Comparative Analysis of Carbohydrate Active Enzymes in the Flammulina velutipes var. lupinicola Genome


1
Department of Medicinal Biosciences, Research Institute for Biomedical & Health Science, College of Biomedical and Health Science, Konkuk University, 268 Chungwon-daero, Chungju-si 27478, Korea
2
Mushroom Research Division, National Institute of Horticultural and Herbal Science, Rural Development Administration, 92, Bisan-ro, Eumseong-gun 27709, Korea
*
Author to whom correspondence should be addressed.
Microorganisms 2021, 9(1), 20; https://doi.org/10.3390/microorganisms9010020
Submission received: 11 November 2020
/
Revised: 17 December 2020
/
Accepted: 18 December 2020
/
Published: 23 December 2020
(This article belongs to the Section Environmental Microbiology)
Abstract
:The purpose of this study was to determine the genome sequence of Flammulina velutipes var. lupinicola based on next-generation sequencing (NGS) and to identify the genes encoding carbohydrate-active enzymes (CAZymes) in the genome. The optimal assembly (71 kmer) based on ABySS de novo assembly revealed a total length of 33,223,357 bp (49.53% GC content). A total of 15,337 gene structures were identified in the F. velutipes var. lupinicola genome using ab initio gene prediction method with Funannotate pipeline. Analysis of the orthologs revealed that 11,966 (96.6%) out of the 15,337 predicted genes belonged to the orthogroups and 170 genes were specific for F. velutipes var. lupinicola. CAZymes are divided into six classes: auxiliary activities (AAs), glycosyltransferases (GTs), carbohydrate esterases (CEs), polysaccharide lyases (PLs), glycoside hydrolases (GHs), and carbohydrate-binding modules (CBMs). A total of 551 genes encoding CAZymes were identified in the F. velutipes var. lupinicola genome by analyzing the dbCAN meta server database (HMMER, Hotpep, and DIAMOND searches), which consisted of 54–95 AAs, 145–188 GHs, 55–73 GTs, 6–19 PLs, 13–59 CEs, and 7–67 CBMs. CAZymes can be widely used to produce bio-based products (food, paper, textiles, animal feed, and biofuels). Therefore, information about the CAZyme repertoire of the F. velutipes var. lupinicola genome will help in understanding the lignocellulosic machinery and in-depth studies will provide opportunities for using this fungus for biotechnological and industrial applications.
1. Introduction
Flammulina velutipes var. lupinicola (Physalacriaceae) was first identified in 1999 by Redhead and Petersen [1]. However, except for the following characteristics, its molecular biology and biological properties were not well known; first, basidiospores are larger (7−14.5 × 3.7–6.5 μm) than those of the typical F. velutipes variety. Secondly, it seems to be limited geographically (from southern to northern California) in ecologically distinctive zone (in costal dunes) largely on a specific host (Lupinus arboreus) native to the region. It has been suggested that F. velutipes var. lupinicola and typical varieties (F. velutipes var. velutipes) have only partial genetic differences. [1].
Enzymes, including carbohydrate esterases (CEs), glycoside hydrolases (GHs), polysaccharide lyases (PLs), glycosyltransferases (GTs), and auxiliary activities (AAs), are collectively known as carbohydrate-active enzymes (CAZymes), and these enzymes are involved in the catabolism of carbohydrates [2]. These enzymes have attracted attention in biotechnological and industrial applications as they can be used to produce bio-based products, including food, paper, textiles, animal feed and, especially, biofuels [3,4,5]. Many fungal species that exist extensively in nature, including basidiomycetes, can efficiently degrade plant lignocellulosic biomass as they possess many types of CAZymes [3,4,5]. This ability allows the fungi to inhabit a variety of natural environments. Among the various fungi present in nature, white-rotting basidiomycetes are generally known to be able to degrade both lignin and polysaccharides from plant sources [3,4,5]. Thus, the discovery and understanding of CAZymes from fungal species, including Basidiomycetes, will enable the use of these enzymes for relevant applications [3,4,5].
Currently, genome sequence analysis of various organisms is actively under way due to the advances in genome sequencing technology such as next-generation sequencing [3]. Out of many organisms that can be sequenced, several fungal species are commonly used for genome sequencing in order to discover various biomass-degrading enzymes and to understand the wood-degrading machinery in the fungal genomes [3]. For example, the genome of Phanerochaete chrysosporium (white rot basidiomycete) has been reported to possess a vast array of genes associated with the lignocellulolytic machinery [6]. In addition, we also previously reported the genome sequences of Flammulina velutipes [7], Flammulina elastica [8], Flammulina fennae [9], and Flammulina ononidis [10], and identified well-developed wood-degrading machineries containing various CAZymes. The study of biomass-degrading enzymes through genome sequence analysis is an active field of research to comprehensively understand the wood-degrading machinery of various fungal species in this genomic era.
In this study, to our knowledge, we have reported, for the first time, the genome sequence and a well-developed wood-degrading machinery of F. velutipes var. lupinicola. This information will potentially facilitate its applicability for biotechnological and industrial applications as well as help in understanding the potential biotechnological and industrial applications of this fungus.
2. Materials and Methods
2.1. Fungal Strain Culture and Genomic DNA Isolation
Flammulina velutipes var. lupinicola ASI4195 was obtained from the Mushroom Research Division, National Institute of Horticultural and Herbal Science (Rural Development Administration, Republic of Korea) and was grown at 25 °C on potato dextrose agar (PDA, 4 g potato starch, 20 g dextrose, 15 g agar per liter) for 15 days. For the genomic DNA extraction, the mycelia were frozen with liquid nitrogen and ground using a mortar and pestle. Extraction buffer (0.25 M Tris-HCl, 100 mM NaCl, 50 mM ethylenediaminetetraacetic acid, 5% SDS), 2 × CTAB buffer (100 mM Tris-HCl pH 8, 20 mM EDTA pH 8, 2% CTAB, 1.4 M NaCl, and 1% polyvinyl pyrrolidone), and phenol-chloroform-isoamylalcohol (25:24:1) were added to the mycelia and mixed. After centrifugation at 13.000 rpm at 4 °C for 5 min, the supernatant was mixed with 0.7 times its volume of isopropanol. This mixture was centrifuged for 15 min at 4 °C. After washing with 70% ethanol, the dried samples were dissolved in TE buffer and then treated with RNase A (Qiagen, Seoul, Korea). Final sample was quantified and validated using the NanoDrop ND1000 (NanoDrop Technologies, Inc., Wilminton, DE, USA) and the Agilent 2100 Bioanalyzer (Agilent Technologies Korea, Ltd., Seoul, Korea).
2.2. Genome Sequencing and Gene Modeling
Next-generation sequencing (NGS)-based genome sequencing of the F. velutipes var. lupinicola was performed using the HiSeq 2000 platform (Illumina, Inc., San Diego, CA, USA). Library preparation was performed using Paired-End DNA Sample Prep Kit (Illumina, Inc., San Diego, CA, USA) according to the manufacturer’s instructions. Raw reads (100 bp paired-end) were processed using FastQC software (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and Trimmomatic (version 0.32) software [11] for quality checks and adapter trimmings. The resultant short-reads were used for de novo assembly using ABySS software (size = 20 to 90 kmer) [12]. Gene prediction and annotation were carried out using the Funannotate pipelines version 1.7.2 (AUGUSTUS, Codingquary, GeneMark, gilmmerhmm, and SNAP for predictions; Pfam, Uniprot, BUSCOS, Protease, and NCBI_NR for annotations) [13], which was trained using the strain, F. velutipes KACC42780 with transcriptome data as hints file. The predicted gene models of F. velutipes var. lupinicola were annotated using DIAMOND [14] software with the non-redundant database from the National Center for Biotechnology Information (NCBI).
