNV in other C6/36 cells suggested that CppDNV was absent in every C6/36 RNA-Seq dataset examined (Supplementary Table S5). This indicates that either CppDNV is so lowly abundant that it is beyond detection, potentially integrated into the genome, or only exists in a handful of laboratory C6/36 isolates. In addition to potential infection of Aedes cells, we identified MDV infections in Ae. albopictus RNA-Seq data from Manassas, USA [90] and the Foshan, China colony (62, 63). The coding-complete genome of MDV from the Ae. albopictus Foshan colony (BioProject: PRJNA275727) is most closely related to Aedes albopictus densovirus 7 isolate GZ07 (Figure 7) [137].
3.8. Evidence of the Jingmen Tick Virus (Flaviviridae) in an Ae. albopictus Mosquito Colony
In 2014, Jingmen tick virus (JMTV), a segmented positive-sense RNA virus, was reported following isolation from Rhipicephalus microplus tick and mosquito pools collected in the Jingmen region of Hubei Province, China [138]. The genome structure of JMTV comprises four polyadenylated segments, two of which share close homology to non-structural (NS) proteins of classical flaviviruses, with segment one encoding an NS5-like RdRp and methyltransferase, and segment three bearing close similarity to the protease (NS3), with the other two segments originating from a yet unidentified ancestor (Figure 8A). JMTV replicates in Ae. albopictus C6/36 and several mammalian cell lines [138,139]. Since the first description of JMTV, there has been evidence of human infections from both Kosovo [140] and China [139]. We assembled an almost complete JMTV strain from the Rimini, Italy colony of Ae. albopictus except for a 27 nt gap in segment 1. Subsequent re-mapping of all libraries from this study indicated that only three out of the 24 libraries had detectable JMTV RNA (BioProject PRJNA493544: SRA Accessions: SRR7907917, SRR7907927, SRR7907936) [141]. Phylogenetic analysis of the conserved Segment 1 and Segment 3 of the Rimini JMTV strain suggests that all segments are most closely related to JMTV strains from ticks sampled from the French Antilles [142] and also Trinidad and Tobago [143] (Figure 8B,C), sharing approximately 92–95% nucleotide identity over the coding genome.
These RNA-Seq data samples are replicate pools of 10–24 fat bodies and heads from 5-day-old females fed on 20% sucrose and water. Given the tissues sampled and that JMTV reads were lowly abundant in these libraries (0.02% of all reads), it appears unlikely that the introduction of JMTV into these samples are due to recent blood-feeding for maintenance. However, given that the Rimini colony was established in 2004 and sampled in November 2017, we cannot rule out the possibility that JMTV may have been introduced from the previous blood-feeding to maintain the colony. Further examination of the incidence and persistence of JMTV in Aedes mosquitoes is required.
3.9. Diversity of the RNAi Response against ISVs of Ae. aegypti and Ae. albopictus
To examine the RNAi response to Aedes ISVs, we mapped reads from small RNA sequencing libraries against our virus database, examining the size distribution of the virus fragments originating from these genomes and the mapping profiles of Aedes viruses (Extended Figures in Supplementary File S2). We demonstrate that ten of these viruses presented with prototypical virus-derived short interfering RNAs (vsiRNAs) as part of the RNAi response with a 21-nucleotide peak targeting both sense and antisense genomes. A summary of the size distribution profile of mapped reads to virus genomes is presented in Figure 9.
The virus-derived small RNA profiles are consistent with published virus RNAi profiles from closely related viruses; for example, San Gabriel mononegavirus has abundant virus-derived P-element Induced Wimpy testis RNAs (vpiRNA) length small RNAs dominant in the population of small RNAs consistent with other studies examining the RNAi response of mononegaviruses [18,144].
We could identify the vsiRNA profile for both segments of the Verdadero virus, a partiti-like virus previously identified in Ae. aegypti colony metatranscriptomics [145]. We also show that the positive-sense RNA virus Wenzhou sobemo-like virus 4 produces an RNAi response in vivo in Ae. albopictus mosquitoes from several libraries [86,87]. We could also identify the RNAi signature from both Renna virus segments [29] (Figure 9).
In addition to virus RNA being targeted and degraded by the RNAi pathway, we also observed that many of these viruses abundantly produce virus-derived RNAs of size 24–30 nt corresponding to vpiRNAs. vpiRNAs can be determined by a ping-pong signature (U1-A10) and 10 nt complementary overlapping reads. For the San Gabriel virus and all segments of Aedes phasma virus [58,146], as well as individual segments of TMTLV and WSBLV-4, we observed a ping-pong signature (U1-A10) and over-represented complementary 10 nt overlapping pairs of sRNA from reads of 24–29 nt from these viruses (Supplementary File S2). The absence of a predominant 21 nt species of vsiRNA targeting CppDNV and WSBLV-4 in Ae. albopictus C6/36 and C7-10, respectively, are consistent with these cell lines RNAi deficient nature. Virus infections within these cell lines do not produce a typical 21 vsiRNA peak. Instead, dsRNA is targeted and processed in C6/36 and C7-10 cells by the piRNA pathway [129,147].
3.10. Differentially Abundant ISVs in Aedes Laboratory Colonies
To understand the composition and incidence of ISVs between different laboratory colonies, we created a database of representative genomes of novel viruses identified here and previously identified viruses known to infect these mosquitoes (n = 94). Libraries originating from one biological sample or group were trimmed, pooled, and mapped to these genomes. Previous studies have used an arbitrary cut-off of 100 mapped reads per million using a total library sub-sample. Some of the viruses presented here either do not have poly-A tails or are unlikely to have poly-A tails. As most of the data examined were produced from transcriptome studies using a poly-A enrichment step, we felt this cut-off might be too stringent. Instead, we used the complete libraries and visually inspected mapping coverage to exclude false-positive mapping. To additionally implicate viruses as being present, we queried our de novo assemblies through BLASTn analysis. We manually assessed hits (E-value < 10−5) as bearing >85% nucleotide identity over a minimum of 500 bp for each virus. It is essential to appreciate that the RNA samples processing, sequencing library preparation, instruments used, and read length of all data used between studies are vastly different. As such, it is challenging to normalise and draw quantitative comparisons between studies. We believe, however, that the analysis presented here reasonably incriminates samples as positively infected. However, we concede that given the limitations of the library preparations, viruses not containing poly-A tails or in huge abundance may be incorrectly characterised as “negative”. Given this limitation, we recommend screening mosquito samples using RT-PCR or RT-qPCR methods. A summary of common ISV infections and their associated BioProject is available in Supplementary Tables S3 and S4.
Examining the general trends between the ISV composition of Ae. aegypti and Ae. albopictus suggests that ISV compositions are entirely separate between mosquitoes, with very few examples of potential contamination between laboratory colonies (Supplementary Tables S3 and S4). Singleton virus infections in colonies are rare, and in most colonies, there exists a common suite of viruses. While the “core virome” describes common virus populations between single species in metatranscriptomics studies [121,146], multiple factors beyond host species identity influence the ecology and diversity of the metazoan virome [148].
In laboratory strains of Ae aegypti that had recently been field-adapted (within 10 generations), almost all BioProjects (57/60) had sufficient ISV RNA identifiable in these libraries, with some harbouring up to 11 individual ISVs. While recently adapted colonies of mosquitoes had the most diverse viromes, there were some Ae. aegypti colonies that we could not detect any virus RNA in their corresponding data and, based on our analysis, are potentially “virus sterile”. The ROCK or Rockefeller strain, which is of Caribbean origin established in 1930 [149], as well as the Rexville-D strain, established from an isofemale line collected in Rexville, Puerto Rico in the early 1990s [150], both appear to have very low, or no mapped reads originating from RNA viruses. Given that the Rexville-D strain was established from a single isofemale, it seems likely that this reduced the virome diversity.
By comparison, the Liverpool reference stain, used to generate the Ae. aegypti genome and maintained at the Liverpool School of Tropical Medicine since 1936 (reviewed in [149]), was identified to have almost three-quarters of BioProjects infected with Aedes aegypti toti-like virus (ATLV-1) (18/24 BioPojects) and up to a third infected with Liverpool tombus-like virus (LTLV; 7/24 BioProjects). While generally Liverpool and Rexville-D RNA-Seq libraries did not appear to be consistently infected with any other viruses, we identified a handful of examples in which specific libraries of these strains were infected with more than ATLV-1 or Liverpool virus (Supplementary Table S3). For instance, one study examining the tRNA fragments of various colonies, including the Rexville, Liverpool, and Trinidad colony, observed a convincing RNAi response to HTV and Renna virus in the Liverpool colony [120]. This suggests active infection in these samples. While lane contamination during sequencing may be responsible for this observation, it is also possible that co-housing different strains of mosquitoes in a facility might cause contamination.
The reduction in virome diversity was similarly observed in the Foshan or Guangdong strain of Ae. albopictus, which has been in culture since 1981 and used to assemble the nuclear genome [69]. While Foshan colonies had a less diverse virome than recently lab-adapted strains, there was evidence of potential MDV infection in 4/8 BioProjects, and a low abundance of AOMV-2 (2/8 BioProject accessions). By comparison, laboratory colonies recently lab-adapted from wild-caught samples had the most diverse viromes [59,86].
3.11. Individual Ae. aegypti and Ae. albopictus Mosquitoes from the Same Colony Harbour Heterogeneous Virus Populations
Transcriptomic studies are increasingly using libraries prepared from single individuals rather than pooled mosquitoes as biological replicates. We set out to analyze individual datasets to explore the heterogeneity of viruses within individual mosquitoes. For this, we used two studies using individual Ae. aegypti samples recently adapted to the laboratory (within ten generations). The first study sequenced the midguts from individual mosquitoes from Thep Na Korn, Thailand midguts [119]. The second study examined 42 whole adult samples from a colony established from wild-caught mosquitoes in Cairns, Australia and sequenced after three generations [108].
Both publications experimentally infected Ae. aegypti with clinical isolates of dengue virus (DENV-1) [119], and (DENV-3) [108]. As the genome of DENV does not have a poly-A tail, we felt that DENV read numbers may be a reasonable detection limit for virus populations. The DENV-3 read numbers in Ae. aegypti Cairns libraries [108] were low but detectable in the DENV positive samples with 6–335 mapped reads ( = 90.27, n = 18). However, in the midgut libraries produced by Raquin et al. 2017 [119], at 24hpi in the individual midgut samples, DENV-1 reads were between 0–82 mapped reads ( = 18.35 n = 17), and at 96hpi there were 298–105442 reads mapped to the DENV-1 genome ( = 25,328.58, n = 17).
Within the recently colonised Thep Na Korn, Thailand strain [119], we identified reads of up to eight viruses within the midgut libraries produced for the study. Only two midgut libraries did not have any detectable reads of any virus and 15/47 midgut libraries had reads mapping to at least one virus (Figure 10A). Most individual libraries harboured multiple virus populations, with 17/47 carrying two, 12/47 carrying three viruses, and one infected with four viruses. The most common virus present in this colony was the Phasi Charoen-like virus (PCLV) with 41/47, followed by Humaita-Tubiacanga virus (HTV) (14/47) samples infected, and both Verdadero virus and Aedes anphevirus (AeAV) in 6/47 midgut samples.
Virus heterogeneity was similar in whole mosquitoes originating from Cairns, Australia. However, the most abundant identified virus was Aedes aegypti toti-like virus 1 (ATLV-1), which was almost fixed in the mosquitoes with ~90% of individuals infected with ATLV-1 (38/42 samples) (Figure 10B). The second most abundant was PCLV (26/42), followed by HTV in 19/42 libraries and APLV-1. In this dataset, every single mosquito was infected with at least one virus with five samples with one virus, 5/42 samples with two ISVs, 13/42 samples with three ISVs, 15/42 samples with four ISVs, and 4/42 samples infected with five ISVs. These data indicate that at least in recent colonies of Aedes mosquitoes, most individual mosquitoes are infected on average with two to three ISVs and can be super-infected with up to five viruses in individual mosquitoes.
3.12. Composition of Viruses in Commonly Used Aedes Cell Lines Reveals Super-Infection of ISVs in Wolbachia Transfected Cell Lines
Cell lines are invaluable tools in arbovirus research, and numerous reports have demonstrated that most harbour persistent infections of ISVs [151]. Early records of the Ae. aegypti Aag2 cell line indicated a persistent infection with an insect-specific flavivirus CFAV [152,153,154]. Aag2 also persistently harbours the negative-sense PCLV (Phenuiviridae) [35,155], and one report of this cell line also suggests infection with AeAV [156]. PCLV is also known to infect the Ae. albopictus RML-12 cell line [157]. It is also well-established that the Culex Y virus (Birnaviridae) is a common laboratory contaminant of Ae. albopictus U4.4 cell line [129,151] and occasionally cross-contaminating Ae. albopictus C7-10 cells and Ae. aegypti Aag2 cells [158].
In addition to RNA virus infection, mosquito cell lines are known to be persistently infected with densoviruses [159]. Aedes albopictus densovirus (AalDNV-2) was first completely sequenced and characterised [160,161] from a persistently infected C6/36 cell line [162]. Previously, we have demonstrated that most published Aag2 cells appear to have a defective Aedes albopictus densovirus (AalDNV-2) genome and a truncated VP gene [163]. Defective AalDNV-2 genomes are exclusively targeted by the vsiRNA response, whereas complete AalDNV-2 infection in Aag2 cells produces both vsiRNA and vpiRNA species [163].
Previously reported ISVs were identified in this study (Supplementary Table S5). However, additional infections of Aedes cell lines were uncovered. For example, we identified Wenzhou Sobemo-like virus 4 (WSBLV-4) in Ae. albopictus C7-10 cells from total-RNA sequencing obtained as part of the Arthropod Cell Line RNA-Seq initiative, Broad Institute (broadinstitute.org), and three independent small RNA sequencing datasets [129,164]. It appears that not all C7-10 cells were infected with WSBLV-4; however, [58] suggesting heterogeneity between sources of C7-10 cell lines. Additionally, we identified a convincing RNAi response (mostly piRNAs) against WSBLV-4 from C6/36 cells (SRA: SRR11252294) from this dataset [58]. The RNAi profile of WSBLV-4 infection in C7-10 and C6/36 cells is in line with the Dicer-2 deficient nature of the cell line (Figure 9).
There was evidence that cell lines transinfected with the endosymbiotic bacterium Wolbachia are super-infected with a range of viruses, including the positive-sense RNA virus WSBLV-4 and three negative-sense RNA viruses San Gabriel virus, PCLV, and AeAV (Figure 11).
The super-infection of WSBLV-4, San Gabriel virus, PCLV, and AeAV was observed in two independent Ae. albopictus C6/36 wMelPop-CLA infected cells [64,65] and Ae. albopictus RML-12 wMelPop-CLA infected cells [62]. San Gabriel virus infecting Wolbachia cell line derivatives (RML-12.wMelPop, Aag2.wMelPop, C6/36.wMelPop) were more closely related to the cell line-identified San Gabriel virus strains (98–99% over 99% of the genome) than to the wild-derived San Gabriel virus from 95.86% over 99% of the genome of the Ae. albopictus colony from the USA. This observation indicates that these infections are likely to have originated from the parental RML-12 cell lines for reasons previously discussed [18].
