
Whole Genome Sequencing Refines Knowledge on the Population Structure of Mycobacterium bovis from a Multi-Host Tuberculosis System

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. M. bovis Isolates Dataset
2.2. Ethical Approval
2.3. DNA Extraction
2.4. Whole-Genome Sequencing and SNP Analysis
2.5. Phylogenetic Analysis
2.6. Phylogeographic Analyses
2.7. Isolation by Distance Dissimilarity Analysis
3. Results
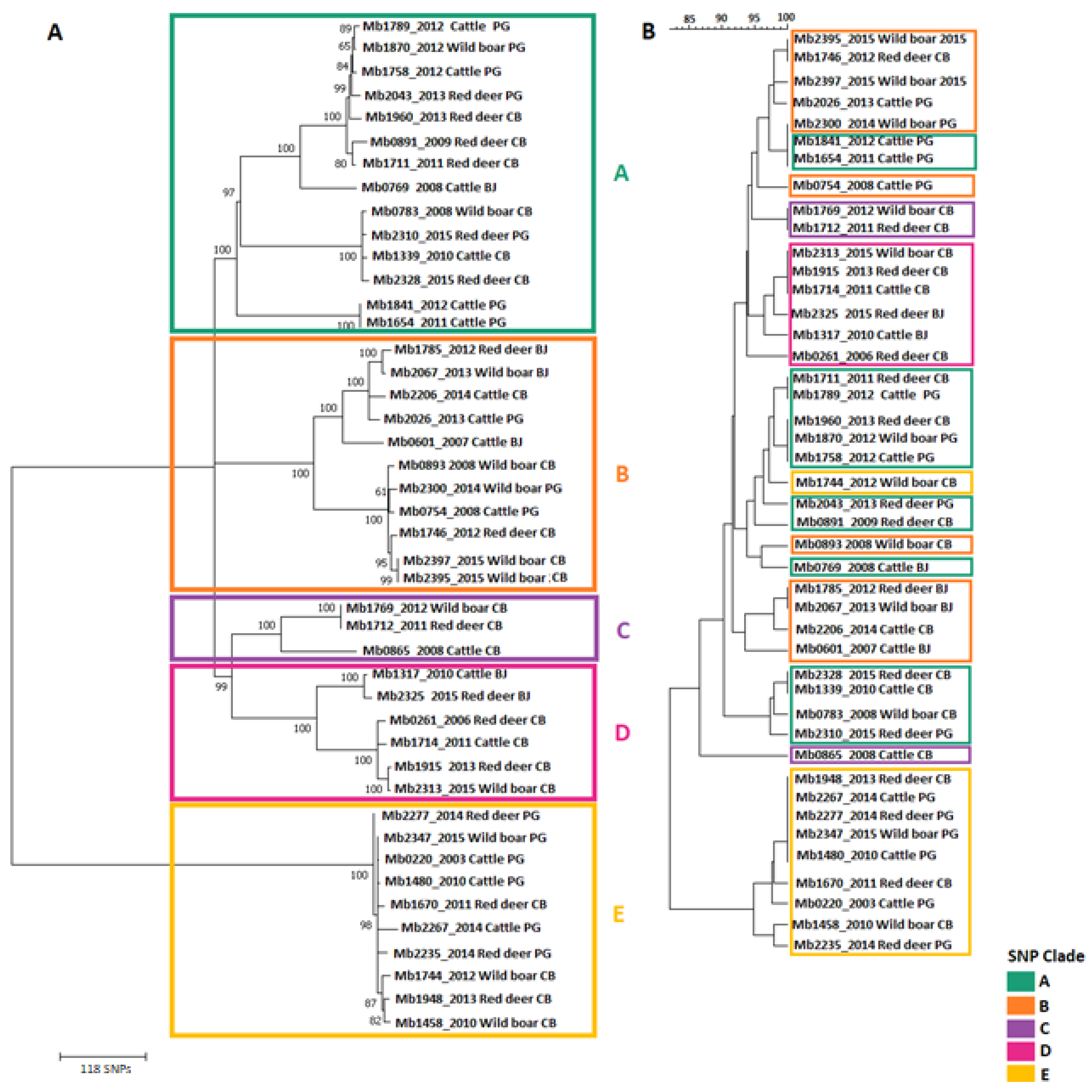
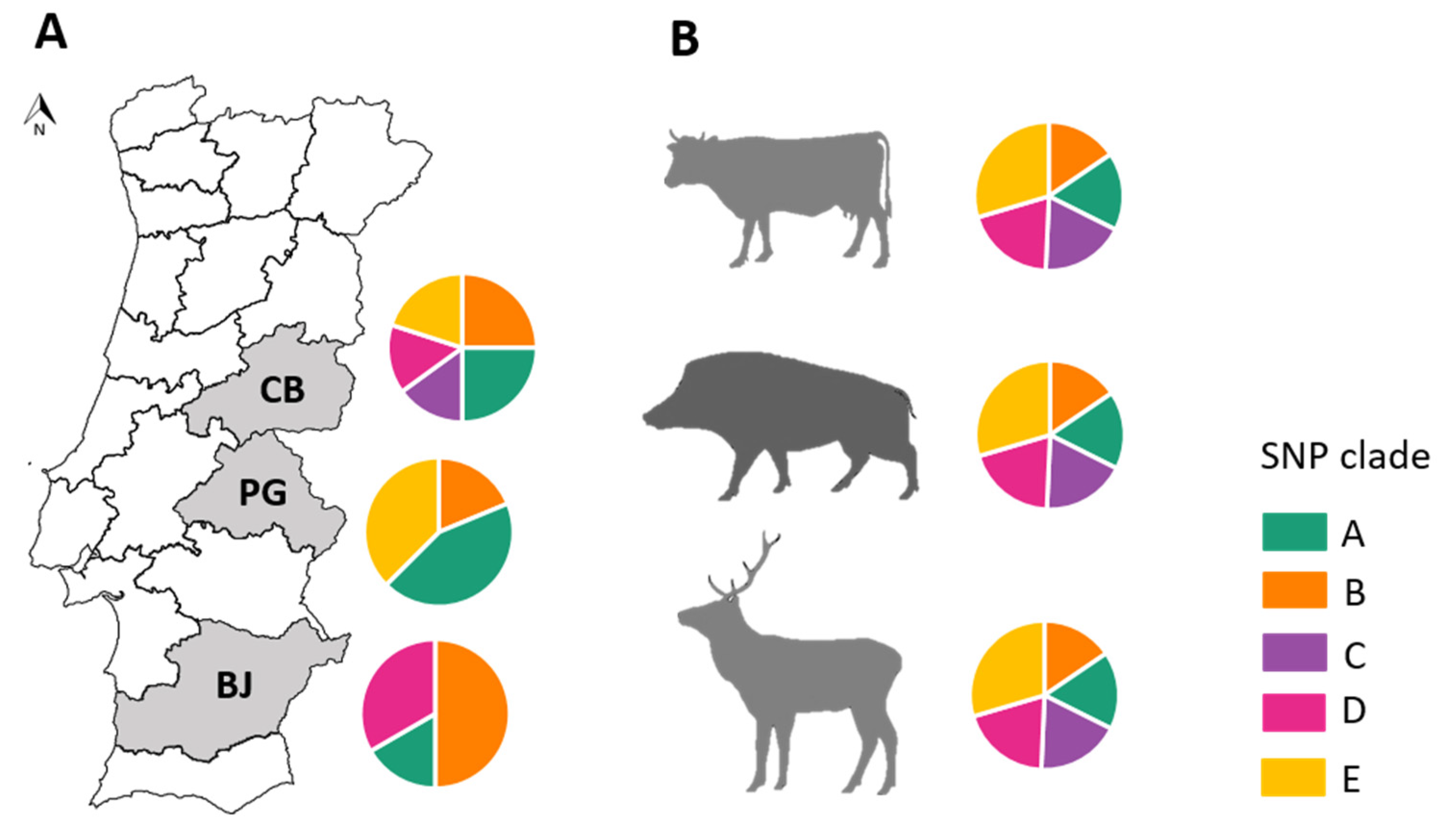
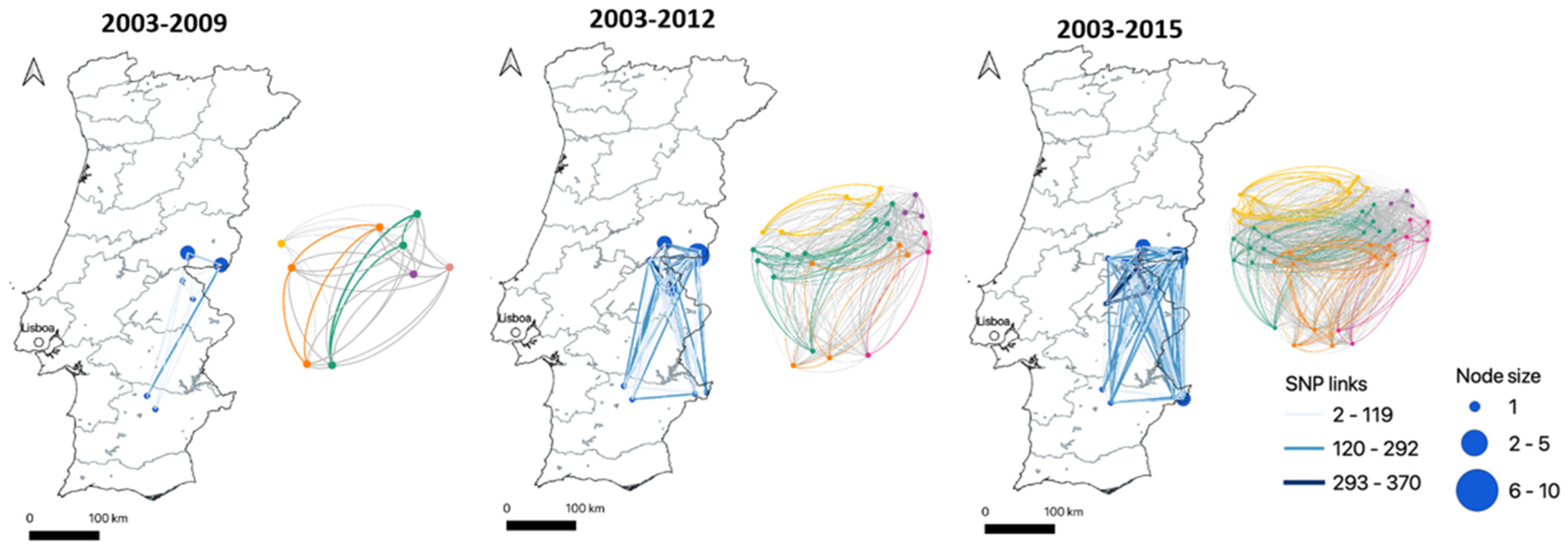
SNP-Based Genotyping and Phylogenetic Analyses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brites, D.; Loiseau, C.; Menardo, F.; Borrell, S.; Boniotti, M.B.; Warren, R.; Dippenaar, A.; Parsons, S.D.C.; Beisel, C.; Behr, M.A.; et al. A New Phylogenetic Framework for the Animal-Adapted Mycobacterium tuberculosis Complex. Front. Microbiol. 2018, 9, 2820. [Google Scholar] [CrossRef] [Green Version]
- Gagneux, S. Ecology and evolution of Mycobacterium tuberculosis. Nat. Rev. Microbiol. 2018, 16, 202–213. [Google Scholar] [CrossRef] [PubMed]