2.3. Ortholog Clustering
F. velutipes var. lupinicola genes were analyzed by OrthoFinder (version 2.3.3) software [15] for orthologous groups clustering with the following fungal species; F. elastica KACC46182 [8], F. fennae KACC46185 [9], F. ononidis KACC46186 [10], F. velutipes KACC42780 [7], Aspergillus nidulans FGSC-A4 [16], Botrytis cinerea B05.10 [17], Laccaria bicolor S238N-H82 [18], Agaricus bisporus var. bisporus H97 [19], Coprinopsis cinerea okayama 7#130 [20], Lentinula edodes [21], Cordyceps militaris CM01 [22], Cryptococcus neoformans var. grubii H99 [23], P. chrysosporium RP78 [6], Saccharomyces cerevisiae S288C [24], Neurospora crassa OR74A [25], Schizophyllum commune H4-8 [26], Trichoderma reesei QM6a [27], and Ustilago maydis 521 [28].
2.4. Prediction of CAZymes and Signal Peptides
The putative genes encoding for the CAZymes, including GH, PL, CE, GT, AA, and CBM genes in F. velutipes var. lupinicola, were identified using the dbCAN meta server (http://bcb.unl.edu/dbCAN2/) including the dbCAN CAZyme domain (by HMMER search), short conserved motifs (by Hotpep search), and CAZy databases (by DIAMOND search) [29]. The predicted genes encoding for CAZymes were further processed using the SignalP 5.0 software [30] to look for signal peptides.
2.5. Data Availability
Raw sequencing reads were deposited in the NCBI Sequence Read Archive (SRA) under the accession number SRR9964157 (SAMN12569251, PRJNA560135).
3. Results and Discussion
3.1. Genome Sequence Assembly, Gene Modeling, and Genome Comparison
The quality checked reads (38,416,296 reads, >Q30) were derived from the raw reads (41,592,600 reads) and were used for de novo assembly using the Abyss software [12]. The resultant optimized assembly (71 kmer) consisted of 2570 sequence contigs with a total length of 33,223,357 bp (49.53% GC content) and N50 length of 48,981 bp. A total of 15,337 gene models, with an average gene length of 1122 bp, were predicted from the assembled contigs using the Funannotate pipeline [13]. The average exon and intron lengths were 24,466 and 5367 nucleotides, respectively. The general features of the F. velutipes var. lupinicola ASI4195 genome are presented in Table 1.
Out of the 15,337 predicted genes, 82.1% (12,600) had sequence similarity (0.001 > e-value) with the genes in NCBI-NR database (Table S1). The total number of genes in F. velutipes var. lupinicola was comparable to that of its closest sequenced species, Flammulina species, as well as to those of other basidiomycetes with a similar genome size (Table 2).
We conducted cluster analysis with other sequenced fungal species and identified 8431 groups containing at least one F. velutipes var. lupinicola protein (Table 3 and Table S2). Analysis of these clusters suggested that 47.2% of F. velutipes var. lupinicola proteins had orthologs belonging to Dikarya, and hence were conserved in basidiomycetes and ascomycetes. Among the set of homologous genes, there were 70 species-specific orthogroups containing 170 species-specific genes in F. velutipes var. lupinicola (Table 3 and Table S2).
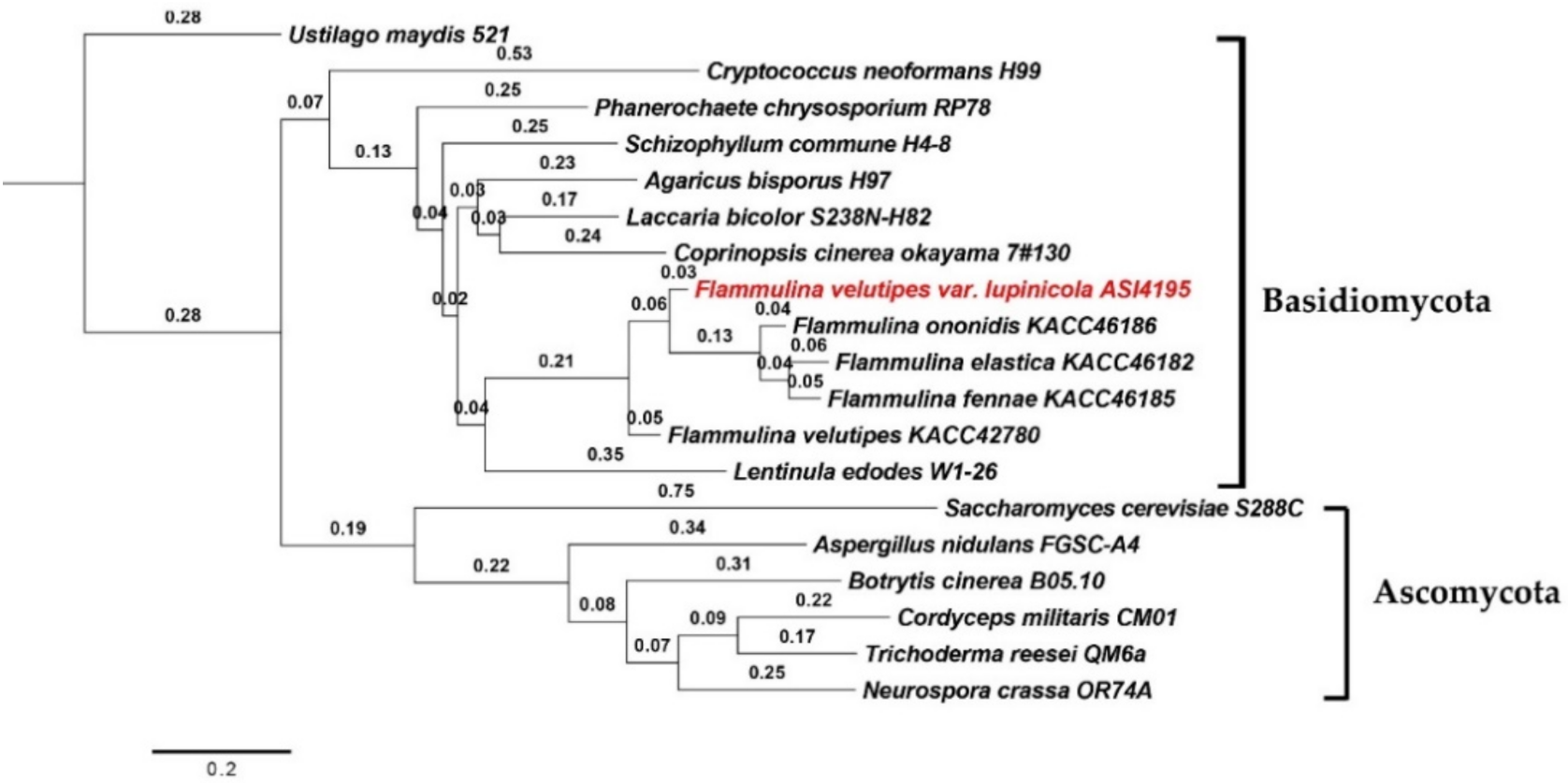
Ortholog analysis also revealed that, F. velutipes var. lupinicola was classified into one group with F. ononidis, F. fennae, and F. elastica and clustered into another group together with and F. velutipes and L. edodes by ortholog-based clustering analysis (Figure 1).