We have previously demonstrated that another mononegavirus, AeAV, persistently infects the RML-12 and Ae. aegypti Aag2.wMelPop-CLA cell lines with up to 1.2 million out of ~22 million small RNA reads (~5.45%) mapping to AeAV [18]. Reanalysis of these cell line data indicated a smaller fraction of vsiRNA reads from the library ~0.54% map to the San Gabriel virus genome in Aag2.wMelPop-CLA cells (119,424/22,078,545 reads). The RNAi profile indicates both the genome and replicative intermediate are targeted by the RNAi response, indicating active infection in these cells. By comparison, we could map no reads from the paired Aag2 cell line. We identified both WSBLV-4 and Saint Gabriel virus in the Ae. albopictus Aa23 cell line and a second Ae. aegypti cell line transcriptome that was stably transinfected with the Wolbachia wAlbB strain from Aa23 cells (Aag2.wAlbB), suggesting contamination of San Gabriel virus from the Aa23 cells during the production of the Aag2.wAlbB cell line [63]. The presence of three negative-sense RNA viruses is consistent with reports that Wolbachia does not confer a refractory virus phenotype to negative-sense RNA viruses [18,165,166]. However, examinations between Wolbachia and the positive sense WSBLV-4 deserve further experimentation.
4. Conclusions
We have identified up to ten novel viruses associated with Ae. aegypti and Ae. albopictus colonies that infect colonies, wild-caught populations, and cell lines worldwide. Phylogenetic analysis of the viruses reported here suggests all these viruses are closely related to other virus families that have recently been described to infect arthropods except for one Ae. albopictus colony potentially infected with the vertebrate infecting Jingmen tick virus. While most of these viruses are likely to have an insect-restricted host range, experimental validation should be undertaken due to the extensive distribution of some of these viruses in Aedes populations. We show that recently established lab colonies harbour up to five viruses in individual mosquitoes and very few colonies of Aedes mosquitoes are “sterile” using our analysis. In addition to identifying novel viruses, we have explored the geographical distribution of previously known ISVs of Ae. aegypti and Ae. albopictus. As we establish the contribution of viruses associated with mosquitoes to the vector competence of arboviruses, understanding the prevalence and distribution of these viruses provides a resource for further assessment of the ecology, evolution, and interaction of ISVs with their mosquito hosts and arboviruses they transmit.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/microorganisms9081653/s1, Supplementary File 1: Annotations and mapping coverage of novel and updated Aedes viruses. Supplementary File 2: Annotations of the RNAi response in Aedes viruses. Table S1: All Ae. aegypti samples analysed in this study and associated publications. Table S2: All Ae. albopictus samples analysed in this study and associated publications. Table S3: Incidence of ISVs in Ae. aegypti colonies and wild-caught samples. Table S4: Incidence of ISVs in Ae. albopictus colonies and wild-caught samples. Table S5: Incidence of ISVs in Ae. aegypti and Ae. albopictus cell lines.
Author Contributions
Conceptualisation, R.P., and S.A.; methodology, R.P., and S.A.; formal analysis, R.P. and M.E.J.; investigation, R.P. and M.E.J.; resources, S.A.; data curation, R.P., and S.A.; writing—original draft preparation, R.P.; writing—review and editing, R.P., M.E.J., S.A.; visualisation, R.P.; supervision, S.A.; project administration, S.A.; funding acquisition, S.A. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Australian Research Council, grant number DP190102048 to Sassan Asgari. Rhys Parry was supported by a University of Queensland Research Higher Degree scholarship.
Institutional Review Board Statement
Ethical review and approval were not applicable for this study as it did not involve humans or animals.
Informed Consent Statement
Not applicable.
Data Availability Statement
All virus assemblies that have not been directly sequenced by the authors from previous projects will be submitted to the Third-Party Annotation (TPA) database and made available for download after manual curation annotation from NCBI staff. All primary sequencing data are available from accessions listed in the Supplementary Tables. The NCBI BioProject ID for this study is PRJNA720639.
Acknowledgments
We want to express our thanks to the numerous scientists and project staff contributing to producing the original data analysed within this publication. Additionally, we appreciate researchers that replied to emails about the provenance of the RNA-Seq data, all of which are too numerous to name individually. We appreciate the technical assistance of the team that operates and maintains the Australian Galaxy service (https://usegalaxy.org.au/) (accessed on 20 April 2020) and members of the Asgari lab for fruitful discussions. Picture of Ae. aegypti and Ae. albopictus from Figure 1 by Ana L. Ramírez.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Kraemer, M.U.; Sinka, M.E.; Duda, K.A.; Mylne, A.Q.; Shearer, F.M.; Barker, C.M.; Moore, C.G.; Carvalho, R.G.; Coelho, G.E.; Van Bortel, W.; et al. The global distribution of the arbovirus vectors Aedes aegypti and Ae. albopictus. eLife 2015, 4, e08347. [Google Scholar] [CrossRef] [PubMed]
- Hawley, W.A. The biology of Aedes albopictus. J. Am. Mosq. Control. Assoc. Suppl. 1988, 1, 1–39. [Google Scholar]
- Li, Y.; Kamara, F.; Zhou, G.; Puthiyakunnon, S.; Li, C.; Liu, Y.; Zhou, Y.; Yao, L.; Yan, G.; Chen, X.G. Urbanization increases Aedes albopictus larval habitats and accelerates mosquito development and survivorship. PLoS Negl. Trop. Dis. 2014, 8, e3301. [Google Scholar] [CrossRef] [Green Version]
- Souza-Neto, J.A.; Powell, J.R.; Bonizzoni, M. Aedes aegypti vector competence studies: A review. Infect. Genet. Evol. 2019, 67, 191–209. [Google Scholar] [CrossRef]
- Gratz, N.G. Critical review of the vector status of Aedes albopictus. Med. Vet. Entomol. 2004, 18, 215–227. [Google Scholar] [CrossRef]
- Vega-Rua, A.; Zouache, K.; Girod, R.; Failloux, A.B.; Lourenco-de-Oliveira, R. High level of vector competence of Aedes aegypti and Aedes albopictus from ten American countries as a crucial factor in the spread of Chikungunya virus. J. Virol. 2014, 88, 6294–6306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouzid, M.; Colon-Gonzalez, F.J.; Lung, T.; Lake, I.R.; Hunter, P.R. Climate change and the emergence of vector-borne diseases in Europe: Case study of dengue fever. BMC Public Health 2014, 14, 781. [Google Scholar] [CrossRef] [Green Version]
- Bhatt, S.; Gething, P.W.; Brady, O.J.; Messina, J.P.; Farlow, A.W.; Moyes, C.L.; Drake, J.M.; Brownstein, J.S.; Hoen, A.G.; Sankoh, O.; et al. The global distribution and burden of dengue. Nature 2013, 496, 504–507. [Google Scholar] [CrossRef]
- Lambrechts, L.; Scott, T.W.; Gubler, D.J. Consequences of the expanding global distribution of Aedes albopictus for dengue virus transmission. PLoS Negl. Trop. Dis. 2010, 4, e646. [Google Scholar] [CrossRef] [PubMed]
- Gardner, C.L.; Ryman, K.D. Yellow fever: A reemerging threat. Clin. Lab. Med. 2010, 30, 237–260. [Google Scholar] [CrossRef] [Green Version]
- Musso, D.; Gubler, D.J. Zika Virus. Clin. Microbiol Rev. 2016, 29, 487–524. [Google Scholar] [CrossRef] [Green Version]
- Metsky, H.C.; Matranga, C.B.; Wohl, S.; Schaffner, S.F.; Freije, C.A.; Winnicki, S.M.; West, K.; Qu, J.; Baniecki, M.L.; Gladden-Young, A.; et al. Zika virus evolution and spread in the Americas. Nature 2017, 546, 411–415. [Google Scholar] [CrossRef] [Green Version]
- Duchemin, J.B.; Mee, P.T.; Lynch, S.E.; Vedururu, R.; Trinidad, L.; Paradkar, P. Zika vector transmission risk in temperate Australia: A vector competence study. Virol. J. 2017, 14, 108. [Google Scholar] [CrossRef] [PubMed]
- Monath, T.P. Yellow fever: An update. Lancet Infect. Dis. 2001, 1, 11–20. [Google Scholar] [CrossRef]
- Kramer, L.D.; Ciota, A.T. Dissecting vectorial capacity for mosquito-borne viruses. Curr. Opin. Virol. 2015, 15, 112–118. [Google Scholar] [CrossRef] [Green Version]
- Sigle, L.T.; McGraw, E.A. Expanding the canon: Non-classical mosquito genes at the interface of arboviral infection. Insect. Biochem. Mol. Biol. 2019, 109, 72–80. [Google Scholar] [CrossRef]
- Hegde, S.; Rasgon, J.L.; Hughes, G.L. The microbiome modulates arbovirus transmission in mosquitoes. Curr. Opin. Virol. 2015, 15, 97–102. [Google Scholar] [CrossRef] [Green Version]
- Parry, R.; Asgari, S. Aedes Anphevirus: An Insect-Specific Virus Distributed Worldwide in Aedes aegypti Mosquitoes That Has Complex Interplays with Wolbachia and Dengue Virus Infection in Cells. J. Virol. 2018, 92, e00224-18. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Asad, S.; Khromykh, A.A.; Asgari, S. Cell fusing agent virus and dengue virus mutually interact in Aedes aegypti cell lines. Sci. Rep. 2017, 7, 6935. [Google Scholar] [CrossRef] [Green Version]
- Hobson-Peters, J.; Yam, A.W.; Lu, J.W.; Setoh, Y.X.; May, F.J.; Kurucz, N.; Walsh, S.; Prow, N.A.; Davis, S.S.; Weir, R.; et al. A new insect-specific flavivirus from northern Australia suppresses replication of West Nile virus and Murray Valley encephalitis virus in co-infected mosquito cells. PLoS ONE 2013, 8, e56534. [Google Scholar] [CrossRef]
- Hall-Mendelin, S.; McLean, B.J.; Bielefeldt-Ohmann, H.; Hobson-Peters, J.; Hall, R.A.; van den Hurk, A.F. The insect-specific Palm Creek virus modulates West Nile virus infection in and transmission by Australian mosquitoes. Parasit Vectors 2016, 9, 414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nasar, F.; Erasmus, J.H.; Haddow, A.D.; Tesh, R.B.; Weaver, S.C. Eilat virus induces both homologous and heterologous interference. Virology 2015, 484, 51–58. [Google Scholar] [CrossRef] [Green Version]
- Baidaliuk, A.; Miot, E.F.; Lequime, S.; Moltini-Conclois, I.; Delaigue, F.; Dabo, S.; Dickson, L.B.; Aubry, F.; Merkling, S.H.; Cao-Lormeau, V.M.; et al. Cell-Fusing Agent Virus Reduces Arbovirus Dissemination in Aedes aegypti Mosquitoes In Vivo. J. Virol. 2019, 93, e00705-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, R.; Kato, F.; Kobayashi, D.; Murota, K.; Takasaki, T.; Tajima, S.; Lim, C.K.; Saijo, M.; Isawa, H.; Sawabe, K. Persistent viruses in mosquito cultured cell line suppress multiplication of flaviviruses. Heliyon 2018, 4, e00736. [Google Scholar] [CrossRef] [Green Version]
- Mosimann, A.L.; Bordignon, J.; Mazzarotto, G.C.; Motta, M.C.; Hoffmann, F.; Santos, C.N. Genetic and biological characterization of a densovirus isolate that affects dengue virus infection. Mem. Inst. Oswaldo Cruz. 2011, 106, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Schultz, M.J.; Frydman, H.M.; Connor, J.H. Dual Insect specific virus infection limits Arbovirus replication in Aedes mosquito cells. Virology 2018, 518, 406–413. [Google Scholar] [CrossRef]
- Shi, M.; Neville, P.; Nicholson, J.; Eden, J.S.; Imrie, A.; Holmes, E.C. High-Resolution Metatranscriptomics Reveals the Ecological Dynamics of Mosquito-Associated RNA Viruses in Western Australia. J. Virol. 2017, 91, e00680-17. [Google Scholar] [CrossRef] [Green Version]
- Webster, C.L.; Waldron, F.M.; Robertson, S.; Crowson, D.; Ferrari, G.; Quintana, J.F.; Brouqui, J.M.; Bayne, E.H.; Longdon, B.; Buck, A.H.; et al. The Discovery, Distribution, and Evolution of Viruses Associated with Drosophila melanogaster. PLoS Biol. 2015, 13, e1002210. [Google Scholar] [CrossRef] [Green Version]
- Fauver, J.R.; Akter, S.; Morales, A.I.O.; Black, W.C.T.; Rodriguez, A.D.; Stenglein, M.D.; Ebel, G.D.; Weger-Lucarelli, J. A reverse-transcription/RNase H based protocol for depletion of mosquito ribosomal RNA facilitates viral intrahost evolution analysis, transcriptomics and pathogen discovery. Virology 2018, 528, 181–197. [Google Scholar] [CrossRef]
- Chandler, J.A.; Liu, R.M.; Bennett, S.N. RNA shotgun metagenomic sequencing of northern California (USA) mosquitoes uncovers viruses, bacteria, and fungi. Front. Microbiol. 2015, 6, 185. [Google Scholar] [CrossRef] [Green Version]
- Sabin, L.R.; Zheng, Q.; Thekkat, P.; Yang, J.; Hannon, G.J.; Gregory, B.D.; Tudor, M.; Cherry, S. Dicer-2 processes diverse viral RNA species. PLoS ONE 2013, 8, e55458. [Google Scholar] [CrossRef]
- Jayachandran, B.; Hussain, M.; Asgari, S. RNA interference as a cellular defense mechanism against the DNA virus baculovirus. J. Virol. 2012, 86, 13729–13734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnard, A.C.; Nijhof, A.M.; Fick, W.; Stutzer, C.; Maritz-Olivier, C. RNAi in Arthropods: Insight into the Machinery and Applications for Understanding the Pathogen-Vector Interface. Genes 2012, 3, 702–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Q.; Luo, Y.; Lu, R.; Lau, N.; Lai, E.C.; Li, W.X.; Ding, S.W. Virus discovery by deep sequencing and assembly of virus-derived small silencing RNAs. Proc. Natl. Acad. Sci. USA 2010, 107, 1606–1611. [Google Scholar] [CrossRef] [Green Version]