- Corner, L.A.L.; Murphy, D.; Gormley, E. Mycobacterium bovis Infection in the Eurasian Badger (Meles meles): The Disease, Pathogenesis, Epidemiology and Control. J. Comp. Pathol. 2011, 144, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, S.D.; Kaneene, J.B. Wildlife Reservoirs of Bovine Tuberculosis Worldwide: Hosts, Pathology, Surveillance, and Control. Vet. Pathol. 2012, 50, 488–499. [Google Scholar] [CrossRef]
- Palmer, M.V. Tuberculosis: A reemerging disease at the interface of domestic animals and wildlife. Curr. Top. Microbiol. Immunol. 2007, 315, 195–215. [Google Scholar] [CrossRef]
- Palmer, M.V.; Thacker, T.C.; Waters, W.R.; Gortázar, C.; Corner, L.A.L. Mycobacterium bovis: A model pathogen at the interface of livestock, wildlife, and humans. Vet. Med. Int. 2012, 2012, 236205. [Google Scholar] [CrossRef] [Green Version]
- Gortázar, C.; Torres, M.J.; Vicente, J.; Acevedo, P.; Reglero, M.; de la Fuente, J.; Negro, J.J.; Aznar-Martín, J. Bovine tuberculosis in Doñana Biosphere Reserve: The role of wild ungulates as disease reservoirs in the last Iberian lynx strongholds. PLoS ONE 2008, 3, e2776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naranjo, V.; Gortázar, C.; Vicentea, J.; de la Fuente, J. Evidence of the role of European wild boar as a reservoir of Mycobacterium tuberculosis complex. Vet. Microbiol. 