The Effect of a High-Grain Diet on the Rumen Microbiome of Goats with a Special Focus on Anaerobic Fungi

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Diet
2.2. Nutritional Composition of AH and HG Diets
2.3. Sample Collection and DNA Extraction
2.4. Characterization of Goat Rumen Microbiome by High-Throughput Sequencing
2.5. Characterization of Anaerobic Fungi Population Using Clone Libraries
2.6. qPCR Analysis
2.7. Statistical Analysis
3. Results
3.1. Influence of Diet on Ruminal pH
3.2. High-Throughput Sequencing of the 16S rRNA Gene V4 Region and the ITS2 Region
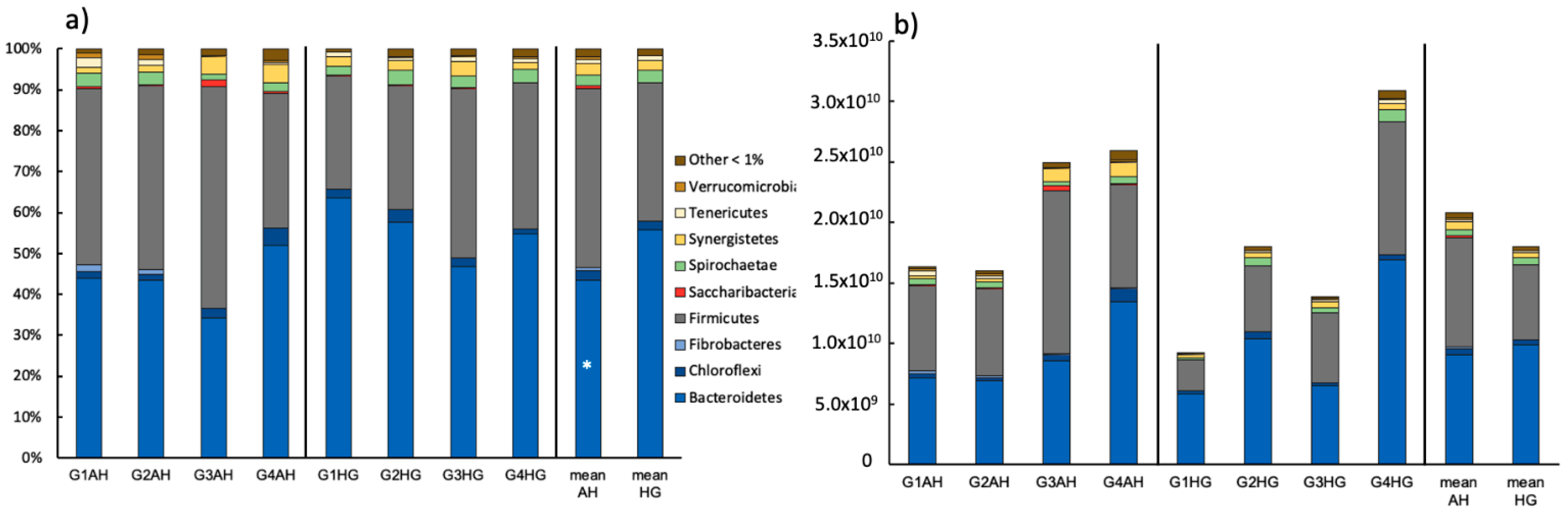
3.2.1. Bacterial Community Composition
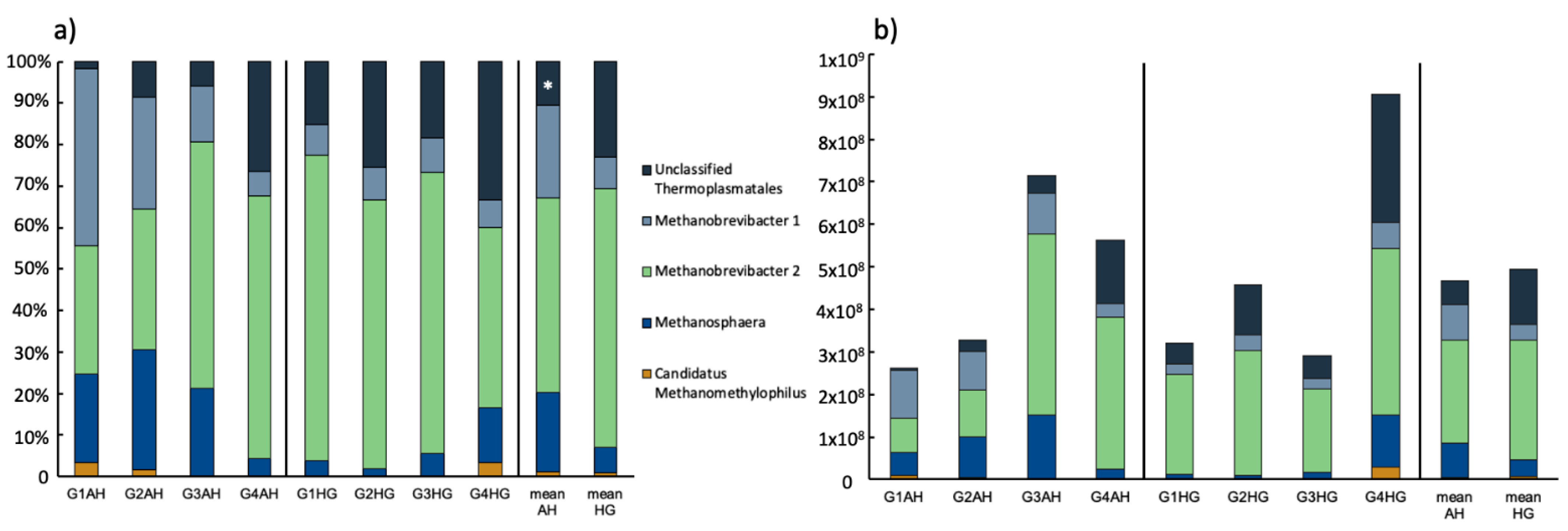
3.2.2. Archaeal Community Composition
3.2.3. Fungal Community Composition
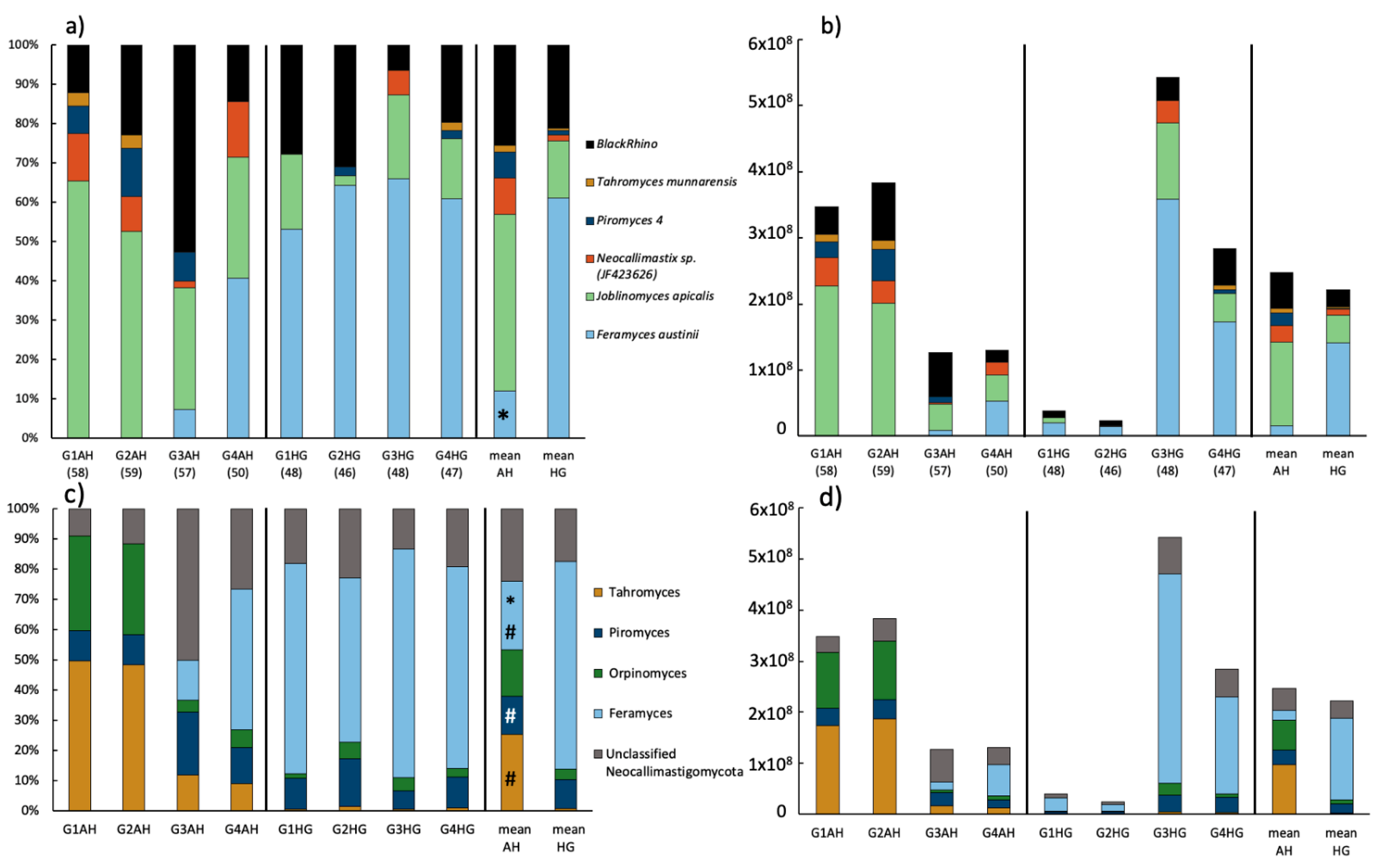
3.3. Community Composition of Neocallimastigomycota Based on HTS of the ITS2
3.4. Community Composition of Neocallimastigomycota Based on ITS1 Clone Libraries
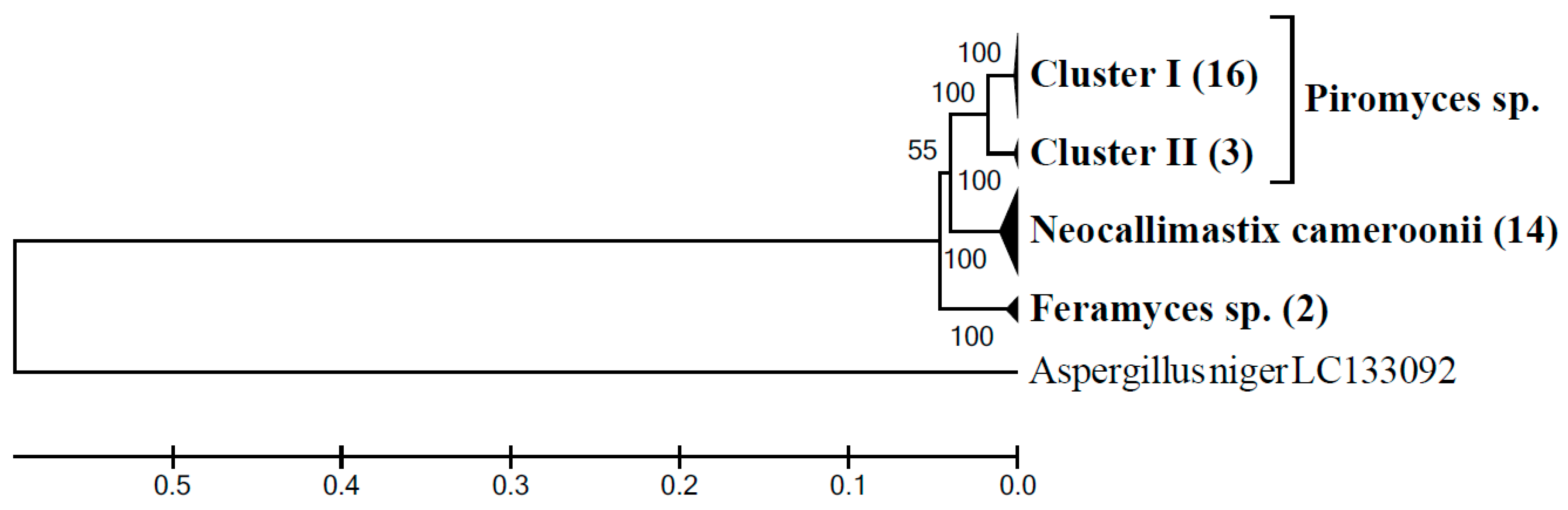
3.5. Community Composition of Neocallimastigomycota in Sample G3AH Based on LSU Clone Library
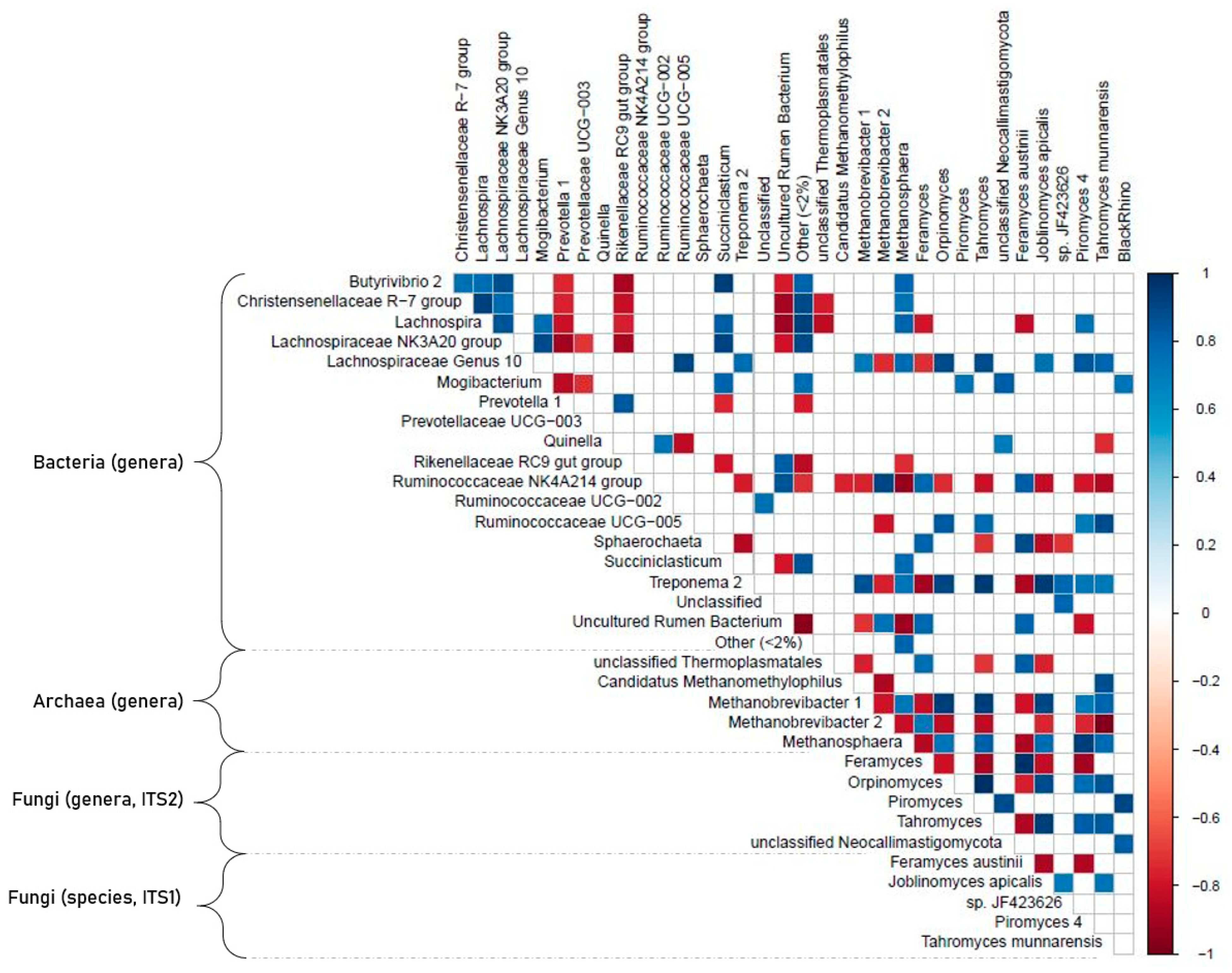
3.6. Correlation Analyses
4. Discussion
4.1. Prokaryotic Response on Diet Shift
4.2. Fungal Response on Diet Shift
4.3. Comparison of the ITS1 and ITS2 as Taxonomic Marker for AF
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Skapetas, B.; Bampidis, V. Goat production in the world: Present situation and trends. Livest. Res. Rural Dev. 2016, 28, 200. [Google Scholar]
- Bi, Y.; Zeng, S.; Zhang, R.; Diao, Q.; Tu, Y. Effects of dietary energy levels on rumen bacterial community composition in Holstein heifers under the same forage to concentrate ratio condition. BMC Microbiol. 2018, 18, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Mao, S.Y.; Zhang, R.Y.; Wang, D.S.; Zhu, W.Y. Impact of subacute ruminal acidosis (SARA) adaptation on rumen microbiota in dairy cattle using pyrosequencing. Anaerobe 2013, 24, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Nagata, R.; Kim, Y.H.; Ohkubo, A.; Kushibiki, S.; Ichijo, T.; Sato, S. Effects of repeated subacute ruminal acidosis challenges on the adaptation of the rumen bacterial community in Holstein bulls. J. Dairy Sci. 2018, 101, 4424–4436. [Google Scholar] [CrossRef]
- Petri, R.M.; Schwaiger, T.; Penner, G.B.; Beauchemin, K.A.; Forster, R.J.; McKinnon, J.J.; McAllister, T.A. Changes in the rumen epimural bacterial diversity of beef cattle as affected by diet and induced ruminal acidosis. Appl. Environ. Microbiol. 2013, 79, 3744–3755. [Google Scholar] [CrossRef] [Green Version]