3.2. F. velutipes var. lupinicola and other Fungal Species CAZymes
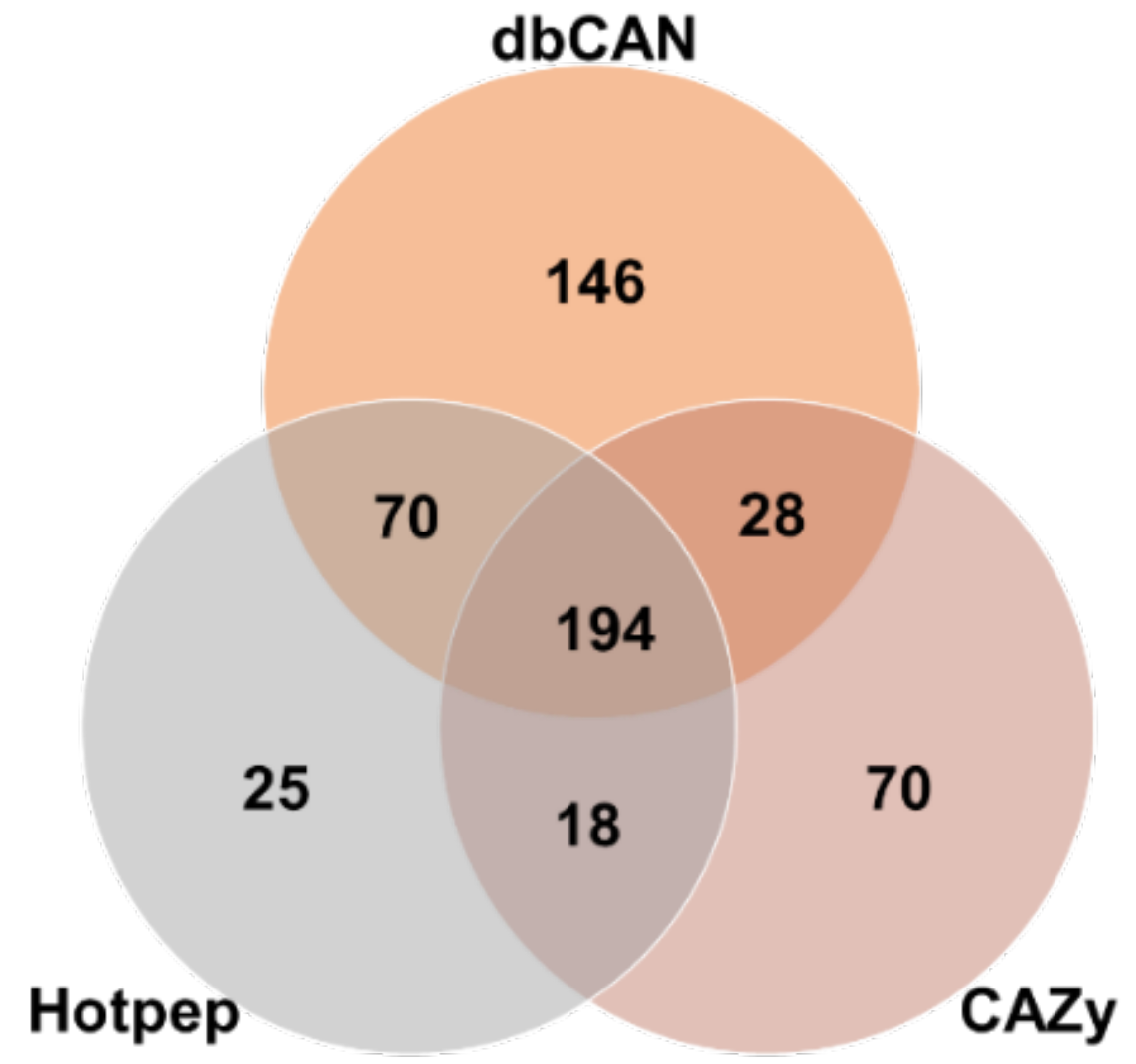
The genome sequence of F. velutipes var. lupinicola revealed a series of genes involved in the assembly (GT) and breakdown (GHs, PLs, CEs) of carbohydrate complexes. In addition, genes related to lignin degradation (auxiliary activity; AA) and carbohydrate binding module (CBM) were identified in the F. velutipes var. lupinicola genome. Annotation of the predicted genes of F. velutipes var. lupinicola using the dbCAN meta server (http://bcb.unl.edu/dbCAN2/) [29] revealed 551 genes encoding for CAZymes, including 439 from dbCAN (HMMER), 360 from Hotpep, and 336 from the CAZy database (DIAMOND) (Figure 2 and Table 4). Among the 551 genes, some genes were annotated as different CAZymes depending on the database or predicted to simultaneously encode for two different CAZymes (Table S3). Therefore, 360–439 CAZymes, including 54–95 AAs, 145–188 GHs, 55–73 GTs, 6–19 PLs, 13–59 CEs, and 7–67 CBMs were identified in the F. velutipes var. lupinicola genome (Table S4).
3.2.1. Glycosyltransferases (GTs)
GT is an enzyme that catalyzes the transfer of glycosyl groups to the specific acceptor molecules and utilizes activated donor sugar phosphates to form glycosidic bond (EC 2.4.x.y), which is involved in the biosynthesis of glycoconjugates, oligosaccharides, and polysaccharides [31,32,33,34].
CAZyme annotation revealed that F. velutipes var. lupinicola contains a total of 55 GT families encoded by 95 genes in its genome sequence (Figure 3 and Table S3). Among the predicted GTs in the F. velutipes var. lupinicola genome, the GT2 family with 10–17 genes was the most prominent one (Figure 3 and Table S4). Several GT2 families have been identified in 18 other fungal genomes, including 12 species of basidiomycetes and six species of ascomycetes. Genome-wide comparisons of GT families indicated that the GT2 family was also prominent, suggesting that the GT2 family is a major component of the GT family in most fungal species (Table S4). Moreover, genome sequencing of bacterial, archaeal, and eukaryotic organisms has revealed that there are a large number of genes encoding GTs (about 1–2% of gene products) in their genomes [2]. Breton et al. [31] suggested that the number of families might increase with the incorporation of newly discovered GT genes, and not all sequences encoding GT were present in public databases. At the time of writing (August 2020), GT2 and GT4 were listed in the CAZy database and account for about half of the total number of GTs, with more than 740,000 classified and 17,000 unclassified GT sequences classified into 111 families (CAZy database; http://www.cazy.org/).
GT, which is a membrane protein located mainly in endoplasmic reticulum and Golgi apparatus, has a C-terminal catalytic domain, an N-terminal cytoplasmic tail, and a signal-anchor domain that consists of a non-cleavable signal peptide [35,36]. Thus, the signal peptides predicted in 6 out of the 95 GT genes in the in F. velutipes var. lupinicola genome suggest that these signatures are likely to act as signal-anchor domains (Table S3).
Previous studies have demonstrated the difficulty in classifying GTs based on sequence similarity as many GTs have different activities even though the GT sequences are very similar [37]. In this study, six genes from the F. velutipes var. lupinicola genome were annotated as GT0 family (not yet assigned to the family) based on amino acid sequence similarities (Table S3). This indicates that further studies based on structural and mutational analysis of these genes annotated with the GT0 family are needed to characterize their enzymatic properties.