2008, 127, 1–9. [Google Scholar] [CrossRef]
- Santos, N.; Correia-Neves, M.; Ghebremichael, S.; Källenius, G.; Svenson, S.B.; Almeida, V. Epidemiology of Mycobacterium bovis infection in wild boar (Sus scrofa) from Portugal. J. Wildl. Dis. 2009, 45, 1048–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vieira-Pinto, M.; Alberto, J.; Aranha, J.; Serejo, J.; Canto, A.; Cunha, M.V.; Botelho, A. Combined evaluation of bovine tuberculosis in wild boar (Sus scrofa) and red deer (Cervus elaphus) from Central-East Portugal. Eur. J. Wildl. Res. 2011, 57, 1189–1201. [Google Scholar] [CrossRef]
- Barasona, J.A.; Torres, M.J.; Aznar, J.; Gortázar, C.; Vicente, J. DNA Detection Reveals Mycobacterium tuberculosis Complex Shedding Routes in Its Wildlife Reservoir the Eurasian Wild Boar. Transbound. Emerg. Dis. 2017, 64, 906–915. [Google Scholar] [CrossRef] [PubMed]
- Cunha, M.V.; Matos, F.; Canto, A.; Albuquerque, T.; Alberto, J.R.; Aranha, J.M.; Vieira-Pinto, M.; Botelho, A. Implications and challenges of tuberculosis in wildlife ungulates in Portugal: A molecular epidemiology perspective. Res. Vet. Sci. 2012, 92, 225–235. [Google Scholar] [CrossRef]