- González, L.A.; Manteca, X.; Calsamiglia, S.; Schwartzkopf-Genswein, K.S.; Ferret, A. Ruminal acidosis in feedlot cattle: Interplay between feed ingredients, rumen function and feeding behavior (a review). Anim. Feed Sci. Technol. 2012, 172, 66–79. [Google Scholar] [CrossRef]
- Khiaosa-ard, R.; Zebeli, Q. Diet-induced inflammation: From gut to metabolic organs and the consequences for the health and longevity of ruminants. Res. Vet. Sci. 2018, 120, 17–27. [Google Scholar] [CrossRef]
- Zebeli, Q.; Metzler-Zebeli, B.U. Interplay between rumen digestive disorders and diet-induced inflammation in dairy cattle. Res. Vet. Sci. 2012, 93, 1099–1108. [Google Scholar] [CrossRef]
- Sun, Y.Z.; Mao, S.Y.; Zhu, W.Y. Rumen chemical and bacterial changes during stepwise adaptation to a high-concentrate diet in goats. Animal 2010, 4, 210–217. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.C.; Zhou, X.L.; Zhao, P.T.; Gao, M.; Han, H.; Hu, H.L. Effects of Increasing Non-Fiber Carbohydrate to Neutral Detergent Fiber Ratio on Rumen Fermentation and Microbiota in Goats. J. Integr. Agric. 2013, 12, 319–326. [Google Scholar] [CrossRef]
- Huo, W.; Zhu, W.; Mao, S. Impact of subacute ruminal acidosis on the diversity of liquid and solid-associated bacteria in the rumen of goats. World J. Microbiol. Biotechnol. 2014, 30, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Wetzels, S.U.; Mann, E.; Metzler-Zebeli, B.U.; Wagner, M.; Klevenhusen, F.; Zebeli, Q.; Schmitz-Esser, S. Pyrosequencing reveals shifts in the bacterial epimural community relative to dietary concentrate amount in goats. J. Dairy Sci. 2015, 98, 5572–5587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.H.; Bian, G.R.; Zhu, W.Y.; Mao, S.Y. High-grain feeding causes strong shifts in ruminal epithelial bacterial community and expression of Toll-like receptor genes in goats. Front. Microbiol. 2015, 6, 167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metzler-Zebeli, B.U.; Schmitz-Esser, S.; Klevenhusen, F.; Podstatzky-Lichtenstein, L.; Wagner, M.; Zebeli, Q. Grain-rich diets differently alter ruminal and colonic abundance of microbial populations and lipopolysaccharide in goats. Anaerobe 2013, 20, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Ye, H.; Liu, J.; Mao, S. High-grain diets altered rumen fermentation and epithelial bacterial community and resulted in rumen epithelial injuries of goats. Appl. Microbiol. Biotechnol. 2017, 101, 6981–6992. [Google Scholar] [CrossRef]