- Aguiar, E.R.; Olmo, R.P.; Paro, S.; Ferreira, F.V.; de Faria, I.J.; Todjro, Y.M.; Lobo, F.P.; Kroon, E.G.; Meignin, C.; Gatherer, D.; et al. Sequence-independent characterization of viruses based on the pattern of viral small RNAs produced by the host. Nucleic Acids Res. 2015, 43, 6191–6206. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [Green Version]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [Green Version]
- Katzourakis, A.; Gifford, R.J. Endogenous viral elements in animal genomes. PLoS Genet. 2010, 6, e1001191. [Google Scholar] [CrossRef]
- Ter Horst, A.M.; Nigg, J.C.; Dekker, F.M.; Falk, B.W. Endogenous Viral Elements Are Widespread in Arthropod Genomes and Commonly Give Rise to PIWI-Interacting RNAs. J. Virol. 2019, 93, e02124-18. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Koonin, E.V. Iterated profile searches with PSI-BLAST–A tool for discovery in protein databases. Trends Biochem. Sci. 1998, 23, 444–447. [Google Scholar] [CrossRef]
- Kosugi, S.; Hasebe, M.; Tomita, M.; Yanagawa, H. Systematic identification of cell cycle-dependent yeast nucleocytoplasmic shuttling proteins by prediction of composite motifs. Proc. Natl. Acad. Sci. USA 2009, 106, 10171–10176. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [Green Version]
- Thorvaldsdottir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [Green Version]
- Parry, R.; Naccache, F.; Ndiaye, E.H.; Fall, G.; Castelli, I.; Luhken, R.; Medlock, J.; Cull, B.; Hesson, J.C.; Montarsi, F.; et al. Identification and RNAi Profile of a Novel Iflavirus Infecting Senegalese Aedes vexans arabiensis Mosquitoes. Viruses 2020, 12, 440. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Capella-Gutierrez, S.; Silla-Martinez, J.M.; Gabaldon, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Le, S.Q.; Gascuel, O. An improved general amino acid replacement matrix. Mol. Biol. Evol. 2008, 25, 1307–1320. [Google Scholar] [CrossRef] [Green Version]
- Minh, B.Q.; Nguyen, M.A.; von Haeseler, A. Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 2013, 30, 1188–1195. [Google Scholar] [CrossRef] [PubMed]
- McBride, C.S.; Baier, F.; Omondi, A.B.; Spitzer, S.A.; Lutomiah, J.; Sang, R.; Ignell, R.; Vosshall, L.B. Evolution of mosquito preference for humans linked to an odorant receptor. Nature 2014, 515, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Sadeghi, M.; Altan, E.; Deng, X.; Barker, C.M.; Fang, Y.; Coffey, L.L.; Delwart, E. Virome of >12 thousand Culex mosquitoes from throughout California. Virology 2018, 523, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Green, T.J.; Cox, R.; Tsao, J.; Rowse, M.; Qiu, S.; Luo, M. Common mechanism for RNA encapsidation by negative-strand RNA viruses. J. Virol. 2014, 88, 3766–3775. [Google Scholar] [CrossRef] [Green Version]
- Gamez, S.; Antoshechkin, I.; Mendez-Sanchez, S.C.; Akbari, O.S. The Developmental Transcriptome of Ae. albopictus, a Major Worldwide Human Disease Vector. G3 2020, 10, 1051–1062. [Google Scholar] [CrossRef] [Green Version]
- Ma, Q.; Srivastav, S.P.; Gamez, S.; Dayama, G.; Feitosa-Suntheimer, F.; Patterson, E.I.; Johnson, R.M.; Matson, E.M.; Gold, A.S.; Brackney, D.E.; et al. A mosquito small RNA genomics resource reveals dynamic evolution and host responses to viruses and transposons. Genome Res. 2021, 31, 512–528. [Google Scholar] [CrossRef]
- Frangeul, L.; Blanc, H.; Saleh, M.C.; Suzuki, Y. Differential Small RNA Responses against Co-Infecting Insect-Specific Viruses in Aedes albopictus Mosquitoes. Viruses 2020, 12, 468. [Google Scholar] [CrossRef] [Green Version]
- Xiao, P.; Han, J.; Zhang, Y.; Li, C.; Guo, X.; Wen, S.; Tian, M.; Li, Y.; Wang, M.; Liu, H.; et al. Metagenomic Analysis of Flaviviridae in Mosquito Viromes Isolated From Yunnan Province in China Reveals Genes From Dengue and Zika Viruses. Front. Cell Infect. Microbiol. 2018, 8, 359. [Google Scholar] [CrossRef]
- Kubacki, J.; Flacio, E.; Qi, W.; Guidi, V.; Tonolla, M.; Fraefel, C. Viral Metagenomic Analysis of Aedes albopictus Mosquitos from Southern Switzerland. Viruses 2020, 12, 929. [Google Scholar] [CrossRef]
- Darby, A.C.; Gill, A.C.; Armstrong, S.D.; Hartley, C.S.; Xia, D.; Wastling, J.M.; Makepeace, B.L. Integrated transcriptomic and proteomic analysis of the global response of Wolbachia to doxycycline-induced stress. ISME J. 2014, 8, 925–937. [Google Scholar] [CrossRef] [Green Version]
- Bishop, C.; Asgari, S. Altered gene expression profile of Wolbachia pipientis wAlbB strain following transinfection from its native host Aedes albopictus to Aedes aegypti cells. Mol. Microbiol. 2020, 115, 1229–1243. [Google Scholar] [CrossRef]
- Woolfit, M.; Algama, M.; Keith, J.M.; McGraw, E.A.; Popovici, J. Discovery of putative small non-coding RNAs from the obligate intracellular bacterium Wolbachia pipientis. PLoS ONE 2015, 10, e0118595. [Google Scholar]
- Teramoto, T.; Huang, X.; Armbruster, P.A.; Padmanabhan, R. Infection of Aedes albopictus Mosquito C6/36 Cells with the wMelpop Strain of Wolbachia Modulates Dengue Virus-Induced Host Cellular Transcripts and Induces Critical Sequence Alterations in the Dengue Viral Genome. J. Virol. 2019, 93, e00581-19. [Google Scholar] [CrossRef] [Green Version]
- Mayoral, J.G.; Etebari, K.; Hussain, M.; Khromykh, A.A.; Asgari, S. Wolbachia infection modifies the profile, shuttling and structure of microRNAs in a mosquito cell line. PLoS ONE 2014, 9, e96107. [Google Scholar]
- Mayoral, J.G.; Hussain, M.; Joubert, D.A.; Iturbe-Ormaetxe, I.; O’Neill, S.L.; Asgari, S. Wolbachia small noncoding RNAs and their role in cross-kingdom communications. Proc. Natl. Acad. Sci. USA 2014, 111, 18721–18726. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.B.; Su, X.H.; Bonizzoni, M.; Zhong, D.B.; Li, Y.J.; Zhou, G.F.; Nguyen, H.; Tong, S.; Yan, G.Y.; Chen, X.G. Comparative transcriptome analysis and RNA interference reveal CYP6A8 and SNPs related to pyrethroid resistance in Aedes albopictus. PLoS Negl. Trop. Dis. 2018, 12, e0006828. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.G.; Jiang, X.T.; Gu, J.B.; Xu, M.; Wu, Y.; Deng, Y.H.; Zhang, C.; Bonizzoni, M.; Dermauw, W.; Vontas, J.; et al. Genome sequence of the Asian Tiger mosquito, Aedes albopictus, reveals insights into its biology, genetics, and evolution. Proc. Natl. Acad. Sci. USA 2015, 112, e5907–e5915. [Google Scholar] [CrossRef] [Green Version]
- Hall, A.B.; Basu, S.; Jiang, X.; Qi, Y.; Timoshevskiy, V.A.; Biedler, J.K.; Sharakhova, M.V.; Elahi, R.; Anderson, M.A.; Chen, X.G.; et al. Sex Determination. A male-determining factor in the mosquito Aedes aegypti. Science 2015, 348, 1268–1270. [Google Scholar] [CrossRef] [Green Version]
- Lombardo, F.; Salvemini, M.; Fiorillo, C.; Nolan, T.; Zwiebel, L.J.; Ribeiro, J.M.; Arca, B. Deciphering the olfactory repertoire of the tiger mosquito Aedes albopictus. BMC Genom. 2017, 18, 770. [Google Scholar] [CrossRef]
- Vedururu, R.K.; Neave, M.J.; Tachedjian, M.; Klein, M.J.; Gorry, P.R.; Duchemin, J.B.; Paradkar, P.N. RNASeq Analysis of Aedes albopictus Mosquito Midguts after Chikungunya Virus Infection. Viruses 2019, 11, 513. [Google Scholar] [CrossRef] [Green Version]
- Shi, M.; Lin, X.D.; Tian, J.H.; Chen, L.J.; Chen, X.; Li, C.X.; Qin, X.C.; Li, J.; Cao, J.P.; Eden, J.S.; et al. Redefining the invertebrate RNA virosphere. Nature 2016, 540, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Lewis, S.H.; Quarles, K.A.; Yang, Y.; Tanguy, M.; Frézal, L.; Smith, S.A.; Sharma, P.P.; Cordaux, R.; Gilbert, C.; Giraud, I. Pan-arthropod analysis reveals somatic piRNAs as an ancestral defence against transposable elements. Nat. Ecol. Evol. 2018, 2, 174–181. [Google Scholar] [CrossRef] [Green Version]
- Bona, A.C.; Chitolina, R.F.; Fermino, M.L.; de Castro Poncio, L.; Weiss, A.; Lima, J.B.; Paldi, N.; Bernardes, E.S.; Henen, J.; Maori, E. Larval application of sodium channel homologous dsRNA restores pyrethroid insecticide susceptibility in a resistant adult mosquito population. Parasit Vectors 2016, 9, 397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camargo, C.; Ahmed-Braimah, Y.H.; Amaro, I.A.; Harrington, L.C.; Wolfner, M.F.; Avila, F.W. Mating and blood-feeding induce transcriptome changes in the spermathecae of the yellow fever mosquito Aedes aegypti. Sci. Rep. 2020, 10, 14899. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Alto, B.W.; Shin, D.; Yu, F. The Effect of Permethrin Resistance on Aedes aegypti Transcriptome Following Ingestion of Zika Virus Infected Blood. Viruses 2018, 10, 470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Liu, Y.; Nie, K.; Du, S.; Qiu, J.; Pang, X.; Wang, P.; Cheng, G. Flavivirus NS1 protein in infected host sera enhances viral acquisition by mosquitoes. Nat. Microbiol. 2016, 1, 16087. [Google Scholar] [CrossRef]
- Ribeiro, J.M.; Martin-Martin, I.; Arca, B.; Calvo, E. A Deep Insight into the Sialome of Male and Female Aedes aegypti Mosquitoes. PLoS ONE 2016, 11, e0151400. [Google Scholar] [CrossRef] [Green Version]
- Kang, D.S.; Barron, M.S.; Lovin, D.D.; Cunningham, J.M.; Eng, M.W.; Chadee, D.D.; Li, J.; Severson, D.W. A transcriptomic survey of the impact of environmental stress on response to dengue virus in the mosquito, Aedes aegypti. PLoS Negl. Trop. Dis. 2018, 12, e0006568. [Google Scholar] [CrossRef]
- Zakrzewski, M.; Rasic, G.; Darbro, J.; Krause, L.; Poo, Y.S.; Filipovic, I.; Parry, R.; Asgari, S.; Devine, G.; Suhrbier, A. Mapping the virome in wild-caught Aedes aegypti from Cairns and Bangkok. Sci. Rep. 2018, 8, 4690. [Google Scholar] [CrossRef] [Green Version]
- Boyles, S.M.; Mavian, C.N.; Finol, E.; Ukhanova, M.; Stephenson, C.J.; Hamerlinck, G.; Kang, S.; Baumgartner, C.; Geesey, M.; Stinton, I.; et al. Under-the-Radar Dengue Virus Infections in Natural Populations of Aedes aegypti Mosquitoes. mSphere 2020, 5, e00316-20. [Google Scholar] [CrossRef]
- Huang, X.; Poelchau, M.F.; Armbruster, P.A. Global Transcriptional Dynamics of Diapause Induction in Non-Blood-Fed and Blood-Fed Aedes albopictus. PLoS Negl. Trop. Dis. 2015, 9, e0003724. [Google Scholar] [CrossRef] [PubMed]
- Tsujimoto, H.; Hanley, K.A.; Sundararajan, A.; Devitt, N.P.; Schilkey, F.D.; Hansen, I.A. Dengue virus serotype 2 infection alters midgut and carcass gene expression in the Asian tiger mosquito, Aedes albopictus. PLoS ONE 2018, 13, e0171345. [Google Scholar] [CrossRef] [Green Version]
- Esquivel, C.J.; Cassone, B.J.; Piermarini, P.M. Transcriptomic evidence for a dramatic functional transition of the malpighian tubules after a blood meal in the Asian tiger mosquito Aedes albopictus. PLoS Negl. Trop. Dis. 2014, 8, e2929. [Google Scholar] [CrossRef] [Green Version]
- Poirier, E.Z.; Goic, B.; Tome-Poderti, L.; Frangeul, L.; Boussier, J.; Gausson, V.; Blanc, H.; Vallet, T.; Loyd, H.; Levi, L.I.; et al. Dicer-2-Dependent Generation of Viral DNA from Defective Genomes of RNA Viruses Modulates Antiviral Immunity in Insects. Cell Host Microbe 2018, 23, 353–365.e8. [Google Scholar] [CrossRef] [Green Version]
- Batz, Z.A.; Goff, A.C.; Armbruster, P.A. MicroRNAs are differentially abundant during Aedes albopictus diapause maintenance but not diapause induction. Insect. Mol. Biol. 2017, 26, 721–733. [Google Scholar] [CrossRef]
- Poelchau, M.F.; Reynolds, J.A.; Denlinger, D.L.; Elsik, C.G.; Armbruster, P.A. A de novo transcriptome of the Asian tiger mosquito, Aedes albopictus, to identify candidate transcripts for diapause preparation. BMC Genom. 2011, 12, 619. [Google Scholar] [CrossRef] [Green Version]
- Poelchau, M.F.; Reynolds, J.A.; Denlinger, D.L.; Elsik, C.G.; Armbruster, P.A. Transcriptome sequencing as a platform to elucidate molecular components of the diapause response in the Asian tiger mosquito Aedes albopictus. Physiol. Entomol. 2013, 38, 173–181. [Google Scholar] [CrossRef] [Green Version]
- Poelchau, M.F.; Reynolds, J.A.; Elsik, C.G.; Denlinger, D.L.; Armbruster, P.A. Deep sequencing reveals complex mechanisms of diapause preparation in the invasive mosquito, Aedes albopictus. Proc. Biol. Sci. 2013, 280, 20130143. [Google Scholar] [CrossRef] [Green Version]
- Bellone, R.; Lequime, S.; Jupille, H.; Goertz, G.P.; Aubry, F.; Mousson, L.; Piorkowski, G.; Yen, P.S.; Gabiane, G.; Vazeille, M.; et al. Experimental adaptation of dengue virus 1 to Aedes albopictus mosquitoes by in vivo selection. Sci. Rep. 2020, 10, 18404. [Google Scholar] [CrossRef]
- Adelman, Z.N.; Anderson, M.A.; Wiley, M.R.; Murreddu, M.G.; Samuel, G.H.; Morazzani, E.M.; Myles, K.M. Cooler temperatures destabilize RNA interference and increase susceptibility of disease vector mosquitoes to viral infection. PLoS Negl. Trop. Dis. 2013, 7, e2239. [Google Scholar] [CrossRef] [Green Version]
- Palatini, U.; Masri, R.A.; Cosme, L.V.; Koren, S.; Thibaud-Nissen, F.; Biedler, J.K.; Krsticevic, F.; Johnston, J.S.; Halbach, R.; Crawford, J.E.; et al. Improved reference genome of the arboviral vector Aedes albopictus. Genome Biol. 2020, 21, 215. [Google Scholar] [CrossRef]
- Marconcini, M.; Pischedda, E.; Houé, V.; Palatini, U.; Lozada-Chávez, N.; Sogliani, D.; Failloux, A.B.; Bonizzoni, M. Profile of Small RNAs, vDNA Forms and Viral Integrations in Late Chikungunya Virus Infection of Aedes albopictus Mosquitoes. Viruses 2021, 13, 553. [Google Scholar] [CrossRef]