- Rivière, J.; Carabin, K.; Le Strat, Y.; Hendrikx, P.; Dufour, B. Bovine tuberculosis surveillance in cattle and free-ranging wildlife in EU Member States in 2013: A survey-based review. Vet. Microbiol. 2014, 173, 323–331. [Google Scholar] [CrossRef] [PubMed]
- DGAV Programme for the Eradication of Bovine Tuberculosis, Bovine Brucellosis or Sheep and Goat Brucellosis (B. melitensis). Submitted for Obtaining EU Cofinancing. 2019. Available online: https://www.dgav.pt/wp-content/uploads/2021/01/Programa-Tuberculose-bovina-2019-ref-14777-.pdf (accessed on 1 May 2021).
- DGAV Relatório Técnico de Sanidade Animal—Tuberculose Bovina (ano 2017). 2017. Available online: https://www.dgav.pt/wp-content/uploads/2021/01/RT_2017_TB-BB-BPR.-atualizado-a-16jul2018_v4-PDF.pdf (accessed on 1 May 2021).
- DGAV Plano de Controlo e Erradicação de Tuberculose em Caça Maior. 2011. Available online: https://www.dgav.pt/wp-content/uploads/2021/03/Plano_controlo_erradicacao_Tuberculose_Caca_Maior.pdf (accessed on 1 May 2021).