- Grilli, D.J.; Fliegerová, K.; Kopečný, J.; Lama, S.P.; Egea, V.; Sohaefer, N.; Pereyra, C.; Ruiz, M.S.; Sosa, M.A.; Arenas, G.N.; et al. Analysis of the rumen bacterial diversity of goats during shift from forage to concentrate diet. Anaerobe 2016, 42, 17–26. [Google Scholar] [CrossRef]
- Liu, K.; Xu, Q.; Wang, L.; Wang, J.; Guo, W.; Zhou, M. The impact of diet on the composition and relative abundance of rumen microbes in goat. Asian-Australas. J. Anim. Sci. 2017, 30, 531–537. [Google Scholar] [CrossRef]
- Zhang, J.; Shi, H.; Wang, Y.; Li, S.; Cao, Z.; Ji, S.; He, Y.; Zhang, H. Effect of dietary forage to concentrate ratios on dynamic profile changes and interactions of ruminal microbiota and metabolites in holstein heifers. Front. Microbiol. 2017, 8, 2206. [Google Scholar] [CrossRef] [Green Version]
- Tedersoo, L.; Sánchez-Ramírez, S.; Kõljalg, U.; Bahram, M.; Döring, M.; Schigel, D.; May, T.; Ryberg, M.; Abarenkov, K. High-level classification of the Fungi and a tool for evolutionary ecological analyses. Fungal Divers. 2018, 90, 135–159. [Google Scholar] [CrossRef] [Green Version]
- Engelking, L.R. Overview of carbohydrate metabolism. In Textbook of Veterinary Physiological Chemistry; Academic Press: Amsterdam, The Netherlands, 2015; pp. 120–124. ISBN 978-0-12-384852-9. [Google Scholar]
- Ljungdahl, L.G. The cellulase/hemicellulase system of the anaerobic fungus Orpinomyces PC-2 and aspects of its applied use. Ann. N. Y. Acad. Sci. 2008, 1125, 308–321. [Google Scholar] [CrossRef]
- Couger, M.B.; Youssef, N.H.; Struchtemeyer, C.G.; Liggenstoffer, A.S.; Elshahed, M.S. Transcriptomic analysis of lignocellulosic biomass degradation by the anaerobic fungal isolate Orpinomyces sp. strain C1A. Biotechnol. Biofuels 2015, 8, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solomon, K.V.; Haitjema, C.H.; Henske, J.K.; Gilmore, S.P.; Borges-Rivera, D.; Lipzen, A.; Brewer, H.M.; Purvine, S.O.; Wright, A.T.; Theodorou, M.K.; et al. Early-branching gut fungi possess large, comprehensive array of biomass-degrading enzymes. Science 2016, 351, 1192–1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orpin, C.G. Anaerobic fungi: Taxonomy, biology and distribution in nature. In Anaerobic Fungi: Biology, Ecology and Function; Mountford, D.O., Orpin, C.G., Eds.; Marcel Dekker: New York, NY, USA, 1994; pp. 1–46. [Google Scholar]