3.2.2. Carbohydrate Esterases (CEs)
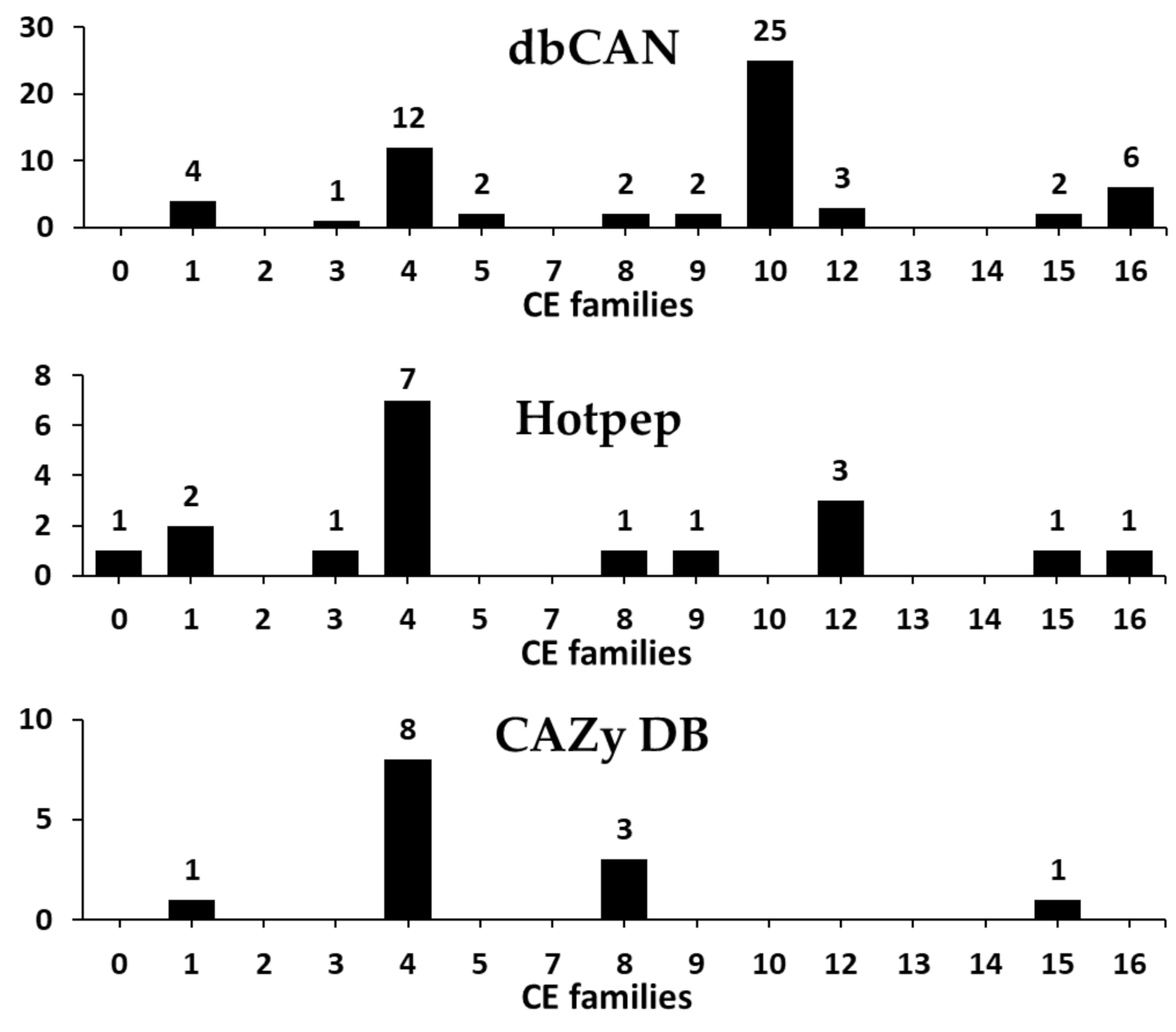
CEs represent a class of esterases which catalyze N-de- or O-deacylation to remove esters from substituted saccharides and are widely used as biocatalysts in industrial processes as well as in biotechnological applications [38,39,40]. CEs have a wide variety of substrate specificities, such as specificity for feruloyl-polysaccharide (feruloyl esterases, EC 3.1.1.73), xylan (acetylxylan esterases, EC 3.1.1.72), acetic ester (acetyl esterases, EC 3.1.1.6), peptidoglycan (poly-N-acetylglucosamine deacetylases, chitin (chitin deacetylases, EC 3.5.1.41), EC 3.5.1.104), and pectin (pectinesterase, EC 3.1.1.11) [41]. These CEs are currently classified into 18 families in the CAZy database (CAZy database; http://www.cazy.org/), including more than 87,000 classified and 1700 non-classified CEs.
Our results revealed a total of 61 predicted CEs classified into 11 families in the F. velutipes var. lupinicola genome based on the dbCAN meta server search (Figure 4 and Table S4). CE10 family was prominent, with 25 CEs, and the CE4 family was the second largest family with 12 CEs in the F. velutipes var. lupinicola genome (Figure 4). Genome-wide comparisons of CEs revealed that the total number of CEs in F. velutipes var. lupinicola was similar to that found in other basidiomycetes, including Flammulina species, C. cinerea, and S. commune, with 57 to 63 CEs (Table S4). In addition, CE1, −4, and −16 families are prominent in several basidiomycetes (Table S4). Furthermore, basidiomycetes and ascomycetes were found to vary in the number of CE families, as only five CEs are known in Cryptococcus neoformans and two in S. cerevisiae (Table S4). Although CAZyme predictions have found a number of genes encoding for the CE10 family members in the F. velutipes var. lupinicola genome, most members of the CE10 family have been reported to act on non-carbohydrate substrates [2,42].
Among the many enzymes identified and classified as CE, some characteristic features have been identified in the amino acid sequence. Members of the CE1, CE4, CE5, and CE7 families have been reported to possess the GXSXG (Gly-Xaa-Ser-Xaa-Gly) conserved motif, as well as the Ser-His-Asp catalytic triad. It has been also reported that CE2 and CE3 family members possess the Gly-Asp-Ser-(Leu) (GDS (L)) motif in their amino acid sequence. In addition, CE16 family members also possess the Ser-Gly-Asn-His catalyst residues and GDS (L) catalytic motif [43]. In the present study, 12 CEs were found to possess conserved motifs, such as GXSXG (Table 5). Some CE family members have been found to have Gly-Xaa-Xaa-Leu (GXXL) motifs, which are generally found in esterases that show high homology to class C β-lactamases [44,45]. In addition, in the present study, 11 of the 12 genes that were predicted to encode CE4 family members were found to have a conserved sequence (Phe-Asp-Asp-Gly-Pro) and further studies are needed to elucidate the biochemical role of this motif (Table 5).