- Duarte, E.L.; Domingos, M.; Amado, A.; Cunha, M.V.; Botelho, A. MIRU-VNTR typing adds discriminatory value to groups of Mycobacterium bovis and Mycobacterium caprae strains defined by spoligotyping. Vet. Microbiol. 2010, 143, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Reis, A.C.; Tenreiro, R.; Albuquerque, T.; Botelho, A.; Cunha, M.V. Long-term molecular surveillance provides clues on a cattle origin for Mycobacterium bovis in Portugal. Sci. Rep. 2020, 10, 20856. [Google Scholar] [CrossRef]
- Biek, R.; O’Hare, A.; Wright, D.; Mallon, T.; Mccormick, C.; Orton, R.J.; Mcdowell, S.; Trewby, H.; Skuce, R.A.; Kao, R.R. Whole Genome Sequencing Reveals Local Transmission Patterns of Mycobacterium bovis in Sympatric Cattle and Badger Populations. PLoS Pathog. 2012, 8, 1003008. [Google Scholar] [CrossRef] [PubMed]
- Glaser, L.; Carstensen, M.; Shaw, S.; Robbe-Austerman, S.; Wunschmann, A.; Grear, D.; Stuber, T.; Thomsen, B. Descriptive Epidemiology and Whole Genome Sequencing Analysis for an Outbreak of Bovine Tuberculosis in Beef Cattle and White-Tailed Deer in Northwestern Minnesota. PLoS ONE 2016, 11, e0145735. [Google Scholar] [CrossRef] [Green Version]
- Price-Carter, M.; Brauning, R.; de Lisle, G.W.; Livingstone, P.; Neill, M.; Sinclair, J.; Paterson, B.; Atkinson, G.; Knowles, G.; Crews, K.; et al. Whole Genome Sequencing for Determining the Source of Mycobacterium bovis Infections in Livestock Herds and Wildlife in New Zealand. Front. Vet. Sci. 2018, 5, 272. [Google Scholar] [CrossRef] [Green Version]
- Trewby, H.; Wright, D.; Breadon, E.L.; Lycett, S.J.; Mallon, T.R.; Mccormick, C.; Johnson, P.; Orton, R.J.; Allen, A.R.; Galbraith, J.; et al. Use of bacterial whole-genome sequencing to investigate local persistence and spread in bovine tuberculosis. Epidemics 2016, 14, 26–35. [Google Scholar] [CrossRef] [Green Version]
- Crispell, J.; Benton, C.H.; Balaz, D.; De Maio, N.; Ahkmetova, A.; Allen, A.; Biek, R.; Presho, E.L.; Dale, J.; Hewinson, G.; et al. Combining genomics and epidemiology to analyse bi-directional transmission of Mycobacterium bovis in a multi-host system. Elife 2019, 8, 45833. [Google Scholar] [CrossRef]
- Crispell, J.; Cassidy, S.; Kenny, K.; McGrath, G.; Warde, S.; Cameron, H.; Rossi, G.; Macwhite, T.; White, P.C.L.; Lycett, S.; et al. Mycobacterium bovis genomics reveals transmission of infection between cattle and deer in Ireland. Microb. Genom. 2020, 6, mgen000388. [Google Scholar] [CrossRef]
- Crispell, J.; Zadoks, R.N.; Harris, S.R.; Paterson, B.; Collins, D.M.; De-Lisle, G.W.; Livingstone, P.; Neill, M.A.; Biek, R.; Lycett, S.J.; et al. Using whole genome sequencing to investigate transmission in a multi-host system: Bovine tuberculosis in New Zealand. BMC Genom. 2017, 18, 180. [Google Scholar] [CrossRef] [Green Version]
- Salvador, L.; O’Brien, D.; Cosgrove, M.; Stuber, T.; Schooley, A.; Crispell, J.; Church, S.; Grohn, Y.; Robbe-Austerman, S.; Kao, R. Disease management at the wildlife-livestock interface: Using whle-genome sequencing to study the role of elk in Mycobacterium bovis transmission in Michigan, USA. Mol. Ecol. 2019, 28, 2192–2205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerra-Assunção, J.; Crampin, A.; Houben, R.; Mzembe, T.; Mallard, K.; Coll, F.; Khan, P.; Banda, L.; Chiwaya, A.; Pereira, R.; et al. Large-scale whole genome sequencing of M. tuberculosis provides insights into transmission in a high prevalence area. Elife 2015, 4, e05166. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows—Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mckenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Depristo, M.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.; Hartl, C.; Philippakis, A.; Rivas, M.; Hanna, M.; Mckenna, A.