- Xu, Y.; Dong, Y.; Xu, Y.; Lai, Z.; Jin, B.; Hao, Y.; Gao, Y.; Sun, Y.; Chen, X.G.; Gu, J. Differentiation of Long Non-Coding RNA and mRNA Expression Profiles in Male and Female Aedes albopictus. Front. Genet. 2019, 10, 975. [Google Scholar] [CrossRef]
- Adelman, Z.N.; Anderson, M.A.; Liu, M.; Zhang, L.; Myles, K.M. Sindbis virus induces the production of a novel class of endogenous siRNAs in Aedes aegypti mosquitoes. Insect. Mol. Biol. 2012, 21, 357–368. [Google Scholar] [CrossRef] [Green Version]
- Hu, W.; Criscione, F.; Liang, S.; Tu, Z. MicroRNAs of two medically important mosquito species: Aedes aegypti and Anopheles stephensi. Insect. Mol. Biol. 2015, 24, 240–252. [Google Scholar] [CrossRef] [Green Version]
- Biedler, J.K.; Hu, W.; Tae, H.; Tu, Z. Identification of early zygotic genes in the yellow fever mosquito Aedes aegypti and discovery of a motif involved in early zygotic genome activation. PLoS ONE 2012, 7, e33933. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Biedler, J.K.; Qi, Y.; Hall, A.B.; Tu, Z. Complete Dosage Compensation in Anopheles stephensi and the Evolution of Sex-Biased Genes in Mosquitoes. Genome Biol. Evol. 2015, 7, 1914–1924. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Fu, X.; Zhu, J. Juvenile hormone-regulated alternative splicing of the taiman gene primes the ecdysteroid response in adult mosquitoes. Proc. Natl. Acad. Sci. USA 2018, 115, e7738–e7747. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.J.; Aliota, M.T.; Mayhew, G.F.; Erickson, S.M.; Christensen, B.M. Dual RNA-seq of parasite and host reveals gene expression dynamics during filarial worm-mosquito interactions. PLoS Negl. Trop. Dis. 2014, 8, e2905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cross, S.T.; Maertens, B.L.; Dunham, T.J.; Rodgers, C.P.; Brehm, A.L.; Miller, M.R.; Williams, A.M.; Foy, B.D.; Stenglein, M.D. Partitiviruses Infecting Drosophila melanogaster and Aedes aegypti Exhibit Efficient Biparental Vertical Transmission. J. Virol. 2020, 94, e01070-20. [Google Scholar] [CrossRef]
- Bonizzoni, M.; Dunn, W.A.; Campbell, C.L.; Olson, K.E.; Marinotti, O.; James, A.A. Complex modulation of the Aedes aegypti transcriptome in response to dengue virus infection. PLoS ONE 2012, 7, e50512. [Google Scholar] [CrossRef]
- Buchman, A.; Gamez, S.; Li, M.; Antoshechkin, I.; Li, H.H.; Wang, H.W.; Chen, C.H.; Klein, M.J.; Duchemin, J.B.; Paradkar, P.N.; et al. Engineered resistance to Zika virus in transgenic Aedes aegypti expressing a polycistronic cluster of synthetic small RNAs. Proc. Natl. Acad. Sci. USA 2019, 116, 3656–3661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Etebari, K.; Osei-Amo, S.; Blomberg, S.P.; Asgari, S. Dengue virus infection alters post-transcriptional modification of microRNAs in the mosquito vector Aedes aegypti. Sci. Rep. 2015, 5, 15968. [Google Scholar] [CrossRef] [Green Version]
- Slonchak, A.; Hugo, L.E.; Freney, M.E.; Hall-Mendelin, S.; Amarilla, A.A.; Torres, F.J.; Setoh, Y.X.; Peng, N.Y.G.; Sng, J.D.J.; Hall, R.A.; et al. Zika virus noncoding RNA suppresses apoptosis and is required for virus transmission by mosquitoes. Nat. Commun. 2020, 11, 2205. [Google Scholar] [CrossRef]
- Wimalasiri-Yapa, B.; Barrero, R.A.; Stassen, L.; Hafner, L.M.; McGraw, E.A.; Pyke, A.T.; Jansen, C.C.; Suhrbier, A.; Yakob, L.; Hu, W.; et al. Temperature modulates immune gene expression in mosquitoes during arbovirus infection. Open Biol. 2021, 11, 200246. [Google Scholar] [CrossRef]
- Koh, C.; Allen, S.L.; Herbert, R.I.; McGraw, E.A.; Chenoweth, S.F. The Transcriptional Response of Aedes aegypti with Variable Extrinsic Incubation Periods for Dengue Virus. Genome Biol. Evol. 2018, 10, 3141–3151. [Google Scholar] [CrossRef] [Green Version]
- Etebari, K.; Hegde, S.; Saldana, M.A.; Widen, S.G.; Wood, T.G.; Asgari, S.; Hughes, G.L. Global Transcriptome Analysis of Aedes aegypti Mosquitoes in Response to Zika Virus Infection. mSphere 2017, 2, e00456-17. [Google Scholar] [CrossRef] [Green Version]
- Saldana, M.A.; Etebari, K.; Hart, C.E.; Widen, S.G.; Wood, T.G.; Thangamani, S.; Asgari, S.; Hughes, G.L. Zika virus alters the microRNA expression profile and elicits an RNAi response in Aedes aegypti mosquitoes. PLoS Negl. Trop. Dis. 2017, 11, e0005760. [Google Scholar] [CrossRef]
- Ferreira, P.G.; Tesla, B.; Horacio, E.C.A.; Nahum, L.A.; Brindley, M.A.; de Oliveira Mendes, T.A.; Murdock, C.C. Temperature Dramatically Shapes Mosquito Gene Expression With Consequences for Mosquito-Zika Virus Interactions. Front. Microbiol. 2020, 11, 901. [Google Scholar] [CrossRef]
- Lee, M.; Etebari, K.; Hall-Mendelin, S.; van den Hurk, A.F.; Hobson-Peters, J.; Vatipally, S.; Schnettler, E.; Hall, R.; Asgari, S. Understanding the role of microRNAs in the interaction of Aedes aegypti mosquitoes with an insect-specific flavivirus. J. Gen. Virol. 2017, 98, 1892–1903. [Google Scholar] [CrossRef]
- Cattel, J.; Haberkorn, C.; Laporte, F.; Gaude, T.; Cumer, T.; Renaud, J.; Sutherland, I.W.; Hertz, J.C.; Bonneville, J.; Arnaud, V.; et al. A genomic amplification affecting a carboxylesterase gene cluster confers organophosphate resistance in the mosquito Aedes aegypti: From genomic characterization to high-throughput field detection. Evol. Appl. 2021, 14, 1009–1022. [Google Scholar] [CrossRef]
- Shrinet, J.; Srivastava, P.; Sunil, S. Transcriptome analysis of Aedes aegypti in response to mono-infections and co-infections of dengue virus-2 and chikungunya virus. Biochem. Biophys. Res. Commun. 2017, 492, 617–623. [Google Scholar] [CrossRef]
- Chowdhury, A.; Modahl, C.M.; Tan, S.T.; Wong Wei Xiang, B.; Misse, D.; Vial, T.; Kini, R.M.; Pompon, J.F. JNK pathway restricts DENV2, ZIKV and CHIKV infection by activating complement and apoptosis in mosquito salivary glands. PLoS Pathog. 2020, 16, e1008754. [Google Scholar] [CrossRef] [PubMed]
- Choy, M.M.; Sessions, O.M.; Gubler, D.J.; Ooi, E.E. Production of Infectious Dengue Virus in Aedes aegypti Is Dependent on the Ubiquitin Proteasome Pathway. PLoS Negl. Trop. Dis. 2015, 9, e0004227. [Google Scholar] [CrossRef] [PubMed]
- Halbach, R.; Miesen, P.; Joosten, J.; Taskopru, E.; Rondeel, I.; Pennings, B.; Vogels, C.B.F.; Merkling, S.H.; Koenraadt, C.J.; Lambrechts, L.; et al. A satellite repeat-derived piRNA controls embryonic development of Aedes. Nature 2020, 580, 274–277. [Google Scholar] [CrossRef] [PubMed]
- Degner, E.C.; Ahmed-Braimah, Y.H.; Borziak, K.; Wolfner, M.F.; Harrington, L.C.; Dorus, S. Proteins, Transcripts, and Genetic Architecture of Seminal Fluid and Sperm in the Mosquito Aedes aegypti. Mol. Cell Proteom. 2019, 18, S6–S22. [Google Scholar] [CrossRef]
- Raquin, V.; Merkling, S.H.; Gausson, V.; Moltini-Conclois, I.; Frangeul, L.; Varet, H.; Dillies, M.A.; Saleh, M.C.; Lambrechts, L. Individual co-variation between viral RNA load and gene expression reveals novel host factors during early dengue virus infection of the Aedes aegypti midgut. PLoS Negl. Trop. Dis. 2017, 11, e0006152. [Google Scholar] [CrossRef]
- Eng, M.W.; Clemons, A.; Hill, C.; Engel, R.; Severson, D.W.; Behura, S.K. Multifaceted functional implications of an endogenously expressed tRNA fragment in the vector mosquito Aedes aegypti. PLoS Negl. Trop. Dis. 2018, 12, e0006186. [Google Scholar] [CrossRef] [Green Version]
- Shi, C.; Beller, L.; Deboutte, W.; Yinda, K.C.; Delang, L.; Vega-Rua, A.; Failloux, A.B.; Matthijnssens, J. Stable distinct core eukaryotic viromes in different mosquito species from Guadeloupe, using single mosquito viral metagenomics. Microbiome 2019, 7, 121. [Google Scholar] [CrossRef]
- Amarasinghe, G.K.; Ayllon, M.A.; Bao, Y.; Basler, C.F.; Bavari, S.; Blasdell, K.R.; Briese, T.; Brown, P.A.; Bukreyev, A.; Balkema-Buschmann, A.; et al. Taxonomy of the order Mononegavirales: Update 2019. Arch. Virol. 2019, 164, 1967–1980. [Google Scholar] [CrossRef] [Green Version]
- Vedururu, R.K.; Neave, M.J.; Sundaramoorthy, V.; Green, D.; Harper, J.A.; Gorry, P.R.; Duchemin, J.B.; Paradkar, P.N. Whole Transcriptome Analysis of Aedes albopictus Mosquito Head and Thorax Post-Chikungunya Virus Infection. Pathogens 2019, 8, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasilakis, N.; Forrester, N.L.; Palacios, G.; Nasar, F.; Savji, N.; Rossi, S.L.; Guzman, H.; Wood, T.G.; Popov, V.; Gorchakov, R.; et al. Negevirus: A proposed new taxon of insect-specific viruses with wide geographic distribution. J. Virol. 2013, 87, 2475–2488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kallies, R.; Kopp, A.; Zirkel, F.; Estrada, A.; Gillespie, T.R.; Drosten, C.; Junglen, S. Genetic characterization of goutanap virus, a novel virus related to negeviruses, cileviruses and higreviruses. Viruses 2014, 6, 4346–4357. [Google Scholar] [CrossRef]
- Fauver, J.R.; Grubaugh, N.D.; Krajacich, B.J.; Weger-Lucarelli, J.; Lakin, S.M.; Fakoli, L.S., 3rd; Bolay, F.K.; Diclaro, J.W., 2nd; Dabire, K.R.; Foy, B.D.; et al. West African Anopheles gambiae mosquitoes harbor a taxonomically diverse virome including new insect-specific flaviviruses, mononegaviruses, and totiviruses. Virology 2016, 498, 288–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webster, C.L.; Longdon, B.; Lewis, S.H.; Obbard, D.J. Twenty-Five New Viruses Associated with the Drosophilidae (Diptera). Evol Bioinform Online 2016, 12, 13–25. [Google Scholar] [CrossRef] [Green Version]
- Adams, M.J.; Adkins, S.; Bragard, C.; Gilmer, D.; Li, D.; MacFarlane, S.A.; Wong, S.M.; Melcher, U.; Ratti, C.; Ryu, K.H.; et al. ICTV Virus Taxonomy Profile: Virgaviridae. J. Gen. Virol. 2017, 98, 1999–2000. [Google Scholar] [CrossRef]
- Morazzani, E.M.; Wiley, M.R.; Murreddu, M.G.; Adelman, Z.N.; Myles, K.M. Production of virus-derived ping-pong-dependent piRNA-like small RNAs in the mosquito soma. PLoS Pathog. 2012, 8, e1002470. [Google Scholar] [CrossRef]
- Nouri, S.; Salem, N.; Falk, B.W. Complete Genome Sequence of Diaphorina citri-associated C virus, a Novel Putative RNA Virus of the Asian Citrus Psyllid, Diaphorina citri. Genome Announc. 2016, 4, e00639-16. [Google Scholar] [CrossRef] [Green Version]
- Kuchibhatla, D.B.; Sherman, W.A.; Chung, B.Y.; Cook, S.; Schneider, G.; Eisenhaber, B.; Karlin, D.G. Powerful sequence similarity search methods and in-depth manual analyses can identify remote homologs in many apparently “orphan” viral proteins. J. Virol. 2014, 88, 10–20. [Google Scholar] [CrossRef] [Green Version]
- Cook, S.; Chung, B.Y.; Bass, D.; Moureau, G.; Tang, S.; McAlister, E.; Culverwell, C.L.; Glucksman, E.; Wang, H.; Brown, T.D.; et al. Novel virus discovery and genome reconstruction from field RNA samples reveals highly divergent viruses in dipteran hosts. PLoS ONE 2013, 8, e80720. [Google Scholar] [CrossRef] [Green Version]
- Johnson, R.M.; Rasgon, J.L. Densonucleosis viruses (‘densoviruses’) for mosquito and pathogen control. Curr. Opin. Insect. Sci. 2018, 28, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Cotmore, S.F.; Agbandje-McKenna, M.; Canuti, M.; Chiorini, J.A.; Eis-Hubinger, A.M.; Hughes, J.; Mietzsch, M.; Modha, S.; Ogliastro, M.; Penzes, J.J.; et al. ICTV Virus Taxonomy Profile: Parvoviridae. J. Gen. Virol. 2019, 100, 367–368. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Hoiczyk, E.; Rasgon, J.L. Viral paratransgenesis in the malaria vector Anopheles gambiae. PLoS Pathog. 2008, 4, e1000135. [Google Scholar] [CrossRef] [PubMed]
- Avila-Bonilla, R.G.; Yocupicio-Monroy, M.; Marchat, L.A.; De Nova-Ocampo, M.A.; del Angel, R.M.; Salas-Benito, J.S. Analysis of the miRNA profile in C6/36 cells persistently infected with dengue virus type 2. Virus Res. 2017, 232, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Dong, Y.; Sun, Y.; Lai, Z.; Zhao, Y.; Liu, P.; Gao, Y.; Chen, X.; Gu, J. A Novel Densovirus Isolated From the Asian Tiger Mosquito Displays Varied Pathogenicity Depending on Its Host Species. Front. Microbiol. 2019, 10, 1549. [Google Scholar] [CrossRef]
- Qin, X.C.; Shi, M.; Tian, J.H.; Lin, X.D.; Gao, D.Y.; He, J.R.; Wang, J.B.; Li, C.X.; Kang, Y.J.; Yu, B.; et al. A tick-borne segmented RNA virus contains genome segments derived from unsegmented viral ancestors. Proc. Natl. Acad. Sci. USA 2014, 111, 6744–6749. [Google Scholar] [CrossRef] [Green Version]
- Jia, N.; Liu, H.B.; Ni, X.B.; Bell-Sakyi, L.; Zheng, Y.C.; Song, J.L.; Li, J.; Jiang, B.G.; Wang, Q.; Sun, Y.; et al. Emergence of human infection with Jingmen tick virus in China: A retrospective study. EBioMedicine 2019, 43, 317–324. [Google Scholar] [CrossRef] [Green Version]
- Emmerich, P.; Jakupi, X.; von Possel, R.; Berisha, L.; Halili, B.; Gunther, S.; Cadar, D.; Ahmeti, S.; Schmidt-Chanasit, J. Viral metagenomics, genetic and evolutionary characteristics of Crimean-Congo hemorrhagic fever orthonairovirus in humans, Kosovo. Infect. Genet. Evol. 2018, 65, 6–11. [Google Scholar] [CrossRef]
- Dittmer, J.; Alafndi, A.; Gabrieli, P. Fat body-specific vitellogenin expression regulates host-seeking behaviour in the mosquito Aedes albopictus. PLoS Biol. 2019, 17, e3000238. [Google Scholar] [CrossRef] [Green Version]