3.2.3. Glycoside Hydrolases (GHs)
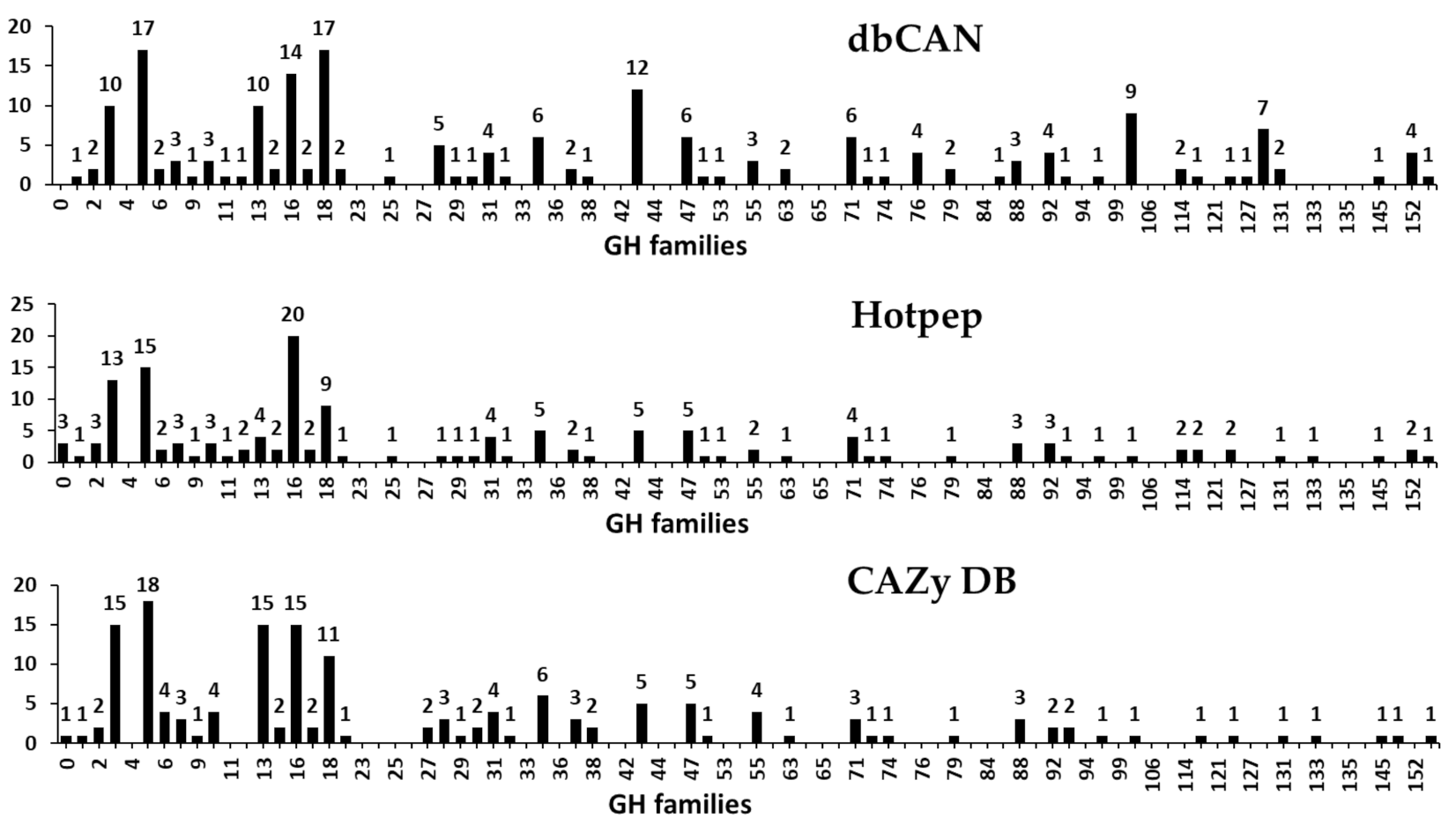
GHs (glycosidases or glycosyl hydrolases, EC 3.2.1.x) catalyze the hydrolysis of glycosidic bonds of complex carbohydrates such as cellulose, hemicellulose, and starch [46,47]. Previously, Henrissat [47] classified GHs into 35 families by comparing 301 amino acid sequences. Currently, 168 families with more than 874,000 classified and 17,000 unclassified GH sequences are listed in the CAZy database (http://www.cazy.org/). In the present study, a total of 246 GHs classified into 55 families were predicted in the F. velutipes var. lupinicola genome based on the dbCAN meta server search (Figure 5 and Table S4). GH family classification also revealed that the GH16 family was the most prominent one with 20 genes in the F. velutipes var. lupinicola genome as those in other fungal species. In addition, multiple copies of GH5 and GH18 found in the F. velutipes var. lupinicola genome were similar to those in other basidiomycetes (Figure 5 and Table S4).
GH16 family comprises a number of enzymes with known activities, such as lichenase (EC 3.2.1.73), xyloglucan xyloglucosyltransferase (EC 2.4.1.207), agarase (EC 3.2.1.81), κ-carrageenase (EC 3.2.1.83), endo-β-1,3-glucanase (EC 3.2.1.39), endo-β-1,3-1,4-glucanase (EC 3.2.1.6), and endo-β-galactosidase (EC 3.2.1.103). Most of these enzymes have the conserved motif, Glu-Xaa-Asp-Xaa-(Xaa)-Glu (EXDX[X]E), and the two glutamic acid (E) residues have been reported to be important for their catalytic activities [48,49,50]. Likewise, all of the predicted GH16 family members in F. velutipes var. lupinicola also possessed this conserved motif (EXDX[X]E) (Table 6).
Although many GHs have been reported to have signal sequences as they are either secreted or targeted to other cellular locations such as the periplasmic space or Golgi apparatus, signal peptide prediction revealed that 80 out of 246 GHs had signal peptides in their amino acid sequence (Table S3). In previous studies, approximately one-third of GH genes have been reported to have no signal sequence, and hence, GHs without signal sequences indicate their cellular location such as the periplasmic space or Golgi apparatus [51].
Polysaccharides in plant cell walls often form complex structures and synergistic action of enzymes is required to efficiently degrade such complex structures. GHs are essential for the processing of polysaccharides (chitin, cellulose, and xylan from plant), which represent a major source of carbon and nitrogen in nature [2,52]. Substrate specificity is one of the distinctive features of these enzymes, which include cellulases (GH5, -6, -7, -8, -9, -12, -44, -45, and -48), xylanases (GH10, -11, and -30), chitinases (GH18, -19, and -85) and they can act on cellulose, xylose, and chitin, respectively. Another group of enzymes, β-glucosidases (GH1 and -3) can convert cellobiose into glucose [2,52]. In this study, CAZyme annotation revealed that F. velutipes var. lupinicola contains a series of genes associated with cellulase (GH5, -6, -7, -9, and -12), xylanase (GH10, -11, and -30), chitinase (GH18 and -85), and β-glucosidases (GH1 and -3) in its genome sequence (Figure 5 and Table S3).
Recently, sequenced bacterial genomes have revealed the variability of GHs and their potential for industrial degradation of biopolymers [53,54,55]. In addition, fungi also show high levels of hydrolytic activity involved in polysaccharide degradation in nature, and the degradation machineries of many species have been characterized to evaluate their potential in biotechnological applications [56,57,58]. Therefore, more than 200 genes encoding various GHs in the F. velutipes var. lupinicola genome suggesting that this fungus has great potential for biotechnological applications.
3.2.4. Polysaccharide Lyases (PLs)
PL (Eliminase, EC 4.2.2.-) cleaves uronic acid-containing polysaccharides through a β-elimination mechanism to produce unsaturated polysaccharides and is currently classified into 40 families in the CAZy database [2,59]. In the present study, a total of 22 PLs classified into 8 families were predicted in the F. velutipes var. lupinicola genome based on the dbCAN meta server search (Figure 6 and Table S4). Among them, the PL1 family was the most prominent, and three families, including PL4, -9, and -26, consisted of only one PL (Figure 6 and Table S4). Some of the PL family members were reported to be phylum specific [60]. Our results showed that while other Basidiomycetes had high numbers of genes encoding for PL14 family members in their genomes, PL20 was only found in ascomycetes, and PL14 appeared to be specific to Basidiomycota. Additionally, although PL5, -15, and -24 family members are Basidiomycota specific, they are present only in a few species of Basidiomycetes (Table S4).