- Silanikove, N. The physiological basis of adaptation in goats to harsh environments. Small Rumin. Res. 2000, 35, 181–193. [Google Scholar] [CrossRef]
- Li, D.B.; Hou, X.Z. Effect of fungal elimination on bacteria and protozoa populations and degradation of straw dry matter in the rumen of sheep and goats. Asian-Australas. J. Anim. Sci. 2007, 20, 70–74. [Google Scholar] [CrossRef]
- Joshi, A.; Lanjekar, V.B.; Dhakephalkar, P.K.; Callaghan, T.M.; Griffith, G.W.; Dagar, S.S. Liebetanzomyces polymorphus gen. et sp. nov., a new anaerobic fungus (Neocallimastigomycota) isolated from the rumen of a goat. MycoKeys 2018, 40, 89–110. [Google Scholar] [CrossRef]
- Hanafy, R.A.; Lanjekar, V.B.; Dhakephalkar, P.K.; Callaghan, T.M.; Dagar, S.S.; Griffith, G.W.; Elshahed, M.S.; Youssef, N.H. Seven new Neocallimastigomycota genera from wild, zoo-housed, and domesticated herbivores greatly expand the taxonomic diversity of the phylum. Mycologia 2020, 112, 1212–1239. [Google Scholar] [CrossRef]
- Hanafy, R.A.; Elshahed, M.S.; Liggenstoffer, A.S.; Griffith, G.W.; Youssef, N.H. Pecoramyces ruminantium, gen. nov., sp. nov., an anaerobic gut fungus from the feces of cattle and sheep. Mycologia 2017, 109, 231–243. [Google Scholar] [CrossRef]
- Hanafy, R.A.; Elshahed, M.S.; Youssef, N.H. Feramyces austinii, gen. nov., sp. nov., an anaerobic gut fungus from rumen and fecal samples of wild barbary sheep and fallow deer. Mycologia 2018, 110, 513–525. [Google Scholar] [CrossRef]
- Stabel, M.; Hanafy, R.A.; Schweitzer, T.; Greif, M.; Aliyu, H.; Flad, V.; Young, D.; Lebuhn, M.; Elshahed, M.S.; Ochsenreither, K.; et al. Aestipascuomyces dupliciliberans gen. nov, sp. nov., the first cultured representative of the uncultured SK4 clade from aoudad sheep and alpaca. Microorganisms 2020, 8, 1734. [Google Scholar] [CrossRef]
- Ho, Y.; Barr, D.J.S.; Abdullah, N.; Jalaludin, S.; Kudo, H. Piromyces spiralis, a new species of anaerobic fungus from the rumen of goat. Mycotaxon 1993, 48, 59–68. [Google Scholar]
- Li, G.J.; Hyde, K.D.; Zhao, R.L.; Hongsanan, S.; Abdel-Aziz, F.A.; Abdel-Wahab, M.A.; Alvarado, P.; Alves-Silva, G.; Ammirati, J.F.; Ariyawansa, H.A.; et al. Fungal diversity notes 253–366: Taxonomic and phylogenetic contributions to fungal taxa. Fungal Divers. 2016, 78, 1–237. [Google Scholar] [CrossRef]
- Hanafy, R.A.; Johnson, B.; Elshahed, M.S.; Youssef, N.H. Anaeromyces contortus, sp. nov., a new anaerobic gut fungal species (Neocallimastigomycota) isolated from the feces of cow and goat. Mycologia 2018, 110, 502–512. [Google Scholar] [CrossRef] [PubMed]