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Van der Auwera, G.; Carneiro, M.O.; Hartl, C.; Poplin, R.; del Angel, G.; Levy-moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinforma. 2014, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2012, 14, 178–192. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paradis, E.; Claude, J.; Strimmer, K. APE: Analyses of Phylogenetics and Evolution in R language. Bioinformatics 2004, 20, 289–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bastian, M.; Heymann, S.; Jacomy, M. Gephi: An Open Source Software for Exploring and Manipulating Networks Visualization and Exploration of Large Graphs. In Proceedings of the International AAAI Conference on Weblogs and Social Media, San Jose, GA, USA, 17–20 May 2009. [Google Scholar]
- Dray, S.; Dufour, A.-B. The ade4 Package: Implementing the Duality Diagram for Ecologists. J. Stat. Softw. 2007, 22, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Campos, S.; Schürch, A.C.; Dale, J.; Lohan, A.J.; Cunha, M.V.; Botelho, A.; De Cruz, K.; Boschiroli, M.L.; Boniotti, M.B.; Pacciarini, M.; et al. European 2—A clonal complex of Mycobacterium bovis dominant in the Iberian Peninsula. Infect. Genet. Evol. 2012, 12, 866–872. [Google Scholar] [CrossRef]
- Zimpel, C.K.; Patané, J.S.L.; Guedes, A.C.P.; de Souza, R.F.; Silva-pereira, T.T.; Camargo, N.C.S.; Filho, A.F.D.S.; Ikuta, C.Y.; Neto, J.S.F.; Setubal, J.C.; et al. Global Distribution and Evolution of Mycobacterium bovis Lineages. Front. Microbiol. 2020, 11, 843. [Google Scholar] [CrossRef]
- Hauer, A.; Michelet, L.; Cochard, T.; Branger, M.; Nunez, J.; Boschiroli, M.L.; Biet, F. Accurate Phylogenetic Relationships Among Mycobacterium bovis Strains Circulating in France Based on Whole Genome Sequencing and Single Nucleotide Polymorphism Analysis. Front. Microbiol. 2019, 10, 955. [Google Scholar] [CrossRef] [Green Version]
- Lasserre, M.; Fresia, P.; Greif, G.; Iraola, G.; Castro-Ramos, M.; Juambeltz, A.; Nuñez, Á.; Naya, H.; Robello, C.; Berná, L. Whole genome sequencing of the monomorphic pathogen Mycobacterium bovis reveals local differentiation of cattle clinical isolates. BMC Genom. 2018, 19, 2. [Google Scholar] [CrossRef] [Green Version]
- Reyes, J.F.; Chan, C.H.S.; Tanaka, M.M. Impact of homoplasy on variable numbers of tandem repeats and spoligotypes in Mycobacterium tuberculosis. Infect. Genet. Evol. 2012, 12, 811–818. [Google Scholar] [CrossRef] [PubMed]
- Comas, I.; Homolka, S.; Niemann, S.; Gagneux, S. Genotyping of genetically monomorphic bacteria: DNA sequencing in Mycobacterium tuberculosis highlights the limitations of current methodologies. PLoS ONE 2009, 4, 7815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearson, T.; Okinaka, R.T.; Foster, J.T.; Keim, P. Phylogenetic understanding of clonal populations in an era of whole genome sequencing. Infect. Genet. Evol. 2009, 9, 1010–1019. [Google Scholar] [CrossRef] [PubMed]
- Maeda, S.; Hijikata, M.; Le Hang, N.T.; Thoung, P.H.; Van Huan, H.; Hoang, N.P.; van Hung, N.; Cuong, V.C.; Miyabayashi, A.; Seto, S.; et al. Genotyping of Mycobacterium tuberculosis spreading in Hanoi, Vietnam using conventional and whole genome sequencing methods. Infect. Genet. Evol. 2020, 78, 104107. [Google Scholar] [CrossRef]
- Hijikata, M.; Keicho, N.; Van Duc, L.; Maeda, S.; Le Hang, N.T.; Matsushita, I.; Kato, S. Spoligotyping and whole-genome sequencing analysis of lineage 1 strains of Mycobacterium tuberculosis in Da Nang, Vietnam. PLoS ONE 2017, 12, e0186800. [Google Scholar] [CrossRef] [Green Version]
- Skuce, R.; Breadon, E.; Allen, A.; Milne, G.; McCormick, C.; Hughes, C.; Rutherford, D.; Smith, G.; Thompson, S.; Graham, J.; et al. Longitudinal dynamics of herd-level Mycobacterium bovis MLVA type surveillance in cattle in Northern Ireland 2003–2016. Infect. Genet. Evol. 2020, 79, 104131. [Google Scholar] [CrossRef] [PubMed]