Pectate (EC 4.2.2.2 and EC 4.2.2.9) or pectin lyases (EC 4.2.2.10) can degrade polygalacturonan (PGA) and pectate lyases are mainly produced by bacterial species [61,62]. However, fungal species produce both pectate and pectin lyases [62]. Genome sequencing of several fungal species, including basidiomycetes, has revealed a number of genes encoding PL, which has led to their potential for use in biotechnological applications. It has been reported that S. commune (basidiomycete) not only produces higher levels of pectinase than Aspergillus niger (ascomycete) in wheat bran, but also high levels of polygalacturonase [26,63].
Pectin and pectate lyases have been classified into 6 PL families, PL1, -2, -3, -9, and -10, in the CAZy database [2]. All of the characterized fungal pectate lyases belong to the families PL1, PL3, and PL9 and the pectin lyases belong to PL1 family [2]. Our results showed that the genes encoding for PL family members, including families 2 and 10, were absent in the F. velutipes var. lupinicola genome or in other fungal species (Figure 6 and Table S4). Additionally, the majority of PLs were pectate lyases (PL1 and -3) in the F. velutipes var. lupinicola genome. Interestingly, most basidiomycetes lack PL family member 9, whereas Flammulina species and S. commune were found to have only the PL9 family (Table S4). These results suggest that F. velutipes var. lupinicola might be a potential candidate for future research focused on polysaccharide lyase as the number of genes encoding PL family members 1, 3, and 9 is similar to that of S. commune.
3.2.5. Auxiliary Activities (AAs)
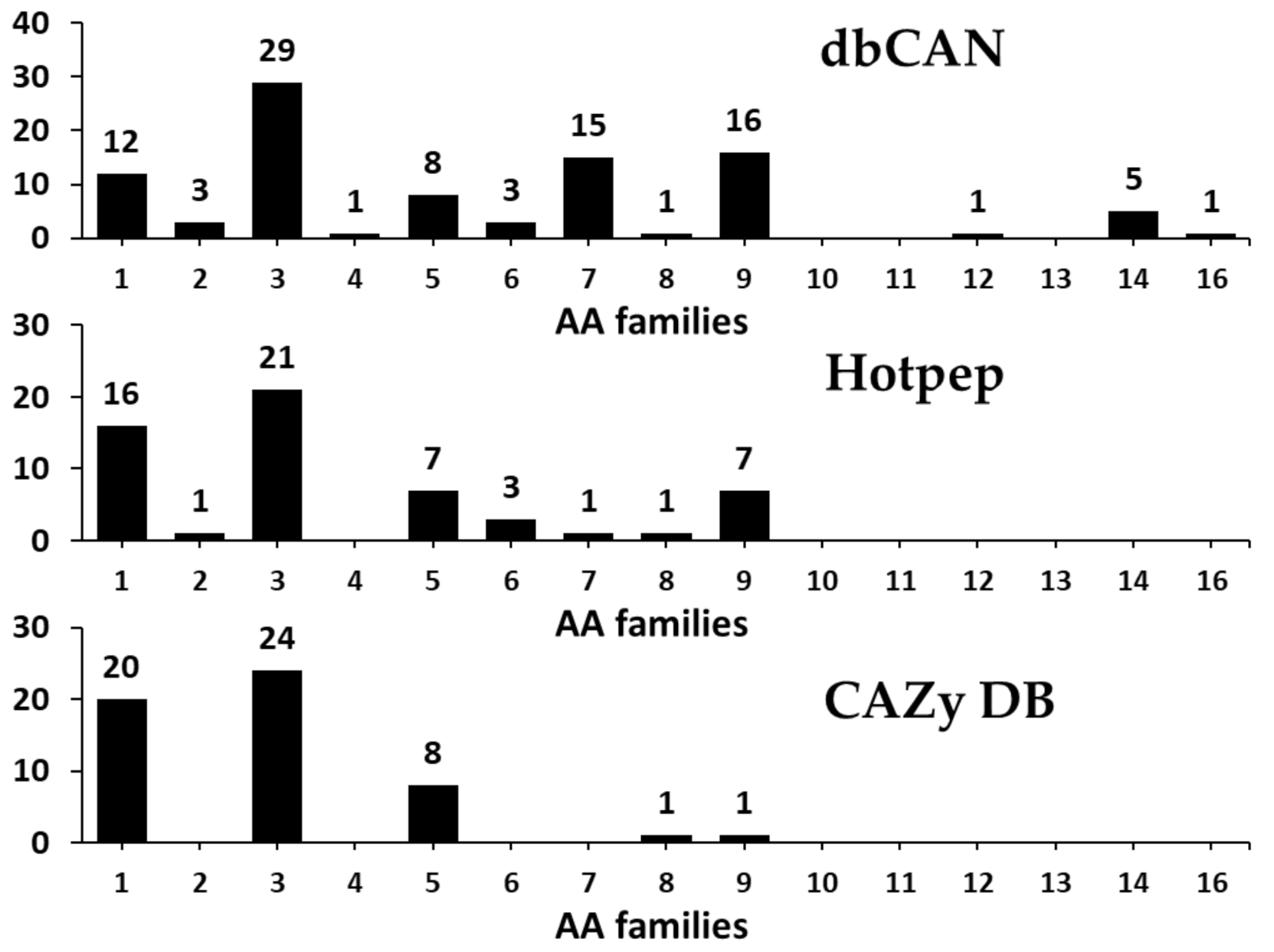
Lignin degradation enzymes such as lytic polysaccharide monooxygenases (LPMOs), involved in depolymerization of non-carbohydrate components (lignin), are classified into AA families [2,64]. In addition, these members originally classified as GH61 and CBM33 have also been found to be involved in the depolymerization of lignin and are now reclassified as AA families [2,64]. AA members are classified into a total of 16 families, including ligninolytic enzymes (9 families) and lytic polysaccharide monooxygenases (6 families), and till date, more than 15,000 classifieds and 50 unclassified AAs have been identified based on amino acid sequence similarities [2].
In the present study, CAZyme annotation revealed that F. velutipes var. lupinicola contains a total of 12 AA families with genes for 104 AAs in its genome sequence (Figure 7 and Table S4). AA family classification also revealed that AA3 is the major representative of the AA family, with 29 AA3 family members (glucose-methanol-choline oxidoreductase, cellobiose dehydrogenase, alcohol oxidase, aryl-alcohol oxidase/glucose oxidase, pyranose oxidase), and AA1 (multicopper oxidases, laccase) comprising the second largest families, with 20 AAs in the F. velutipes var. lupinicola genome (Figure 7 and Table S4). Interestingly, the total number of AAs in the F. velutipes var. lupinicola genome was found to be similar to C. cinerea but different from other Flammulina species, and there were also more AA1 families than other basidiomycetes (Table S4).
In previous studies, some of the AAs were found to possess the conserved motifs required for interaction with the substrate, and this was particularly seen in case of laccases (EC 1.10.3.2, AA1 family), which were found to have His-Xaa-His (HXH), His-Xaa-His-Gly (HXHG), His-Xaa-Xaa-His-Xaa-His (HXXHXH), and His-Cys-His-Xaa3-His-Xaa4-Met/Leu/Phe (HCHXXXHXXXXM/L/F) motifs in their amino acid sequences [65]. Similarly, GMC oxidoreductase proteins (AA3 family) have also been reported to possess a conserved motif such as β-α-β dinucleotide binding-motif (Gly-Xaa-Gly-Xaa-Xaa-Gly-Xaa18-Glu) that interacts with the flavin adenine dinucleotide cofactor [66,67,68]. Our results also showed to that copper-binding and β-α-β dinucleotide-binding motifs are present in the 9 (AA1 families) and 16 genes (AA3 families) of F. velutipes var. lupinicola, respectively, indicating that these genes may act as laccases and GMC oxidoreductases (Table 7).