- Ha, J.K.; Lee, S.S.; Gao, Z.; Kim, C.H.; Kim, S.W.; Ko, J.Y.; Cheng, K.J. The effect of saturates fatty acid on cellulose digestion by the rumen anaerobic fungus, Neocallimastix frontalis C5-1. Asian-Australas. J. Anim. Sci. J. Anim. Sci 2001, 14, 941–946. [Google Scholar] [CrossRef]
- Nagpal, R.; Puniya, A.K.; Sehgal, J.P.; Singh, K. In vitro fibrolytic potential of anaerobic rumen fungi from ruminants and non-ruminant herbivores. Mycoscience 2011, 52, 31–38. [Google Scholar] [CrossRef]
- Thareja, A.; Puniya, A.K.; Goel, G.; Nagpal, R.; Sehgal, J.P.; Singh, P.K.; Singh, K. In vitro degradation of wheat straw by anaerobic fungi from small ruminants. Arch. Anim. Nutr. 2006, 60, 412–417. [Google Scholar] [CrossRef]
- Leis, S.; Dresch, P.; Peintner, U.; Fliegerová, K.; Sandbichler, A.M.; Insam, H.; Podmirseg, S.M. Finding a robust strain for biomethanation: Anaerobic fungi (Neocallimastigomycota) from the Alpine ibex (Capra ibex) and their associated methanogens. Anaerobe 2014, 29, 34–43. [Google Scholar] [CrossRef]
- Liggenstoffer, A.S.; Youssef, N.H.; Couger, M.B.; Elshahed, M.S. Phylogenetic diversity and community structure of anaerobic gut fungi (phylum Neocallimastigomycota) in ruminant and non-ruminant herbivores. ISME J. 2010, 4, 1225–1235. [Google Scholar] [CrossRef]
- Vinzelj, J.; Joshi, A.; Insam, H.; Podmirseg, S.M. Employing anaerobic fungi in biogas production: Challenges & opportunities. Bioresour. Technol. 2020, 300, 122687. [Google Scholar] [CrossRef]
- Kok, C.M.; Sieo, C.C.; Tan, H.Y.; Saad, W.Z.; Liang, J.B.; Ho, Y.W. Anaerobic cellulolytic rumen fungal populations in goats fed with and without Leucaena leucocephala hybrid, as determined by real-time PCR. J. Microbiol. 2013, 51, 700–703. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Luo, H.; Meng, K.; Wang, Y.; Huang, H.; Shi, P.; Pan, X.; Yang, P.; Diao, Q.; Zhang, H.; et al. High genetic diversity and different distributions of glycosyl hydrolase family 10 and 11 xylanases in the goat rumen. PLoS ONE 2011, 6, e16731. [Google Scholar] [CrossRef] [Green Version]
- FASS. Guide for the Care and Use of Agricultural Animals in Research and Teaching, 3rd ed.; Swanson, J.C., McGlone, J.J., Eds.; Federation of Animal Science Societies Inc.: Champaign, IL, USA, 2010. [Google Scholar]
- National Research Council (U.S.). Nutrient Requirements of Small Ruminants: Sheep, Goats, Cervids, and New World Camelids; National Academies Press: Washington, DC, USA, 2007.