- Temmam, S.; Bigot, T.; Chretien, D.; Gondard, M.; Perot, P.; Pommelet, V.; Dufour, E.; Petres, S.; Devillers, E.; Hoem, T.; et al. Insights into the Host Range, Genetic Diversity, and Geographical Distribution of Jingmenviruses. mSphere 2019, 4, e00645-19. [Google Scholar] [CrossRef] [Green Version]
- Sameroff, S.; Tokarz, R.; Charles, R.A.; Jain, K.; Oleynik, A.; Che, X.; Georges, K.; Carrington, C.V.; Lipkin, W.I.; Oura, C. Viral Diversity of Tick Species Parasitizing Cattle and Dogs in Trinidad and Tobago. Sci. Rep. 2019, 9, 10421. [Google Scholar] [CrossRef] [Green Version]
- Ruckert, C.; Prasad, A.N.; Garcia-Luna, S.M.; Robison, A.; Grubaugh, N.D.; Weger-Lucarelli, J.; Ebel, G.D. Small RNA responses of Culex mosquitoes and cell lines during acute and persistent virus infection. Insect. Biochem. Mol. Biol. 2019, 109, 13–23. [Google Scholar] [CrossRef]
- Batson, J.; Dudas, G.; Haas-Stapleton, E.; Kistler, A.L.; Li, L.M.; Logan, P.; Ratnasiri, K.; Retallack, H. Single mosquito metatranscriptomics identifies vectors, emerging pathogens and reservoirs in one assay. eLife 2021, 10, e68353. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Zhao, L.; Atoni, E.; Zeng, W.; Hu, X.; Matthijnssens, J.; Yuan, Z.; Xia, H. Stability of the Virome in Lab- and Field-Collected Aedes albopictus Mosquitoes across Different Developmental Stages and Possible Core Viruses in the Publicly Available Virome Data of Aedes Mosquitoes. mSystems 2020, 5, e00640-20. [Google Scholar] [CrossRef]
- Brackney, D.E.; Scott, J.C.; Sagawa, F.; Woodward, J.E.; Miller, N.A.; Schilkey, F.D.; Mudge, J.; Wilusz, J.; Olson, K.E.; Blair, C.D.; et al. C6/36 Aedes albopictus cells have a dysfunctional antiviral RNA interference response. PLoS Negl. Trop. Dis. 2010, 4, e856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obbard, D.J. Expansion of the metazoan virosphere: Progress, pitfalls, and prospects. Curr. Opin. Virol. 2018, 31, 17–23. [Google Scholar] [CrossRef] [Green Version]
- Kuno, G. Early history of laboratory breeding of Aedes aegypti (Diptera: Culicidae) focusing on the origins and use of selected strains. J. Med. Entomol. 2010, 47, 957–971. [Google Scholar] [CrossRef] [Green Version]
- Miller, B.R.; Mitchell, C.J. Genetic selection of a flavivirus-refractory strain of the yellow fever mosquito Aedes aegypti. Am. J. Trop. Med. Hyg. 1991, 45, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Weger-Lucarelli, J.; Ruckert, C.; Grubaugh, N.D.; Misencik, M.J.; Armstrong, P.M.; Stenglein, M.D.; Ebel, G.D.; Brackney, D.E. Adventitious viruses persistently infect three commonly used mosquito cell lines. Virology 2018, 521, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Stollar, V.; Thomas, V.L. An agent in the Aedes aegypti cell line (Peleg) which causes fusion of Aedes albopictus cells. Virology 1975, 64, 367–377. [Google Scholar] [CrossRef]
- Igarashi, A.; Harrap, K.A.; Casals, J.; Stollar, V. Morphological, Biochemical, and Serological Studies on a Viral Agent (Cfa) Which Replicates in and Causes Fusion of Aedes-Albopictus (Singh) Cells. Virology 1976, 74, 174–187. [Google Scholar] [CrossRef]
- Cammisa-Parks, H.; Cisar, L.A.; Kane, A.; Stollar, V. The complete nucleotide sequence of cell fusing agent (CFA): Homology between the nonstructural proteins encoded by CFA and the nonstructural proteins encoded by arthropod-borne flaviviruses. Virology 1992, 189, 511–524. [Google Scholar] [CrossRef]
- Maringer, K.; Yousuf, A.; Heesom, K.J.; Fan, J.; Lee, D.; Fernandez-Sesma, A.; Bessant, C.; Matthews, D.A.; Davidson, A.D. Proteomics informed by transcriptomics for characterising active transposable elements and genome annotation in Aedes aegypti. BMC Genom. 2017, 18, 101. [Google Scholar] [CrossRef] [Green Version]
- Di Giallonardo, F.; Audsley, M.D.; Shi, M.; Young, P.R.; McGraw, E.A.; Holmes, E.C. Complete genome of Aedes aegypti anphevirus in the Aag2 mosquito cell line. J. Gen. Virol. 2018, 99, 832–836. [Google Scholar] [CrossRef]
- McLean, B.J.; Dainty, K.R.; Flores, H.A.; O’Neill, S.L. Differential suppression of persistent insect specific viruses in trans-infected wMel and wMelPop-CLA Aedes-derived mosquito lines. Virology 2019, 527, 141–145. [Google Scholar] [CrossRef]
- Franzke, K.; Leggewie, M.; Sreenu, V.B.; Jansen, S.; Heitmann, A.; Welch, S.R.; Brennan, B.; Elliott, R.M.; Tannich, E.; Becker, S.C.; et al. Detection, infection dynamics and small RNA response against Culex Y virus in mosquito-derived cells. J. Gen. Virol. 2018, 99, 1739–1745. [Google Scholar] [CrossRef]
- O’Neill, S.; Kittayapong, P.; Braig, H.; Andreadis, T.; Gonzalez, J.; Tesh, R. Insect densoviruses may be widespread in mosquito cell lines. J. Gen. Virol. 1995, 76, 2067–2074. [Google Scholar] [CrossRef]
- Jousset, F.X.; Barreau, C.; Boublik, Y.; Cornet, M. A parvo-like virus persistently infecting a C6/36 clone of Aedes albopictus mosquito cell line and pathogenic for Aedes aegypti larvae. Virus Res. 1993, 29, 99–114. [Google Scholar] [CrossRef]
- Boublik, Y.; Jousset, F.X.; Bergoin, M. Complete nucleotide sequence and genomic organization of the Aedes albopictus parvovirus (AaPV) pathogenic for Aedes aegypti larvae. Virology 1994, 200, 752–763. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, A. Isolation of a singhs Aedes albopictus cell Clone sensitive to Dengue and chikungunya viruses. J. Gen. Virol. 1978, 40, 531–544. [Google Scholar] [CrossRef] [PubMed]
- Parry, R.; Bishop, C.; De Hayr, L.; Asgari, S. Density-dependent enhanced replication of a densovirus in Wolbachia-infected Aedes cells is associated with production of piRNAs and higher virus-derived siRNAs. Virology 2019, 528, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Skalsky, R.L.; Vanlandingham, D.L.; Scholle, F.; Higgs, S.; Cullen, B.R. Identification of microRNAs expressed in two mosquito vectors, Aedes albopictus and Culex quinquefasciatus. BMC Genom. 2010, 11, 119. [Google Scholar] [CrossRef] [Green Version]
- Schultz, M.J.; Isern, S.; Michael, S.F.; Corley, R.B.; Connor, J.H.; Frydman, H.M. Variable Inhibition of Zika Virus Replication by Different Wolbachia Strains in Mosquito Cell Cultures. J. Virol. 2017, 91, e00339-17. [Google Scholar] [CrossRef] [Green Version]
- Schnettler, E.; Sreenu, V.B.; Mottram, T.; McFarlane, M. Wolbachia restricts insect-specific flavivirus infection in Aedes aegypti cells. J. Gen. Virol. 2016, 97, 3024–3029. [Google Scholar] [CrossRef]
Figure 1.
Virus discovery and geographic sample incidence in Aedes aegypti and Aedes albopictus. (A) The bioinformatics pipeline for virus discovery and virus incidence of Ae. aegypti and Ae. albopictus viruses. (B) Sample locations of Ae. aegypti (top) and Ae. albopictus (bottom). Samples are coloured based on the geographic region of the original catch location. The size of the circle indicates the number of independent catch locations and are coloured as follows: African samples (red), Australian samples (blue), Asian samples (green), American samples (gold), and European samples (pink).
Figure 1.
Virus discovery and geographic sample incidence in Aedes aegypti and Aedes albopictus. (A) The bioinformatics pipeline for virus discovery and virus incidence of Ae. aegypti and Ae. albopictus viruses. (B) Sample locations of Ae. aegypti (top) and Ae. albopictus (bottom). Samples are coloured based on the geographic region of the original catch location. The size of the circle indicates the number of independent catch locations and are coloured as follows: African samples (red), Australian samples (blue), Asian samples (green), American samples (gold), and European samples (pink).

Figure 2.
Novel negative-sense RNA viruses of Ae. aegypti and Ae. albopictus mosquitoes. (A) Genome structure and organisation of novel mononegaviruses. NP, nucleocapsid; GP, Glycoprotein; MET, methyltransferase. (B) Phylogenetic placement of novel mononegaviruses constructed based on an alignment of the L protein sequence. Branches have been coloured as per ICTV’s Mononegavirales taxonomy [122] with the family Nyamiviridae (orange), Xinmoviridae (red) and Rhabdoviridae, genus Vesiculovirus (green). Consensus maximum-likelihood trees were generated using IQTree with the LG + G4 amino acid substitution model with 50,000 Ultrafast bootstraps and bootstrap support indicated on the node. Each TSA accession is shown in blue. For clarity, the tree is midpoint rooted. Branch length indicates the number of amino acid substitutions per site. Novel viruses are marked with arrowheads. Nodes with a mosquito indicate clades containing viruses identified from mosquito species.
Figure 2.
Novel negative-sense RNA viruses of Ae. aegypti and Ae. albopictus mosquitoes. (A) Genome structure and organisation of novel mononegaviruses. NP, nucleocapsid; GP, Glycoprotein; MET, methyltransferase. (B) Phylogenetic placement of novel mononegaviruses constructed based on an alignment of the L protein sequence. Branches have been coloured as per ICTV’s Mononegavirales taxonomy [122] with the family Nyamiviridae (orange), Xinmoviridae (red) and Rhabdoviridae, genus Vesiculovirus (green). Consensus maximum-likelihood trees were generated using IQTree with the LG + G4 amino acid substitution model with 50,000 Ultrafast bootstraps and bootstrap support indicated on the node. Each TSA accession is shown in blue. For clarity, the tree is midpoint rooted. Branch length indicates the number of amino acid substitutions per site. Novel viruses are marked with arrowheads. Nodes with a mosquito indicate clades containing viruses identified from mosquito species.

Figure 3.
Novel negative-sense RNA viruses of Ae. aegypti and Ae. albopictus mosquitoes. (A) Genome structure of the Aedes orthomyxo-like virus 2 (AOMLV-2), PB1/PB2, Polymerase basic 1/2 protein; PA, polymerase acidic protein; GP, putative glycoprotein; NP, nucleoprotein. (B) Phylogenetic placement within Orthomyxoviridae constructed using aligned PB1 protein of orthomyxoviruses. Clades are coloured as per the current Orthomyxoviridae taxonomy (ICTV 2018b Release) with influenza viruses (orange) and genus Quaranjavirus (red). Consensus maximum-likelihood trees were made using IQTree with the LG + G4 amino acid substitution model with 50,000 Ultrafast bootstraps and bootstrap support indicated on the node. Each TSA accession is shown in blue. For clarity, the tree is midpoint rooted. Branch length indicates the number of amino acid substitutions per site. Novel viruses are marked with arrowheads. Nodes with a mosquito indicate clades containing viruses identified from mosquito species.
Figure 3.
Novel negative-sense RNA viruses of Ae. aegypti and Ae. albopictus mosquitoes. (A) Genome structure of the Aedes orthomyxo-like virus 2 (AOMLV-2), PB1/PB2, Polymerase basic 1/2 protein; PA, polymerase acidic protein; GP, putative glycoprotein; NP, nucleoprotein. (B) Phylogenetic placement within Orthomyxoviridae constructed using aligned PB1 protein of orthomyxoviruses. Clades are coloured as per the current Orthomyxoviridae taxonomy (ICTV 2018b Release) with influenza viruses (orange) and genus Quaranjavirus (red). Consensus maximum-likelihood trees were made using IQTree with the LG + G4 amino acid substitution model with 50,000 Ultrafast bootstraps and bootstrap support indicated on the node. Each TSA accession is shown in blue. For clarity, the tree is midpoint rooted. Branch length indicates the number of amino acid substitutions per site. Novel viruses are marked with arrowheads. Nodes with a mosquito indicate clades containing viruses identified from mosquito species.

Figure 4.
Novel positive-sense RNA viruses of Ae. albopictus and Ae. aegypti. (A) Genome organisation of Aedes negeviruses and bi-negeviruses. Vmet, Viral methyltransferase; FtsJ, Ftsj-like methyltransferase; HEL, helicase, and (B) phylogenetic placement of novel negeviruses based on concatenated MET, HEL, and RdRp_2 domains. Clades are coloured, indicating the mosquito nelorpivirus (red) and sandewavirus (purple) negevirus clades, as well as the Virgaviridae (green) and Kitaviridae (pink) groupings, a divergent mosquito negevirus clade is indicated in orange. Branch length indicates the number of amino acid substitutions per site. Consensus maximum-likelihood trees are produced using IQTree as per Figure 2.
Figure 4.
Novel positive-sense RNA viruses of Ae. albopictus and Ae. aegypti. (A) Genome organisation of Aedes negeviruses and bi-negeviruses. Vmet, Viral methyltransferase; FtsJ, Ftsj-like methyltransferase; HEL, helicase, and (B) phylogenetic placement of novel negeviruses based on concatenated MET, HEL, and RdRp_2 domains. Clades are coloured, indicating the mosquito nelorpivirus (red) and sandewavirus (purple) negevirus clades, as well as the Virgaviridae (green) and Kitaviridae (pink) groupings, a divergent mosquito negevirus clade is indicated in orange. Branch length indicates the number of amino acid substitutions per site. Consensus maximum-likelihood trees are produced using IQTree as per Figure 2.

Figure 5.