Degradation of wood by fungi usually begins with the depolymerization of lignin, which accelerates further degradation of wood polymers due to highly reactive lignin radicals [69,70]. Therefore, our results, including extensive identification of the AA family in the of F. velutipes var. lupinicola genome, suggest that this fungus can be potentially used for the production of biomaterials such as biofuels in the future.
3.2.6. Carbohydrate-Binding Modules (CBMs)
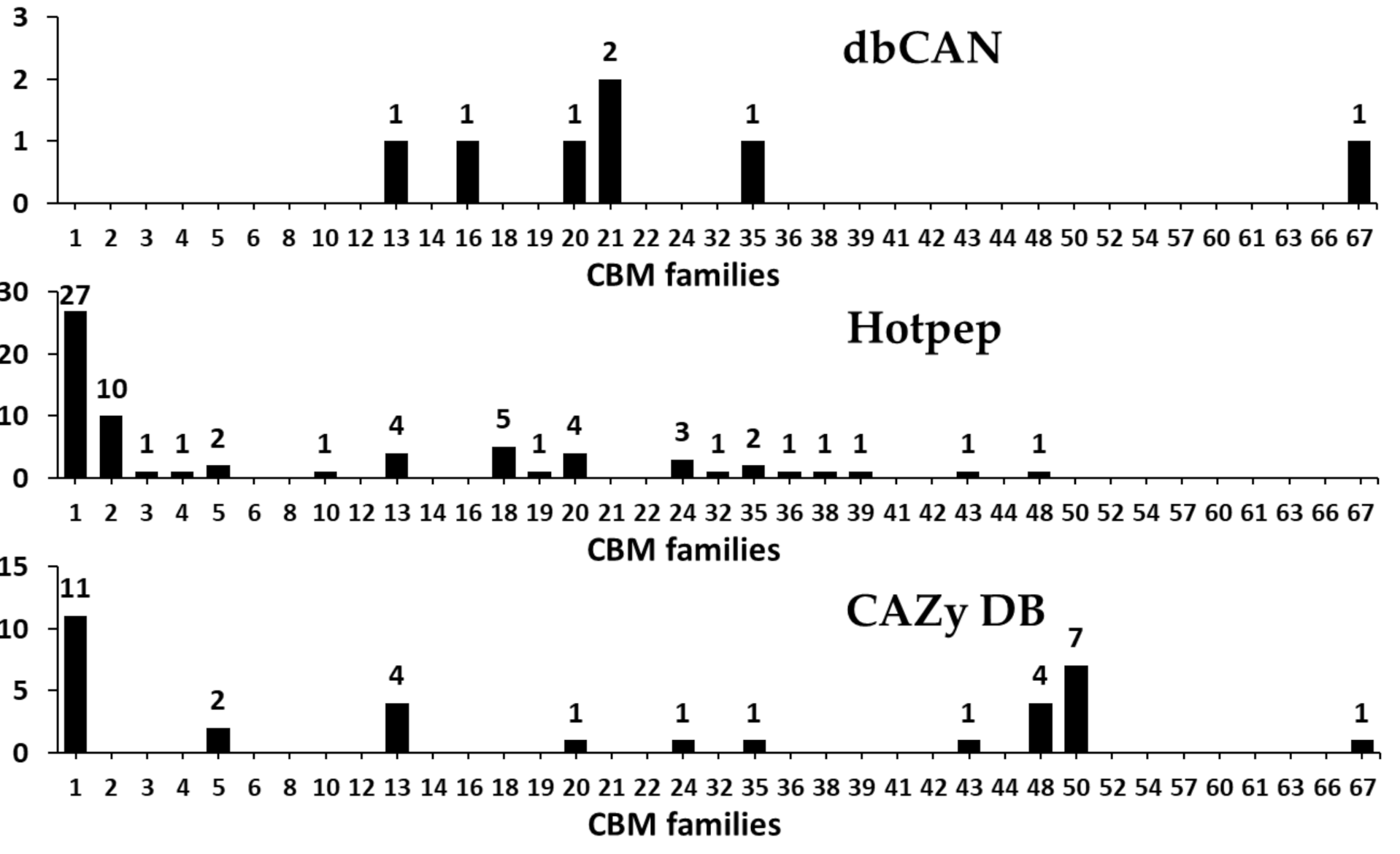
Generally, the amino acid sequences which possesses carbohydrate binding activity in a carbohydrate-active enzyme is known as CBM, and this can bind to the carbohydrate ligands in order to enhance the catalytic activity of a carbohydrate-active enzyme such as GH, PL, and GT [71,72]. Moreover, CBM is often found in proteins without hydrolytic activity, which are known as scaffoldins and they help in organizing the catalytic subunits into non-covalent multi-protein complexes (cellulosome) [72]. Till date, CBMs have been classified into 87 families in the CAZy database, which includes more than 237,000 classifications and 800 non-classified CBM sequences [2].
In the present study, we found that a total of 22 CBM families with genes for 80 CBMs in the F. velutipes var. lupinicola genome based on the dbCAN meta server search (Figure 8 and Table S4). The distribution of CBM, along with multiple copies of CBM1, -13 and -50 family members, is similar to that found in other fungal species (Figure 8 and Table S4). In addition, we found differences in the abundance of some CBM family members between basidiomycetes and ascomycetes, and particularly, CBM 18 family members were found in ascomycetes more than in other basidiomycetes, as well as CBM 12 family members were not observed in all ascomycetes such as in F. velutipes var. lupinicola (Figure 8 and Table S4). These results are consistent with the results of a previous study by Zhao et al. [60], which reported that ascomycetes generally have fewer members of CBM 5 and -12 family than basidiomycetes, while they have more members of CBM 18 family. CBMs have been reported to be required for the activity of cellobiohydrolases classified into the GH6 and -7 families [60]. In the present study, CBM 1 family members were found in the GH6 or GH7 members identified in the F. velutipes var. lupinicola genome (Table S3). In addition, other GH families were found to possess CBMs in their genes, suggesting that these CAZymes may require CBM to efficiently degrade their substrates (Table S3).
4. Conclusions
In the present study, we extensively investigated the lignocellulolytic machinery in basidiomycete fungus F. velutipes var. lupinicola.
We sequenced the genome of F. velutipes var. lupinicola and identified the following genes involved in lignocellulosic biomass degradation; 54~95 auxiliary activities enzymes, 145–188 glycoside hydrolases, 55–73 glycosyltransferases, 6–19 polysaccharide lyases, 1–59 carbohydrate esterases, and 7–67 carbohydrate binding-modules. Although more detailed studies are needed, this CAZyme repertoire of F. velutipes var. lupinicola suggests that this fungus might be applied to produce various biomaterials, including bioethanol through consolidated bioprocessing (CBP), an effective processing method for production of bioethanol from lignocellulosic biomass.
Supplementary Materials
The following are available online at https://www.mdpi.com/2076-2607/9/1/20/s1, Table S1: Gene prediction and annotation of Flammulina velutipes var. lupinicola, Table S2: Ortholog analysis of Flammulina velutipes var. lupinicola with other sequenced fungal species, Table S3: Flammulina velutipes var. lupinicola CAZymes identified from three different databases, Table S4: Distribution of CAZymes in the Flammulina velutipes var. lupinicola genome and in other fungal genomes.