- AOAC International. Official Methods of AOAC International, 17th ed.; Horwitz, W., Latimer, G.W., Eds.; AOAC International: Gaithersburg, MA, USA, 2005; ISBN 0-935584-77-3. [Google Scholar]
- Goering, H.K.; van Soest, P.J. Forage Fiber Analysis (Apparatus Reagents, Procedures and Some Applications); Agriculture Handbook; United States Department of Agriculture, Government Printing Office: Washington, DC, USA, 1970. [Google Scholar]
- Van Soest, P.J.; Robertson, J.B.; Lewis, B.A. Methods for Dietary Fiber, Neutral Detergent Fiber, and Nonstarch Polysaccharides in Relation to Animal Nutrition. J. Dairy Sci. 1991, 74, 3583–3597. [Google Scholar] [CrossRef]
- Crampton, E.W.; Maynard, L.A. The relation of cellulose and lignin content to the nutritive value of animal feeds. J. Nutr. 1938, 15, 383–395. [Google Scholar] [CrossRef]
- Aguilera, J.F. Contributions to the knowledge of the energy nutrition of small ruminants, with particular reference to the goat. Arch. Zootec. 2001, 50, 565–596. [Google Scholar]
- Yu, Z.; Morrison, M. Improved extraction of PCR-quality community DNA from digesta and fecal samples. Biotechniques 2004, 36, 808–812. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 2011, 108, 4516–4522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, T.J.; Bruns, T.D.; Lee, S.B.; Taylor, J.W. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In PCR Protocols: A Guide to Methods and Applications; Innis, M.A., Gelfand, D.H., Sninsky, J.J., White, T.J., Eds.; Academic Press: London, UK, 1990; pp. 315–322. [Google Scholar]
- Hupfauf, S.; Etemadi, M.; Fernández-Delgado Juárez, M.; Gómez-Brandón, M.; Insam, H.; Podmirseg, S.M. CoMA—An intuitive and user-friendly pipeline for amplicon-sequencing data analysis. PLoS ONE 2020, 15, e0243241. [Google Scholar] [CrossRef] [PubMed]
- Pauvert, C.; Buée, M.; Laval, V.; Edel-Hermann, V.; Fauchery, L.; Gautier, A.; Lesur, I.; Vallance, J.; Vacher, C. Bioinformatics matters: The accuracy of plant and soil fungal community data is highly dependent on the metabarcoding pipeline. Fungal Ecol. 2019, 41, 23–33. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Abarenkov, K.; Zirk, A.; Piirmann, T.; Pöhönen, R.; Ivanov, F.; Nilsson, R.; Henrik, R.H.; Kõljalg, U. UNITE General FASTA Release for Fungi 2; Version 04.02.2020; UNITE Community: London, UK, 2020. [Google Scholar]
- Lee, M. Happy Belly Bioinformatics: An open-source resource dedicated to helping biologists utilize bioinformatics. J. Open Source Educ. 2019, 2, 53. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 1–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardes, M.; Bruns, T.D. ITS primers with enhanced specificity for Basidiomycetes—Application to the identification of mycorrhizas and rusts. Mol. Ecol. 1993, 2, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.E.; Kingston-Smith, A.H.; Jimenez, H.R.; Huws, S.A.; Skøt, K.P.; Griffith, G.W.; McEwan, N.R.; Theodorou, M.K. Dynamics of initial colonization of nonconserved perennial ryegrass by anaerobic fungi in the bovine rumen. FEMS Microbiol. Ecol. 2008, 66, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Mura, E.; Edwards, J.; Kittelmann, S.; Kaerger, K.; Voigt, K.; Mrázek, J.; Moniello, G.; Fliegerova, K. Anaerobic fungal communities differ along the horse digestive tract. Fungal Biol. 2019, 123, 240–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagler, M.; Podmirseg, S.M.; Griffith, G.W.; Insam, H.; Ascher-Jenull, J. The use of extracellular DNA as a proxy for specific microbial activity. Appl. Microbiol. Biotechnol. 2018, 102, 2885–2898. [Google Scholar] [CrossRef] [Green Version]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Koetschan, C.; Kittelmann, S.; Lu, J.; Al-Halbouni, D.; Jarvis, G.N.; Müller, T.; Wolf, M.; Janssen, P.H. Internal transcribed spacer 1 secondary structure analysis reveals a common core throughout the anaerobic fungi (Neocallimastigomycota). PLoS ONE 2014, 9, e91928. [Google Scholar] [CrossRef] [Green Version]
- Øvreås, L.; Forney, L.; Daae, F.L.; Torsvik, V. Distribution of bacterioplankton in meromictic lake Saelenvannet, as determined by denaturing gradient gel electrophoresis of PCR-amplified gene fragments coding for 16S rRNA. Appl. Environ. Microbiol. 1997, 63, 3367–3373. [Google Scholar] [CrossRef] [Green Version]
- Stahl, D.A.; Amann, R. Development and application of nucleic acid probes in bacterial systematics. In Nucleic Acid Techniques in Bacterial Systematics; Stackebrandt, E., Goodfellow, M., Eds.; Wiley & Sons Ltd.: Chichester, UK, 1991; pp. 205–248. [Google Scholar]
- Kittelmann, S.; Naylor, G.E.; Koolaard, J.P.; Janssen, P.H. A proposed taxonomy of anaerobic fungi (class neocallimastigomycetes) suitable for large-scale sequence-based community structure analysis. PLoS ONE 2012, 7, e36866. [Google Scholar] [CrossRef]
- Henderson, G.; Cox, F.; Ganesh, S.; Jonker, A.; Young, W.; Janssen, P.H.; Abecia, L.; Angarita, E.; Aravena, P.; Arenas, G.N.; et al. Rumen microbial community composition varies with diet and host, but a core microbiome is found across a wide geographical range. Sci. Rep. 2015, 5, 14567. [Google Scholar] [CrossRef] [PubMed]