Novel positive-sense RNA viruses of Ae. albopictus and Ae. aegypti. Genome organisation of novel tombus-like viruses identified in this study (A) and phylogenetic placement (B) of viruses from aligned RdRp3 domains. Consensus maximum-likelihood trees were made using IQTree with the LG + G4 amino acid substitution model with 50,000 Ultrafast bootstraps with bootstrap support labelled on the node. Each TSA accession is shown in blue, members of the Carmovirus genus are indicated in green. Novel viruses are indicated with arrowheads. Branch length indicates the number of amino acid substitutions per site. For clarity, the tree is midpoint rooted.
Figure 5.
Novel positive-sense RNA viruses of Ae. albopictus and Ae. aegypti. Genome organisation of novel tombus-like viruses identified in this study (A) and phylogenetic placement (B) of viruses from aligned RdRp3 domains. Consensus maximum-likelihood trees were made using IQTree with the LG + G4 amino acid substitution model with 50,000 Ultrafast bootstraps with bootstrap support labelled on the node. Each TSA accession is shown in blue, members of the Carmovirus genus are indicated in green. Novel viruses are indicated with arrowheads. Branch length indicates the number of amino acid substitutions per site. For clarity, the tree is midpoint rooted.

Figure 6.
Novel dsRNA viruses of Ae. aegypti. Genome organisation (A), and phylogenetic placement (B) of Aedes partiti-like virus 1 (APLV-1) within the Partitiviridae family based on RdRp_1 domain alignment. ICTV accepted genus members from Alphapartitivirus (purple), Betapartitivirus (green), Deltapartitivirus (red), and Gammapartitivirus (orange). (C) Genome organisation of Aedes orbi-like virus (AOLV), and (D) phylogenetic position of AOLV based on aligned VP1 RdRp domain within the Reoviridae family. Members of the Orbivirus genus are indicated in red. Consensus maximum-likelihood trees were made with IQTree under the LG + G4 amino acid substitution model with 50,000 Ultrafast bootstraps. For clarity, the tree is midpoint rooted. Each TSA accession is shown in blue. Novel viruses are indicated with arrowheads. Nodes with an adjacent mosquito indicate virus clades identified from mosquito species. Branch length indicates the number of amino acid substitutions per site.
Figure 6.
Novel dsRNA viruses of Ae. aegypti. Genome organisation (A), and phylogenetic placement (B) of Aedes partiti-like virus 1 (APLV-1) within the Partitiviridae family based on RdRp_1 domain alignment. ICTV accepted genus members from Alphapartitivirus (purple), Betapartitivirus (green), Deltapartitivirus (red), and Gammapartitivirus (orange). (C) Genome organisation of Aedes orbi-like virus (AOLV), and (D) phylogenetic position of AOLV based on aligned VP1 RdRp domain within the Reoviridae family. Members of the Orbivirus genus are indicated in red. Consensus maximum-likelihood trees were made with IQTree under the LG + G4 amino acid substitution model with 50,000 Ultrafast bootstraps. For clarity, the tree is midpoint rooted. Each TSA accession is shown in blue. Novel viruses are indicated with arrowheads. Nodes with an adjacent mosquito indicate virus clades identified from mosquito species. Branch length indicates the number of amino acid substitutions per site.

Figure 7.
Phylogenetic relationship of Dipteran brevidensovirus strains identified in this study. Consensus tree constructed using NS1/NS2 and VP whole coding regions of virus genomes aligned using MAFFT. Ambiguous alignment regions were removed using GBLOCKS. Consensus maximum-likelihood trees were constructed with IQTree under the TIM2 + F+G4 nucleotide substitution model with 50,000 Ultrafast bootstraps. Bootstrapping support was conducted using 10,000 ultrafast bootstraps. Culex pipiens pallens densovirus and Culex densovirus nodes were collapsed for simplicity. The tree is midpoint rooted, and Genbank numbers corresponding to published virus strains are indicated on the label; novel strains of mosquito brevidensovirus are marked with arrowheads, along with the BioProject accession number.
Figure 7.
Phylogenetic relationship of Dipteran brevidensovirus strains identified in this study. Consensus tree constructed using NS1/NS2 and VP whole coding regions of virus genomes aligned using MAFFT. Ambiguous alignment regions were removed using GBLOCKS. Consensus maximum-likelihood trees were constructed with IQTree under the TIM2 + F+G4 nucleotide substitution model with 50,000 Ultrafast bootstraps. Bootstrapping support was conducted using 10,000 ultrafast bootstraps. Culex pipiens pallens densovirus and Culex densovirus nodes were collapsed for simplicity. The tree is midpoint rooted, and Genbank numbers corresponding to published virus strains are indicated on the label; novel strains of mosquito brevidensovirus are marked with arrowheads, along with the BioProject accession number.

Figure 8.