- Guerra-Assunção, J.A.; Houben, R.M.G.J.; Crampin, A.C.; Mzembe, T.; Mallard, K.; Coll, F.; Khan, P.; Banda, L.; Chiwaya, A.; Pereira, R.P.A.; et al. Recurrence due to Relapse or Reinfection With Mycobacterium tuberculosis: A Whole-Genome Sequencing Approach in a Large, Population- Based Cohort With a High HIV Infection Prevalence and Active Follow-up. J. Infect. Dis. 2015, 211, 1154–1163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meehan, C.J.; Moris, P.; Kohl, T.A.; Pečerska, J.; Akter, S.; Merker, M.; Utpatel, C.; Beckert, P.; Gehre, F.; Lempens, P.; et al. The relationship between transmission time and clustering methods in Mycobacterium tuberculosis epidemiology. EBioMedicine 2018, 37, 410–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almaw, G.; Mekonnen, G.A.; Mihret, A.; Aseffa, A.; Taye, H.; Conlan, A.J.K.; Gumi, B.; Zewude, A.; Aliy, A.; Tamiru, M.; et al. Population structure and transmission of Mycobacterium bovis in Ethiopia. Microb. Genom. 2021, 7, 000539. [Google Scholar] [CrossRef]
- Walker, T.M.; Ip, C.L.C.; Harrell, R.H.; Evans, J.T.; Kapatai, G.; Dedicoat, M.J.; Eyre, D.W.; Wilson, D.J.; Hawkey, P.M.; Crook, D.W.; et al. Whole-genome sequencing to delineate Mycobacterium tuberculosis outbreaks: A retrospective observational study. Lancet Infect. Dis. 2013, 13, 137–146. [Google Scholar] [CrossRef] [Green Version]
- Roetzer, A.; Diel, R.; Kohl, T.A.; Rückert, C.; Nübel, U.; Blom, J.; Wirth, T.; Jaenicke, S.; Schuback, S.; Rüsch-Gerdes, S.; et al. Whole Genome Sequencing versus Traditional Genotyping for Investigation of a Mycobacterium tuberculosis Outbreak: A Longitudinal Molecular Epidemiological Study. PLoS Med. 2013, 10, e1001387. [Google Scholar] [CrossRef]
- Rossi, G.; Crispell, J.; Brough, T.; Lycett, S.J.; White, P.C.L.; Allen, A.; Ellis, R.J.; Gordon, S.V.; Harwood, R.; Presho, E.L.; et al. Phylodynamic analysis of an emergent Mycobacterium bovis outbreak in an area with no previously known wildlife infections. bioRxiv 2020. [Google Scholar] [CrossRef]
- Akhmetova, A.; Guerrero, J.; McAdam, P.; Salvador, L.C.; Crispell, J.; Presho, E.; Kao, R.R.; Biek, R.; Menzies, F.; Trimble, N.; et al. Genomic epidemiology of Mycobacterium bovis infection in sympatric badger and cattle populations in Northern Ireland. bioRxiv 2021. [Google Scholar] [CrossRef]
- Reis, A.C.; Albuquerque, T.; Botelho, A.; Cunha, M.V. Polyclonal infection as a new scenario in Mycobacterium caprae epidemiology. Vet. Microbiol. 2020, 240, 108533. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SNP Clade | Total SNP Sites | Clade-Defining SNP Sites (a) | Clade-Monomorphic SNP Sites (b) |
|---|---|---|---|
| A (n = 14) | 622 | 108 | - |
| B (n = 11) | 611 | 133 | - |
| C (n = 3) | 320 | 184 | 49 |
| D (n = 6) | 372 | 217 | 82 |
| E (n = 10) | 431 | 360 | 352 |
| A to D (n = 34) | 1419 | 1411 | 106 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reis, A.C.; Salvador, L.C.M.; Robbe-Austerman, S.; Tenreiro, R.; Botelho, A.; Albuquerque, T.; Cunha, M.V. Whole Genome Sequencing Refines Knowledge on the Population Structure of Mycobacterium bovis from a Multi-Host Tuberculosis System. Microorganisms 2021, 9, 1585. https://doi.org/10.3390/microorganisms9081585
Reis AC, Salvador LCM, Robbe-Austerman S, Tenreiro R, Botelho A, Albuquerque T, Cunha MV. Whole Genome Sequencing Refines Knowledge on the Population Structure of Mycobacterium bovis from a Multi-Host Tuberculosis System. Microorganisms. 2021; 9(8):1585. https://doi.org/10.3390/microorganisms9081585
Chicago/Turabian StyleReis, Ana C., Liliana C. M. Salvador, Suelee Robbe-Austerman, Rogério Tenreiro, Ana Botelho, Teresa Albuquerque, and Mónica V. Cunha. 2021. "Whole Genome Sequencing Refines Knowledge on the Population Structure of Mycobacterium bovis from a Multi-Host Tuberculosis System" Microorganisms 9, no. 8: 1585. https://doi.org/10.3390/microorganisms9081585