- Gruninger, R.J.; Ribeiro, G.O.; Cameron, A.; McAllister, T.A. Invited review: Application of meta-omics to understand the dynamic nature of the rumen microbiome and how it responds to diet in ruminants. Animal 2019, 13, 1843–1854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guevara, J.C.; Grünwaldt, E.G.; Estevez, O.R.; Bisigato, A.J.; Blanco, L.J.; Biurrun, F.N.; Ferrando, C.A.; Chirino, C.C.; Morici, E.; Fernández, B.; et al. Range and livestock production in the Monte Desert, Argentina. J. Arid Environ. 2009, 73, 228–237. [Google Scholar] [CrossRef]
- Allegretti, L.; Sartor, C.; Paez Lama, S.; Egea, V.; Fucili, M.; Passera, C. Effect of the physiological state of Criollo goats on the botanical composition of their diet in NE Mendoza, Argentina. Small Rumin. Res. 2012, 103, 152–157. [Google Scholar] [CrossRef]
- Klevenhusen, F.; Hollmann, M.; Podstatzky-Lichtenstein, L.; Krametter-Frötscher, R.; Aschenbach, J.R.; Zebeli, Q. Feeding barley grain-rich diets altered electrophysiological properties and permeability of the ruminal wall in a goat model. J. Dairy Sci. 2013, 96, 2293–2302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khafipour, E.; Li, S.; Plaizier, J.C.; Krause, D.O. Rumen microbiome composition determined using two nutritional models of subacute ruminal acidosis. Appl. Environ. Microbiol. 2009, 75, 7115–7124. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.; Jeong, S.; Seo, J.; Seo, S. Changes in the ruminal fermentation and bacterial community structure by a sudden change to a high-concentrate diet in Korean domestic ruminants. Asian-Australas. J. Anim. Sci. 2019, 32, 92–102. [Google Scholar] [CrossRef] [Green Version]
- Han, X.; Yang, Y.; Yan, H.; Wang, X.; Qu, L.; Chen, Y. Rumen bacterial diversity of 80 to 110-day-old goats using 16S rRNA sequencing. PLoS ONE 2015, 10, e0117811. [Google Scholar] [CrossRef]
- Wang, L.; Xu, Q.; Kong, F.; Yang, Y.; Wu, D.; Mishra, S.; Li, Y. Exploring the goat rumen microbiome from seven days to two years. PLoS ONE 2016, 11, e0154354. [Google Scholar] [CrossRef] [Green Version]
- Hua, C.; Tian, J.; Tian, P.; Cong, R.; Luo, Y.; Geng, Y.; Tao, S.; Ni, Y.; Zhao, R. Feeding a high concentration diet induces unhealthy alterations in the composition and metabolism of ruminal microbiota and host response in a goat model. Front. Microbiol. 2017, 8, 138. [Google Scholar] [CrossRef] [Green Version]
- Cunha, I.S.; Barreto, C.C.; Costa, O.Y.A.; Bomfim, M.A.; Castro, A.P.; Kruger, R.H.; Quirino, B.F. Bacteria and Archaea community structure in the rumen microbiome of goats (Capra hircus) from the semiarid region of Brazil. Anaerobe 2011, 17, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Elekwachi, C.O.; Jiao, J.; Wang, M.; Tang, S.; Zhou, C.; Tan, Z.; Forster, R.J. Investigation and manipulation of metabolically active methanogen community composition during rumen development in black goats. Sci. Rep. 2017, 7, 422. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi, S.H.; Valizade, R.; Heidarian Miri, V. Rumen microbial community of Saanen goats adapted to a high-fiber diet in the Northeast of Iran. Iran. J. Appl. Anim. Sci. 2018, 8, 271–279. [Google Scholar]
- Kumar, S.; Indugu, N.; Vecchiarelli, B.; Pitta, D.W. Associative patterns among anaerobic fungi, methanogenic archaea, and bacterial communities in response to changes in diet and age in the rumen of dairy cows. Front. Microbiol. 2015, 6, 781. [Google Scholar] [CrossRef] [Green Version]
- Jin, W.; Cheng, Y.F.; Mao, S.Y.; Zhu, W.Y. Isolation of natural cultures of anaerobic fungi and indigenously associated methanogens from herbivores and their bioconversion of lignocellulosic materials to methane. Bioresour. Technol. 2011, 102, 7925–7931. [Google Scholar] [CrossRef]
- Belanche, A.; Kingston-Smith, A.H.; Griffith, G.W.; Newbold, C.J. A multi-kingdom study reveals the plasticity of the rumen microbiota in response to a shift from non-grazing to grazing diets in sheep. Front. Microbiol. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Deusch, S.; Camarinha-Silva, A.; Conrad, J.; Beifuss, U.; Rodehutscord, M.; Seifert, J. A structural and functional elucidation of the rumen microbiome influenced by various diets and microenvironments. Front. Microbiol. 2017, 8, 1605. [Google Scholar] [CrossRef] [Green Version]
- Ishaq, S.L.; AlZahal, O.; Walker, N.; McBride, B. An investigation into rumen fungal and protozoal diversity in three rumen fractions, during high-fiber or grain-induced sub-acute ruminal acidosis conditions, with or without active dry yeast supplementation. Front. Microbiol. 2017, 8, 1943. [Google Scholar] [CrossRef] [Green Version]
- Alexander, H.M. Disease in natural plant populations, communities, and ecosystems: Insights into ecological and evolutionary processes. Plant Dis. 2010, 94, 492–503. [Google Scholar] [CrossRef] [Green Version]
- Gallo, A.; Giuberti, G.; Frisvad, J.C.; Bertuzzi, T.; Nielsen, K.F. Review on mycotoxin issues in ruminants: Occurrence in forages, effects of mycotoxin ingestion on health status and animal performance and practical strategies to counteract their negative effects. Toxins 2015, 7, 3057–3111. [Google Scholar] [CrossRef]