Evidence and phylogenetic placement of a Jingmen tick virus (JMTV) strain in the Rimini, Italy Ae. albopictus mosquito colony. (A) Coverage of the four segments of the Rimini JMTV strain originating from the head and fat body samples. Consensus trees constructed using (B) Segment 1 and (C) Segment 3 whole coding regions of virus genomes aligned using MAFFT and ambiguous alignment regions removed using GBLOCKS. Consensus maximum-likelihood trees were constructed with IQTree under the TIM2 + F+G4 nucleotide substitution model with 50,000 Ultrafast bootstraps. The trees are rooted on the Alongshan virus (ALSV) outgroup.
Figure 8.
Evidence and phylogenetic placement of a Jingmen tick virus (JMTV) strain in the Rimini, Italy Ae. albopictus mosquito colony. (A) Coverage of the four segments of the Rimini JMTV strain originating from the head and fat body samples. Consensus trees constructed using (B) Segment 1 and (C) Segment 3 whole coding regions of virus genomes aligned using MAFFT and ambiguous alignment regions removed using GBLOCKS. Consensus maximum-likelihood trees were constructed with IQTree under the TIM2 + F+G4 nucleotide substitution model with 50,000 Ultrafast bootstraps. The trees are rooted on the Alongshan virus (ALSV) outgroup.

Figure 9.
RNAi responses to Aedes viruses. Bar graphs represent extracted small-RNA reads (18–32 nt) mapped to the representative genomes of Aedes viruses presented in this study. Reads originating from the genomic sense are indicated in blue, and anti-genome sense reads are red. Virus segments that have both a characteristic piRNA signature (adenine at position 10, A10, for sense RNA; Uridine at position 1, U1 for antisense RNA), as well as overlapping 10 nt signature, are indicated with an asterisk (*). Viruses grouped by presumed Baltimore classification scheme with (A) IV: (+) ssRNA viruses, (B) V: (−) ssRNA viruses, (C) II: ssDNA virus, and (D) III: dsRNA viruses.
Figure 9.
RNAi responses to Aedes viruses. Bar graphs represent extracted small-RNA reads (18–32 nt) mapped to the representative genomes of Aedes viruses presented in this study. Reads originating from the genomic sense are indicated in blue, and anti-genome sense reads are red. Virus segments that have both a characteristic piRNA signature (adenine at position 10, A10, for sense RNA; Uridine at position 1, U1 for antisense RNA), as well as overlapping 10 nt signature, are indicated with an asterisk (*). Viruses grouped by presumed Baltimore classification scheme with (A) IV: (+) ssRNA viruses, (B) V: (−) ssRNA viruses, (C) II: ssDNA virus, and (D) III: dsRNA viruses.

Figure 10.
Abundance of viruses from individual Aedes aegypti mosquitoes. mRNA-enriched libraries prepared from midguts from Raquin et al. 2017 [119], (A), and whole mosquitoes from Koh et al. 2018 [108]. (B) Abundance is represented as log10(RPKM) mapped reads per million per kilobase. Hierarchical clustering was performed using Pearson complete distance measurement method. Heat map produced using pheatmap R package. RNNV: Renna virus, HTV: Humaita-Tubiacanga virus, APLV-1: Aedes partiti-like virus 1, VeV: Verdadero virus, DENV-1: dengue virus 1 (KDH0030A), DENV-3: dengue virus 3 (JN406515.1), AeAV: Aedes anphevirus, PCLV: Phasi Charoen-like virus, CFAV: Cell fusing agent virus, ATLV-1: Aedes toti-like virus 1, ATV: Aedes toti virus.
Figure 10.
Abundance of viruses from individual Aedes aegypti mosquitoes. mRNA-enriched libraries prepared from midguts from Raquin et al. 2017 [119], (A), and whole mosquitoes from Koh et al. 2018 [108]. (B) Abundance is represented as log10(RPKM) mapped reads per million per kilobase. Hierarchical clustering was performed using Pearson complete distance measurement method. Heat map produced using pheatmap R package. RNNV: Renna virus, HTV: Humaita-Tubiacanga virus, APLV-1: Aedes partiti-like virus 1, VeV: Verdadero virus, DENV-1: dengue virus 1 (KDH0030A), DENV-3: dengue virus 3 (JN406515.1), AeAV: Aedes anphevirus, PCLV: Phasi Charoen-like virus, CFAV: Cell fusing agent virus, ATLV-1: Aedes toti-like virus 1, ATV: Aedes toti virus.

Figure 11.
Evidence of ISV super-infection in Wolbachia-infected C6/36 cells. Heat map of data from Teramoto et al. 2019 [65]. Abundance is represented as log10(RPKM) mapped reads per million per kilobase. Hierarchical clustering was performed using Pearson complete distance measurement method. Heat map produced using pheatmap R package. Labels are: PCLV: Phasi Charoen-like virus, WSBL4: Wenzhou-Sobemo-like virus 4, AeAV: Aedes aegypti anphevirus, SGM: San Gabriel Mononegavirus.
Figure 11.
Evidence of ISV super-infection in Wolbachia-infected C6/36 cells. Heat map of data from Teramoto et al. 2019 [65]. Abundance is represented as log10(RPKM) mapped reads per million per kilobase. Hierarchical clustering was performed using Pearson complete distance measurement method. Heat map produced using pheatmap R package. Labels are: PCLV: Phasi Charoen-like virus, WSBL4: Wenzhou-Sobemo-like virus 4, AeAV: Aedes aegypti anphevirus, SGM: San Gabriel Mononegavirus.


{kind=link}
{kind=link}
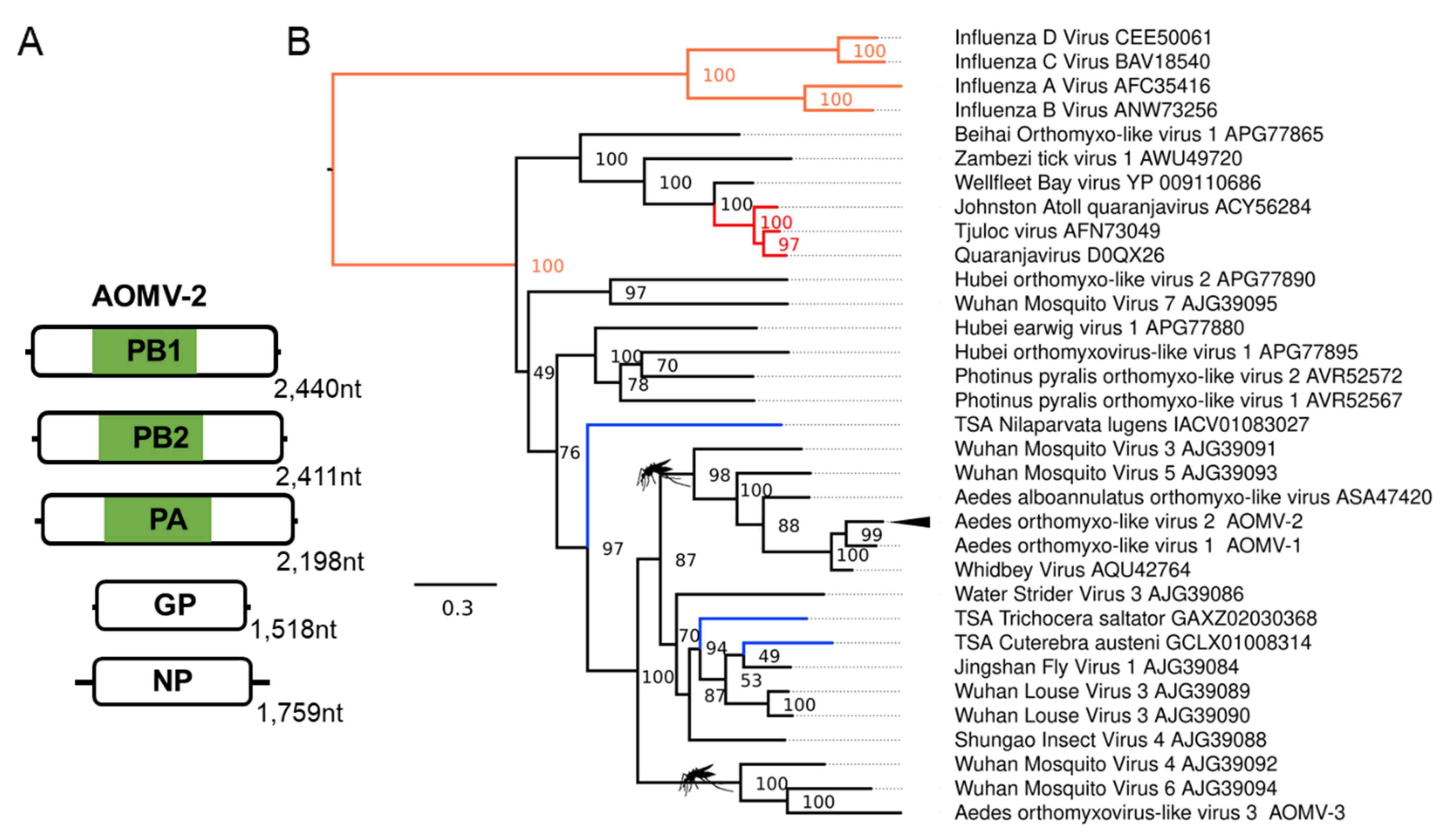{kind=link}
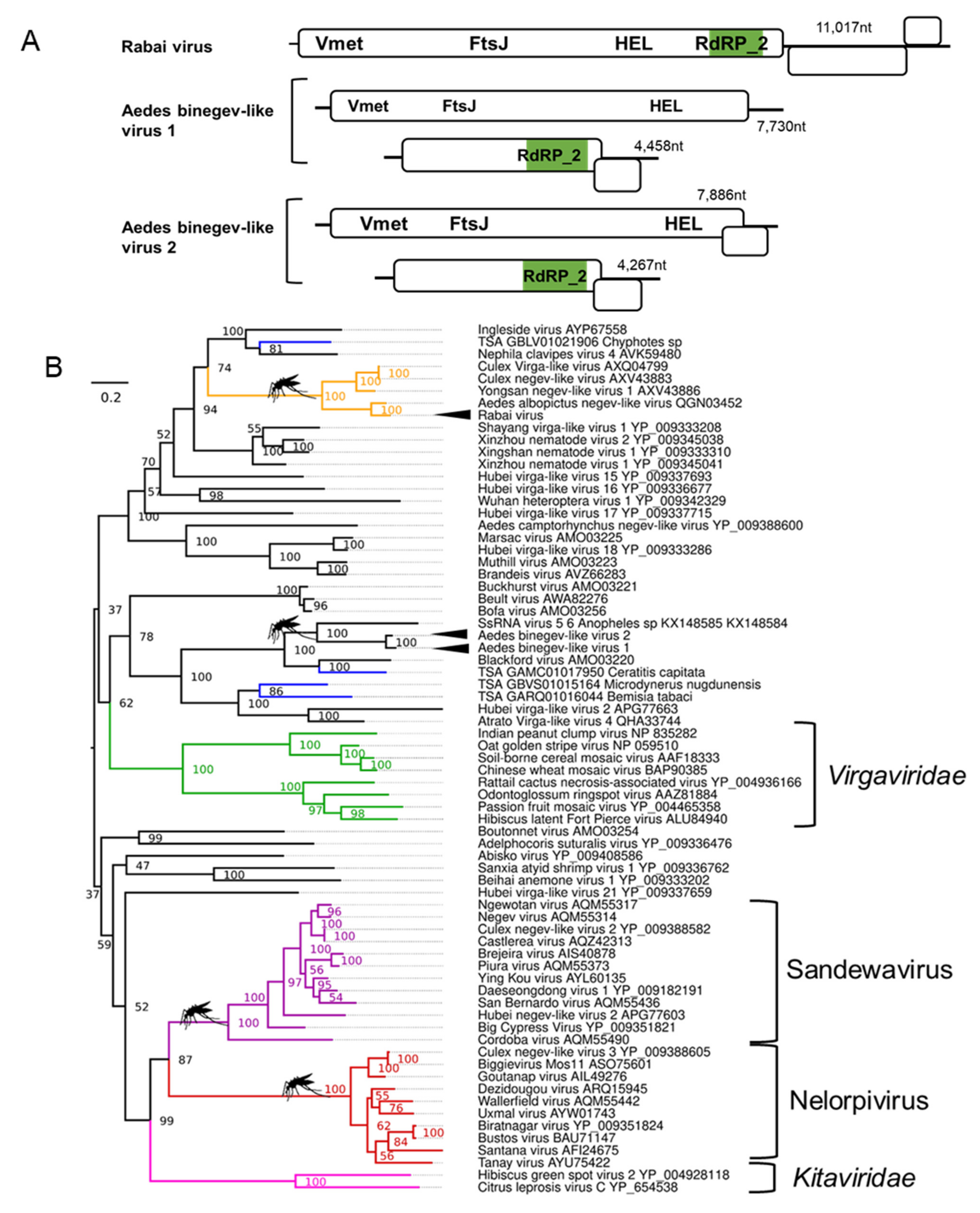{kind=link}
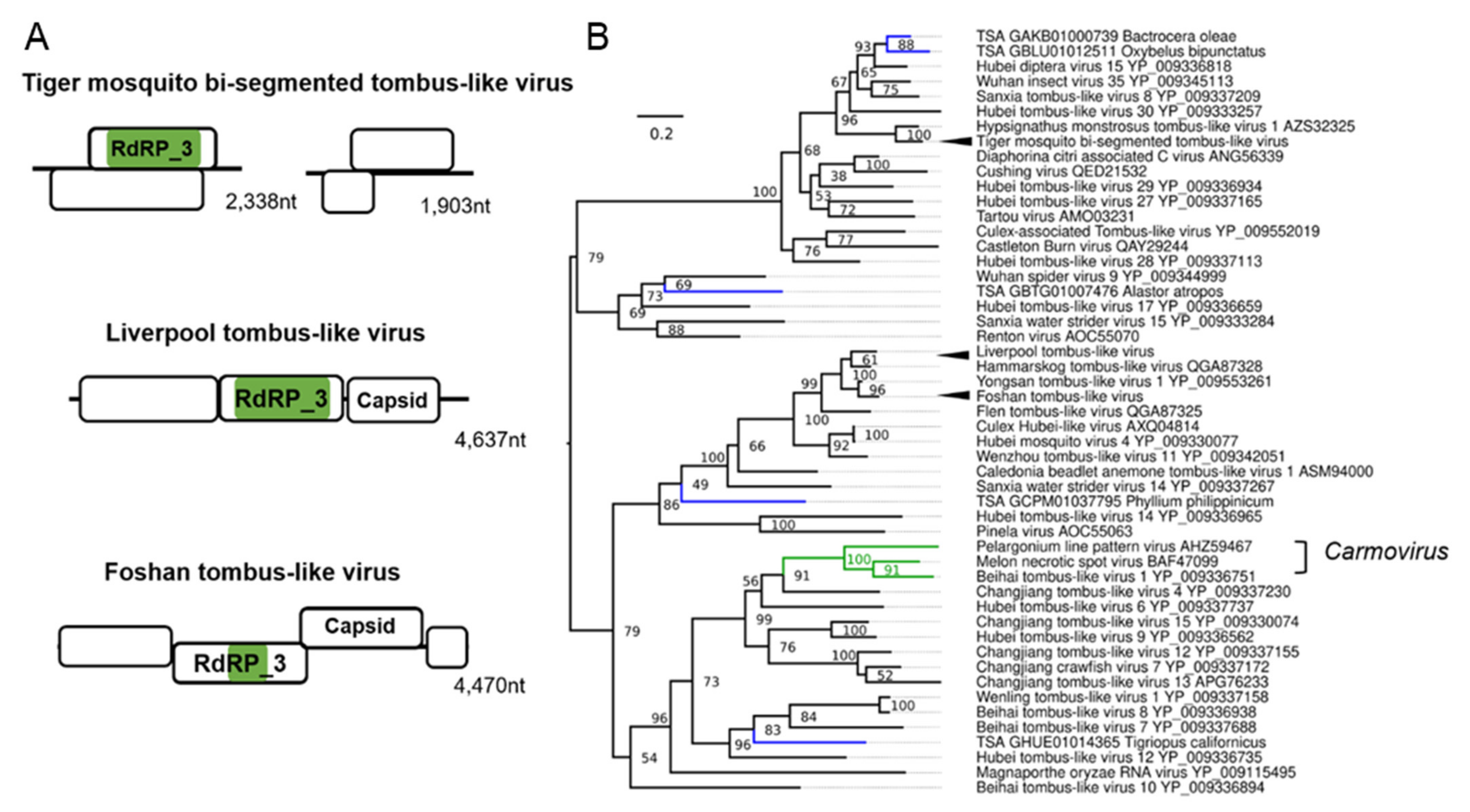{kind=link}
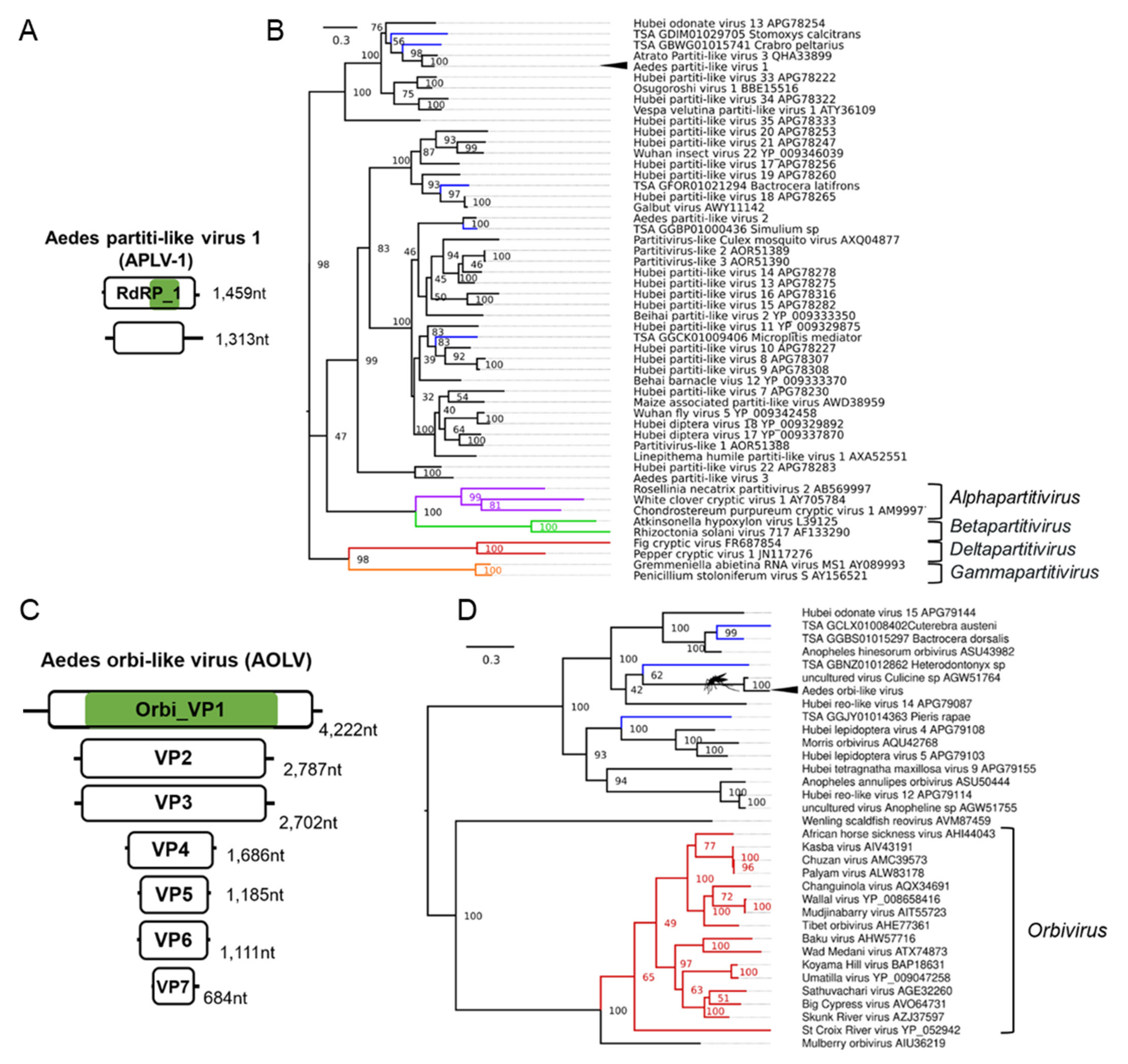{kind=link}
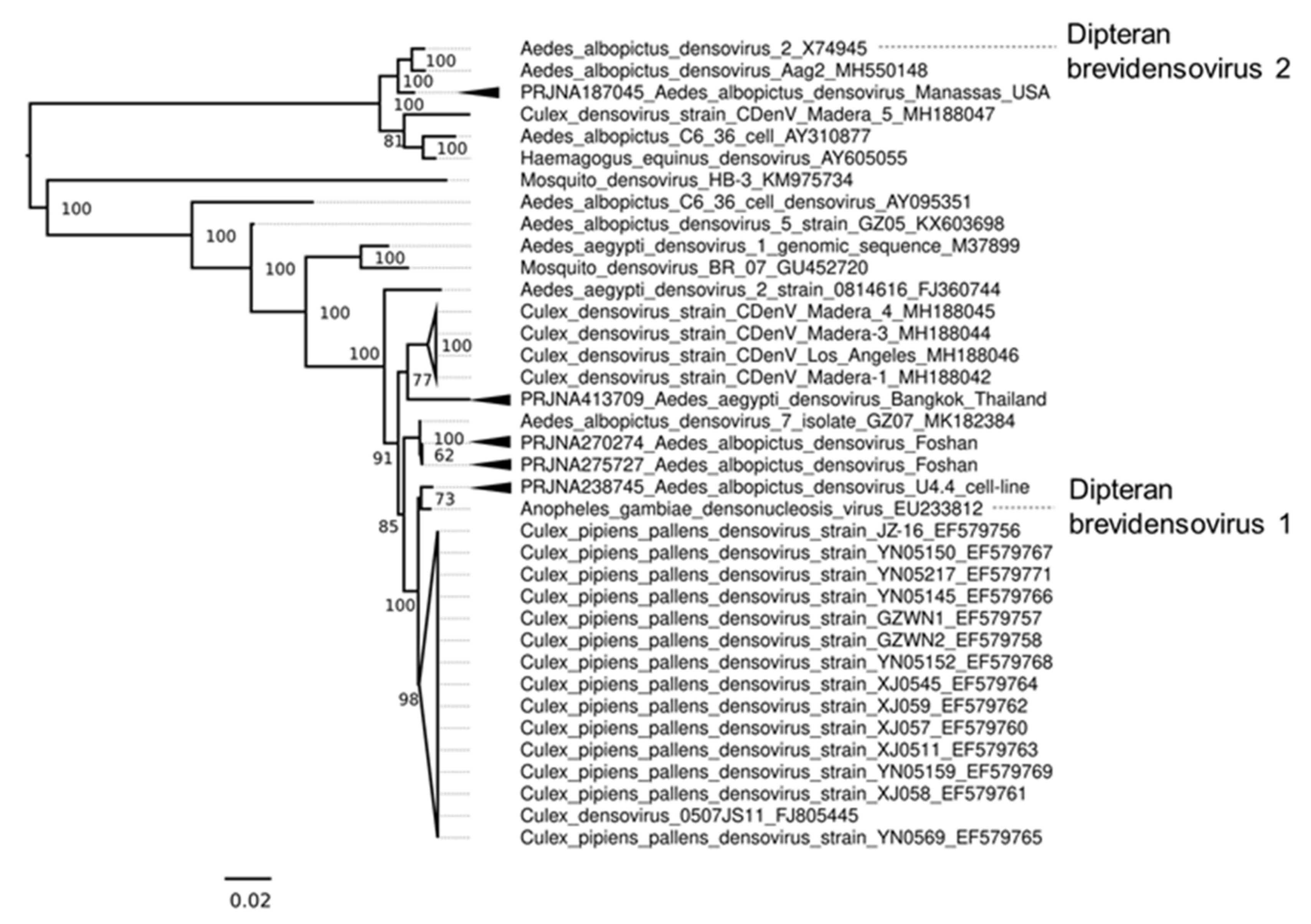{kind=link}
{kind=link}
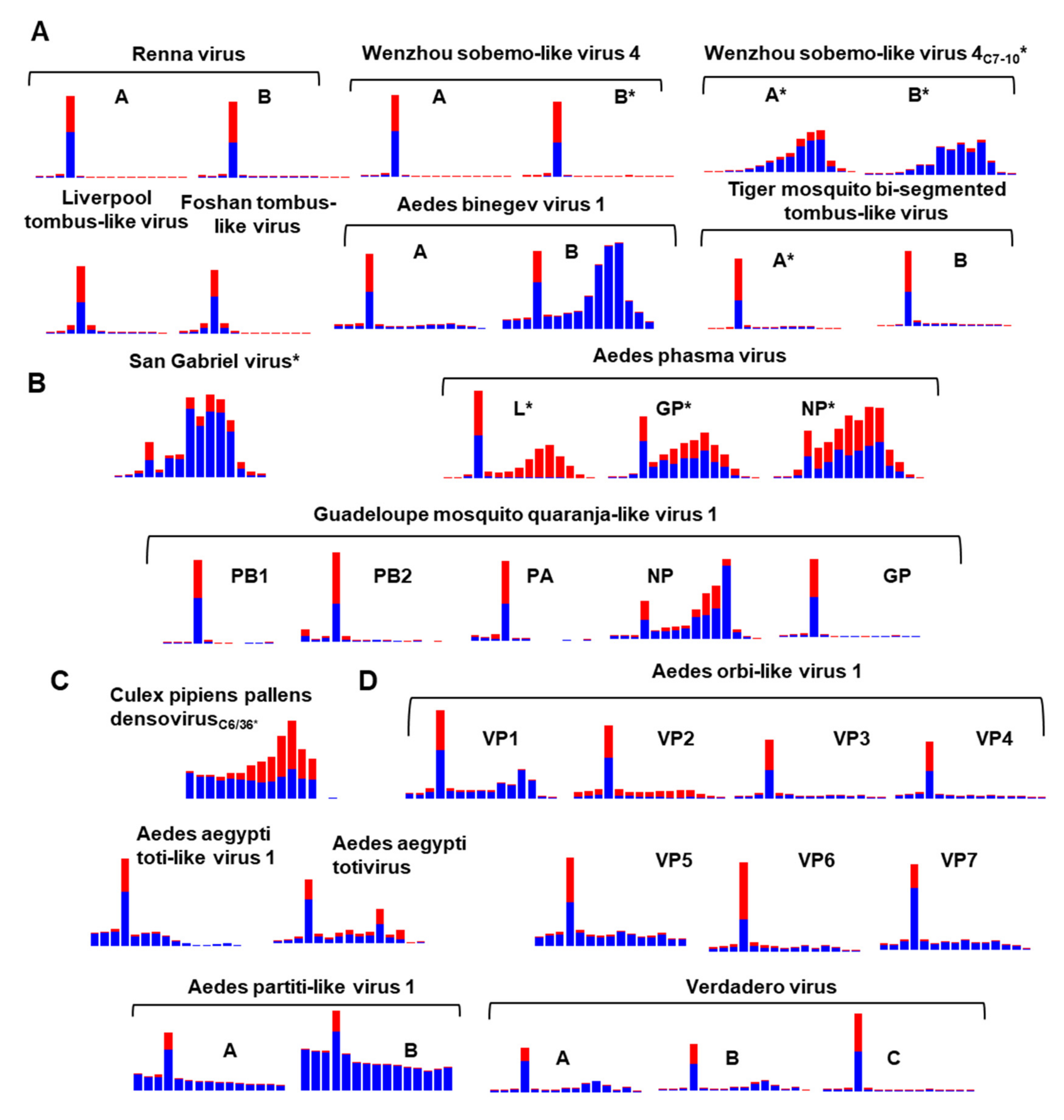{kind=link}
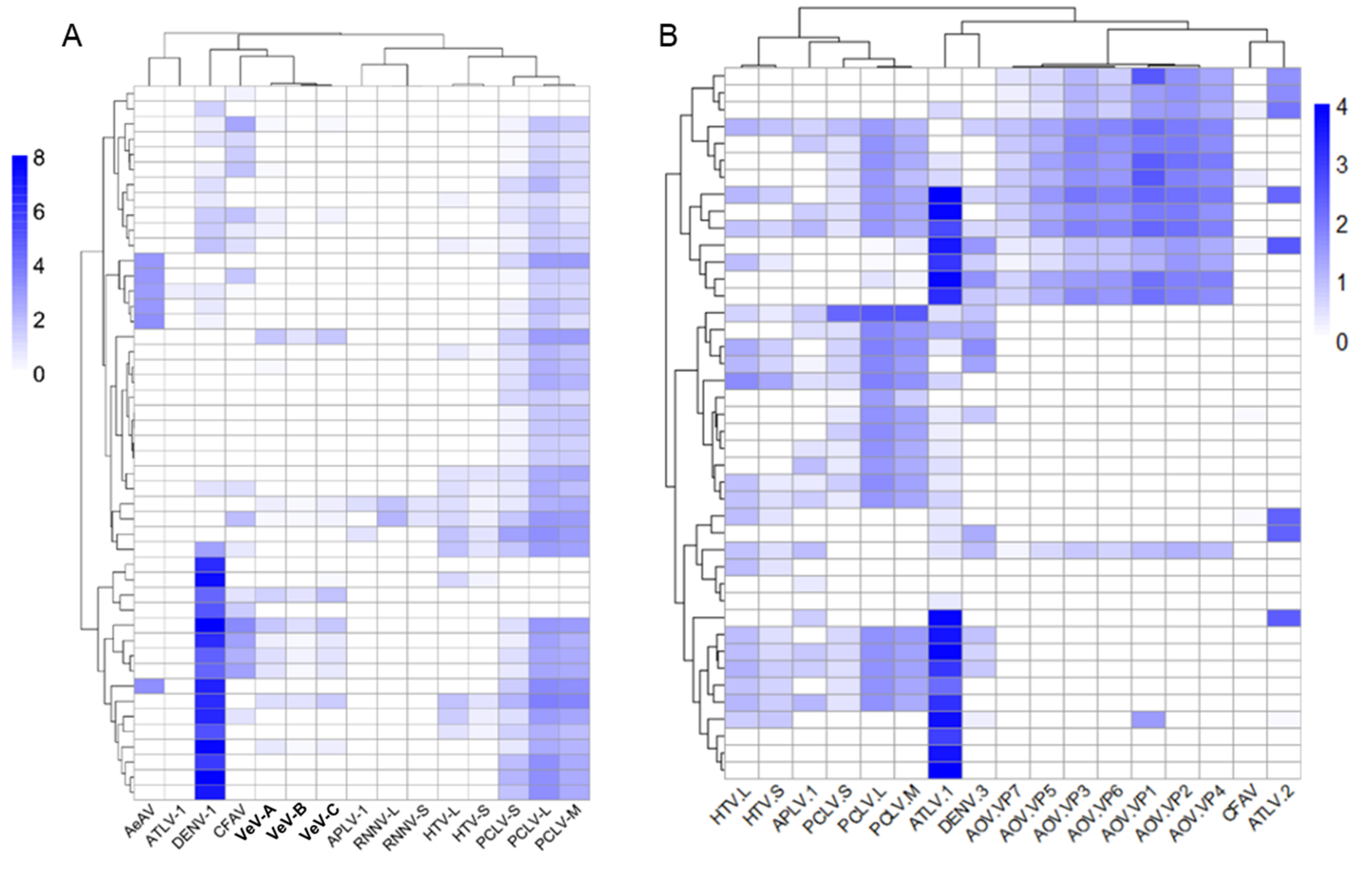{kind=link}
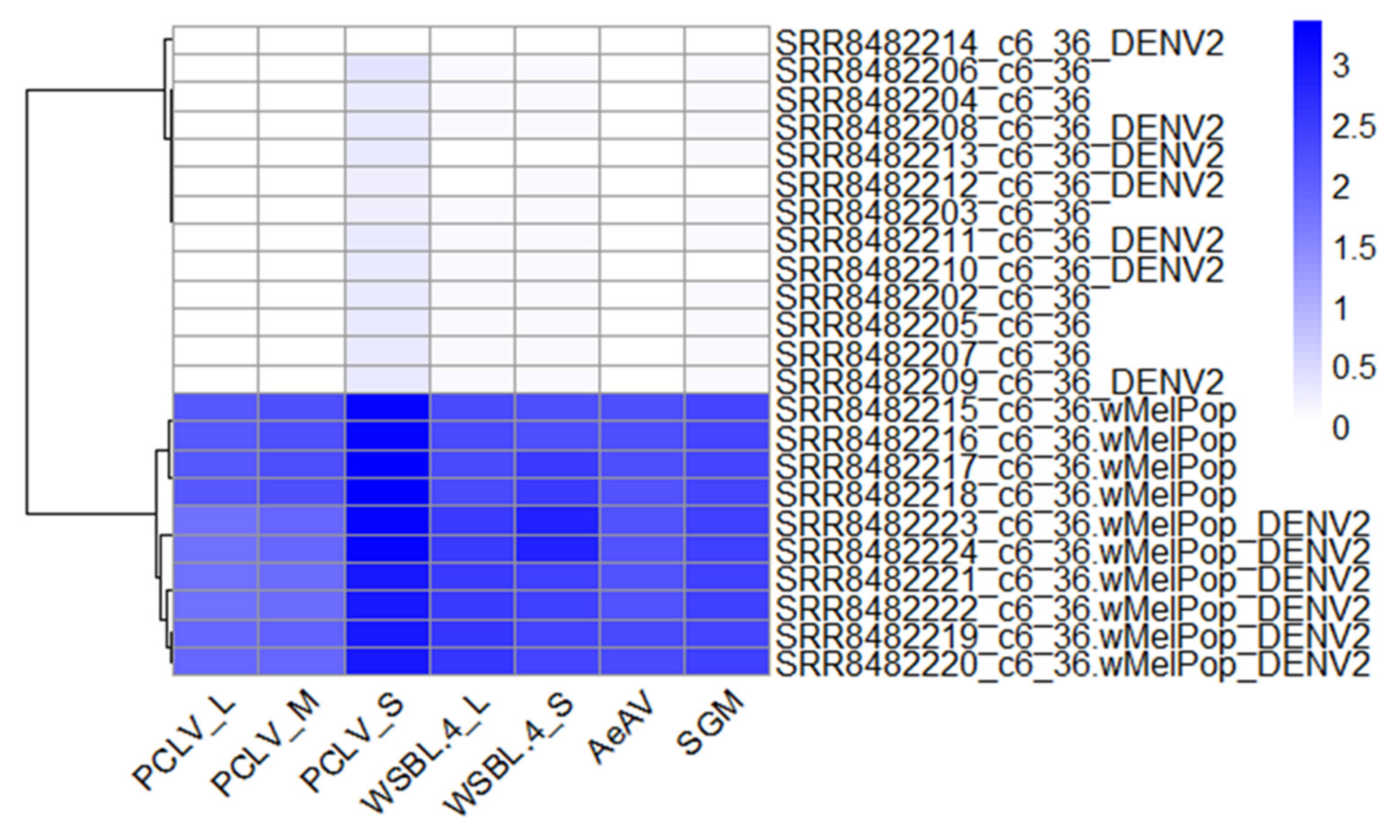{kind=link}
Table 1.
Novel and updated viruses of Aedes mosquitoes.
| Provisional Virus Name; Host | Closest Relative; Genbank ID; Host | Classification: Order (O); Family (F); Genus (G) | Geographical Distribution |
|---|---|---|---|
| Formosus virus; Aedes aegypti | Isfahan virus; YP_007641386.1; Phlebotomus papatasi | O: Mononegavirales; F: Rhabdoviridae | Laboratory colonies: Bundibugyo, Uganda (U30) [54] |
| San Gabriel virus; Aedes albopictus | Wuhan ant virus; YP_009304559.1; Camponotus japonicus | O: Mononegavirales; F: Rhabdoviridae | Laboratory colonies: San Gabriel Valley, Los Angeles County USA [57,58]; Kawasaki, Kanagawa Prefecture, Japan [59]; Wild-caught mosquitoes: Yunnan, China [60]; Ticino, Muzzano, Switzerland [61] Cell lines: Aedes albopictus cells: RML-12.wMelPop [62], Aa23 cells and derivatives [63], C6/36.wMelPop [64,65] Aedes aegypti cells: Aag2.wMelPop [66,67] |
| Longgang virus (LMCV); Aedes albopictus | Shayang fly virus 1; YP_009300663.1; Atherigona orientalis | O: Mononegavirales; likely Chuvirus | Laboratory colonies: Longgang district, Shenzhen, China [68]. |
| Aedes orthomyxo-like virus 2 (AOMV-2); Aedes albopictus | Whidbey virus; AQU42764.1; Aedes dorsalis | F: Orthomyxoviridae | Laboratory colonies: Foshan, China [69,70]; Italy, Rome [71]; Torres strait island, Australia [72]. Wild-caught mosquitoes: Zhejiang, China [73] |
| Rabai virus; Aedes aegypti | Yongsan negev-like virus 1; AXV43886.1; Culex inatomii | Unclassified Negevirus taxon related to F: Virgaviridae | Laboratory colonies: Rabai, Kenya (K2, K4) [54] |
| Aedes binegev-like virus 1 (AeBNV-1); Aedes aegypti | ssRNA virus-like 6 genomic sequence KX148585.1; Anopheles gambiae | Unclassified Negevirus taxon related to F: Virgaviridae | Laboratory colonies: Miami, Florida, USA [74]; Nova Iguaçu Rio de Janeiro, Brazil [75]; Bangkok, Thailand [76]; Key West & Orlando, USA [77]; Liverpool Colony [78,79]; Curepe, Trinidad [80] Wild-caught: Bangkok, Thailand [81]; Manatee County, USA [82]; Miami, USA [12]. |
| Aedes binegev-like virus 2; (AeBNV-2) Aedes albopictus | ssRNA virus-like 6 genomic sequence KX148585.1; Anopheles gambiae | Unclassified Negevirus taxon related to F: Virgaviridae | Laboratory colonies: Longgang District, Shenzhen, China [68]; Manassas, USA [83]. |
| Tiger mosquito bi-segmented tombus-like virus (TMTLV); Aedes albopictus | Culex mosquito virus 1; AXQ04816.1; Culex sp. | Related to the arthropod infecting F: Nodaviridae and F: Tombusviridae | Laboratory colonies: Gainesville (MRA-804), USA [84,85]; Phu Hoa, Binh Duong Province, Vietnam [59,86]; Manassas, USA [83,87,88,89,90]; Nice, France [91]; Wise County, Virginia, USA [92]; Foshan, China [93,94,95]; Kawasaki, Kanagawa Prefecture, Japan [59]. |
| Liverpool tombus-like virus (LTLV) Aedes albopictus, Aedes aegypti | Hammarskog tombus-like virus; QGA87328.1 Coquillettidia richiardii | Related to the arthropod infecting F: Nodaviridae and F: Tombusviridae | Laboratory colonies: Liverpool strains [70,96,97,98,99,100,101], Poza Rica, Mexico [102]; Chetumal (CTM), Mexico [103]; Higgs White Eye (HWE) strain (Variant of Rex-D), Puerto Rico [104]. |
| Aedes orbi-like virus (AOLV); Aedes aegypti | uncultured virus; AGW51764.1; Culicine sp. | F: Reoviridae G: Orbivirus | Laboratory colonies: Cairns, Innisfail, and North QLD Australia [105,106,107,108] Wild-caught: Cairns, Australia [81] |
| Aedes partiti-like virus 1 (APLV-1); Aedes aegypti | Hubei partiti-like virus 34; APG78322.1; Chinese land snail | F: Partitiviridae | Laboratory colonies: Rabai, Kenya (K2, K4, K14) [54]; Galveston, USA [109,110]; Tapachula, Mexico [111]; Cairns, Innisfail, Townsville, Australia [105,106,107,108,112]; Laos [113]; New Delhi, India [114]; Singapore [115,116]; Kamphaeng Phet, Thep Na Korn and Bangkok, Thailand [117,118,119]; Curepe, Trinidad [80,120] Wild-caught: Bangkok, Thailand [81], Cairns, Australia [81]; Manatee County, USA [82]; Miami, USA [12]; Les Abymes & Petit-Bourg, Guadeloupe [121] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Parry, R.; James, M.E.; Asgari, S. Uncovering the Worldwide Diversity and Evolution of the Virome of the Mosquitoes Aedes aegypti and Aedes albopictus. Microorganisms 2021, 9, 1653. https://doi.org/10.3390/microorganisms9081653
AMA Style
Parry R, James ME, Asgari S. Uncovering the Worldwide Diversity and Evolution of the Virome of the Mosquitoes Aedes aegypti and Aedes albopictus. Microorganisms. 2021; 9(8):1653. https://doi.org/10.3390/microorganisms9081653
Chicago/Turabian StyleParry, Rhys, Maddie E James, and Sassan Asgari. 2021. "Uncovering the Worldwide Diversity and Evolution of the Virome of the Mosquitoes Aedes aegypti and Aedes albopictus" Microorganisms 9, no. 8: 1653. https://doi.org/10.3390/microorganisms9081653
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.