- Denman, S.E.; Nicholson, M.J.; Brookman, J.L.; Theodorou, M.K.; McSweeney, C.S. Detection and monitoring of anaerobic rumen fungi using an ARISA method. Lett. Appl. Microbiol. 2008, 47, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Tapio, I.; Fischer, D.; Blasco, L.; Tapio, M.; Wallace, R.J.; Bayat, A.R.; Ventto, L.; Kahala, M.; Negussie, E.; Shingfield, K.J.; et al. Taxon abundance, diversity, co-occurrence and network analysis of the ruminal microbiota in response to dietary changes in dairy cows. PLoS ONE 2017, 12, 1–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boots, B.; Lillis, L.; Clipson, N.; Petrie, K.; Kenny, D.A.; Boland, T.M.; Doyle, E. Responses of anaerobic rumen fungal diversity (phylum Neocallimastigomycota) to changes in bovine diet. J. Appl. Microbiol. 2013, 114, 626–635. [Google Scholar] [CrossRef] [PubMed]
- Hanafy, R.A.; Johnson, B.; Youssef, N.H.; Elshahed, M.S. Assessing anaerobic gut fungal diversity in herbivores using D1/D2 large ribosomal subunit sequencing and multi-year isolation. Environ. Microbiol. 2020, 22, 3883–3908. [Google Scholar] [CrossRef]
- Paul, S.S.; Bu, D.; Xu, J.; Hyde, K.D.; Yu, Z. A phylogenetic census of global diversity of gut anaerobic fungi and a new taxonomic framework. Fungal Divers. 2018, 89, 253–266. [Google Scholar] [CrossRef]
- Edwards, J.E.; Forster, R.J.; Callaghan, T.M.; Dollhofer, V.; Dagar, S.S.; Cheng, Y.; Chang, J.; Kittelmann, S.; Fliegerova, K.; Puniya, A.K.; et al. PCR and omics based techniques to study the diversity, ecology and biology of anaerobic fungi: Insights, challenges and opportunities. Front. Microbiol. 2017, 8, 1657. [Google Scholar] [CrossRef] [Green Version]
- Trinci, A.P.J.; Davies, D.R.; Gull, K.; Lawrence, M.I.; Bonde Nielsen, B.; Rickers, A.; Theodorou, M.K. Anaerobic fungi in herbivorous animals. Mycol. Res. 1994, 98, 129–152. [Google Scholar] [CrossRef]
- Orpin, C.G.; Joblin, K.N. The rumen anaerobic fungi. In The Rumen Microbial Ecosystem; Hobson, P.N., Stewart, C.S., Eds.; Springer: Dordrecht, Germany, 1997; pp. 140–195. ISBN 978-94-009-1453-7. [Google Scholar]
- Schoch, C.L.; Seifert, K.A.; Huhndorf, S.; Robert, V.; Spouge, J.L.; Levesque, C.A.; Chen, W.; Bolchacova, E.; Voigt, K.; Crous, P.W.; et al. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc. Natl. Acad. Sci. USA 2012, 109, 6241–6246. [Google Scholar] [CrossRef] [Green Version]
- Blaalid, R.; Kumar, S.; Nilsson, R.H.; Abarenkov, K.; Kirk, P.M.; Kauserud, H. ITS1 versus ITS2 as DNA metabarcodes for fungi. Mol. Ecol. Resour. 2013, 13, 218–224. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.C.; Liu, C.; Huang, L.; Bengtsson-Palme, J.; Chen, H.; Zhang, J.H.; Cai, D.; Li, J.Q. ITS1: A DNA barcode better than ITS2 in eukaryotes? Mol. Ecol. Resour. 2015, 15, 573–586. [Google Scholar] [CrossRef]
- Edwards, J.E.; Hermes, G.D.A.; Kittelmann, S.; Nijsse, B.; Smidt, H. Assessment of the Accuracy of High-Throughput Sequencing of the ITS1 Region of Neocallimastigomycota for Community Composition Analysis. Front. Microbiol. 2019, 10, 2317. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.H.; Su, J.H.; Shang, J.J.; Wu, Y.Y.; Li, Y.; Bao, D.P.; Yao, Y.J. Evaluation of the ribosomal DNA internal transcribed spacer (ITS), specifically ITS1 and ITS2, for the analysis of fungal diversity by deep sequencing. PLoS ONE 2018, 13, e0206428. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Diets | ||
|---|---|---|
| Alfalfa Hay | High Grain | |
| Total daily intake (g DM) | 1061 ± 173 a | 1252 ± 193 a |
| Daily food intake (g DM): | ||
| Alfalfa Hay | 1061 ± 173 a | 900 ± 193 a |
| Corn | - | 352 |
| Nutrient intake (g DM): | ||
| CP | 168 ± 27 a | 171 ± 30 a |
| NDF | 361 ± 59 a | 335 ± 65 a |
| ADF | 228 ± 37 a | 204 ± 41 a |
| ADL | 42 ± 7 a | 39 ± 7 a |
| Hemicellulose | 133 ± 22 a | 132 ± 24 a |
| Cellulose | 183 ± 30 a | 163 ± 33 a |
| Starch | 27 ± 4 a | 244 ± 5 b |
| Daily intake of ME (MCal kg DM−1) | 2.08 ± 0.34 a | 2.78 ± 0.38 b |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fliegerova, K.O.; Podmirseg, S.M.; Vinzelj, J.; Grilli, D.J.; Kvasnová, S.; Schierová, D.; Sechovcová, H.; Mrázek, J.; Siddi, G.; Arenas, G.N.; et al. The Effect of a High-Grain Diet on the Rumen Microbiome of Goats with a Special Focus on Anaerobic Fungi. Microorganisms 2021, 9, 157. https://doi.org/10.3390/microorganisms9010157
Fliegerova KO, Podmirseg SM, Vinzelj J, Grilli DJ, Kvasnová S, Schierová D, Sechovcová H, Mrázek J, Siddi G, Arenas GN, et al. The Effect of a High-Grain Diet on the Rumen Microbiome of Goats with a Special Focus on Anaerobic Fungi. Microorganisms. 2021; 9(1):157. https://doi.org/10.3390/microorganisms9010157
Chicago/Turabian StyleFliegerova, Katerina O., Sabine M. Podmirseg, Julia Vinzelj, Diego J. Grilli, Simona Kvasnová, Dagmar Schierová, Hana Sechovcová, Jakub Mrázek, Giuliana Siddi, Graciela N. Arenas, and et al. 2021. "The Effect of a High-Grain Diet on the Rumen Microbiome of Goats with a Special Focus on Anaerobic Fungi" Microorganisms 9, no. 1: 157. https://doi.org/10.3390/microorganisms9010157