Author Contributions
Conceptualization and methodology, Y.-J.P.; formal analysis, H.-W.Y., J.-H.I., and W.-S.K.; investigation and data curation, Y.-J.P., H.-W.Y., J.-H.I., and W.-S.K.; software; Y.-J.P. and H.-W.Y.; writing—original draft preparation, Y.-J.P. and H.-W.Y.; writing—review and editing, Y.-J.P.; supervision, Y.-J.P.; project administration, Y.-J.P.; funding acquisition, Y.-J.P. All authors have read and agreed to the published version of the manuscript.
Funding
This work was carried out with the support of “Cooperative Research Program for Agriculture Science & Technology Development (Project No. PJ015164032020)” Rural Development Administration, Republic of Korea.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Redhead, S.A.; Petersen, R.H. New species, varieties and combinations in the genus Flammulina. Mycotaxon 1999, 71, 285–294. [Google Scholar]
- Lombard, V.; Golaconda Ramulu, H.; Drula, E.; Coutinho, P.M.; Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014, 42, D490–D495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sista Kameshwar, A.K.; Qin, W. Comparative study of genome-wide plant biomass-degrading CAZymes in white rot, brown rot and soft rot fungi. Mycology 2018, 9, 93–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rytioja, J.; Hildén, K.; Yuzon, J.; Hatakka, A.; de Vries, R.P.; Mäkelä, M.R. Plant-polysaccharide-degrading enzymes from basidiomycetes. Microbiol. Mol. Biol. Rev. 2014, 78, 614–649. [Google Scholar] [CrossRef] [Green Version]
- Eriksson, K.; Blanchette, R.A.; Ander, P. Morphological aspects of wood degradation by fungi and bacteria. In Microbial and Ezymatic Degradation of Wood and Wood Components; Springer: Berlin/Heidelberg, Germany, 1990; pp. 1–87. ISBN 978-3-642-46687-8. [Google Scholar]
- Martinez, D.; Larrondo, L.F.; Putnam, N.; Sollewijn Gelpke, M.D.; Huang, K.; Chapman, J.; Helfenbein, K.G.; Ramaiya, P.; Detter, J.C.; Larimer, F.; et al. Genome sequence of the lignocellulose degrading fungus Phanerochaete chrysosporium strain RP78. Nat. Biotechnol. 2004, 22, 695–700. [Google Scholar] [CrossRef]
- Park, Y.J.; Baek, J.H.; Lee, S.; Kim, C.; Rhee, H.; Kim, H.; Seo, J.S.; Park, H.R.; Yoon, D.E.; Nam, J.Y.; et al. Whole genome and global gene expression analyses of the model mushroom Flammulina velutipes reveal a high capacity for lignocellulose degradation. PLoS ONE 2014, 9, e93560. [Google Scholar] [CrossRef]
- Park, Y.J.; Jeong, Y.U.; Kong, W.S. Genome sequencing and carbohydrate-active enzyme (CAZyme) repertoire of the white rot fungus Flammulina elastica. Int. J. Mol. Sci. 2018, 19, 2379. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.S.; Kong, W.S.; Park, Y.J. Genome sequencing and genome-wide identification of carbohydrate-active enzymes (CAZymes) in the white rot fungus Flammulina fennae. Microbiol. Biotechnol. Lett. 2018, 46, 300–312. [Google Scholar] [CrossRef]
- Park, Y.J.; Kong, W.S. Genome-wide comparison of carbohydrate-active enzymes (CAZymes) repertoire of Flammulina ononidis. Mycobiology 2018, 46, 349–360. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Jackman, S.D.; Vandervalk, B.P.; Mohamadi, H.; Chu, J.; Yeo, S.; Hammond, S.A.; Jahesh, G.; Khan, H.; Coombe, L.; Warren, R.L.; et al. ABySS 2.0: Resource-efficient assembly of large genomes using a Bloom filter. Genome Res. 2017, 27, 768–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmer, J.M.; Stajich, J.E. Funannotate: Eukaryotic Genome Annotation Pipeline. Available online: http://funannotate.readthedocs.io (accessed on 5 April 2020).
- Buchfink, B.; Xie, C.; Huson, D. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Emms, D.; Kelly, S. OrthoFinder: Solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 2015, 16, 157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galagan, J.E.; Calvo, S.E.; Cuomo, C.; Ma, L.J.; Wortman, J.R.; Batzoglou, S.; Lee, S.I.; Basturkmen, M.; Spevak, C.C.; Clutterbuck, J.; et al. Sequencing of Aspergillus nidulans and comparative analysis with A. fumigatus and A. oryzae. Nature 2005, 438, 1105–1115. [Google Scholar] [CrossRef] [PubMed]
- Staats, M.; van Kan, J.A. Genome update of Botrytis cinerea strains B05. 10 and T4. Eukaryot. Cell 2012, 11, 1413–1414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, F.; Aerts, A.; Ahrén, D.; Brun, A.; Danchin, E.G.J.; Duchaussoy, F.; Gibon, J.; Kohler, A.; Lindquist, E.; Pereda, V.; et al. The genome of Laccaria bicolor provides insights into mycorrhizal symbiosis. Nature 2008, 452, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Morin, E.; Kohler, A.; Baker, A.R.; Foulongne-Oriol, M.; Lombard, V.; Nagye, L.G.; Ohm, R.A.; Patyshakuliyeva, A.; Brun, A.; Aerts, A.L.; et al. Genome sequence of the button mushroom Agaricus bisporus reveals mechanisms governing adaptation to a humic-rich ecological niche. Proc. Natl. Acad. Sci. USA 2012, 109, 17501–17506. [Google Scholar] [CrossRef] [Green Version]
- Stajich, J.E.; Wilke, S.K.; Ahrén, D.; Au, C.H.; Birren, B.W.; Borodovsky, M.; Burns, C.; Canbäck, B.; Casselton, L.A.; Cheng, C.K.; et al. Insights into evolution of multicellular fungi from the assembled chromosomes of the mushroom Coprinopsis cinerea (Coprinus cinereus). Proc. Natl. Acad. Sci. USA 2010, 107, 11889–11894. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Gong, Y.; Cai, Y.; Liu, W.; Zhou, Y.; Xiao, Y.; Xu, Z.; Liu, Y.; Lei, X.; Wang, G.; et al. Genome sequence of the edible cultivated mushroom Lentinula edodes (Shiitake) reveals insights into lignocellulose degradation. PLoS ONE 2016, 11, e0160336. [Google Scholar] [CrossRef]
- Zheng, P.; Xia, Y.; Xiao, G.; Xiong, C.; Hu, X.; Zhang, S.; Zheng, H.; Huang, Y.; Zhou, Y.; Wang, S.; et al. Genome sequence of the insect pathogenic fungus Cordyceps militaris, a valued traditional Chinese medicine. Genome Biol. 2011, 12, R116. [Google Scholar] [CrossRef] [Green Version]
- Janbon, G.; Ormerod, K.L.; Paulet, D.; Byrnes, E.J., III; Yadav, V.; Chatterjee, G.; Mullapudi, N.; Hon, C.-C.; Billmyre, R.B.; Brunel, F.; et al. Analysis of the genome and transcriptome of Cryptococcus neoformans var. grubii reveals complex RNA expression and microevolution leading to virulence attenuation. PLOS Genet. 2014, 10, e1004261. [Google Scholar] [CrossRef]
- Fisk, D.G.; Ball, C.A.; Dolinski, K.; Engel, S.R.; Hong, E.L.; Issel-Tarver, L.; Schwartz, K.; Sethuraman, A.; Botstein, D.; Cherry, J.M. Saccharomyces cerevisiae S288C genome annotation: A working hypothesis. Yeast 2006, 23, 857–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galagan, J.E.; Calvo, S.E.; Borkovich, K.A.; Selker, E.U.; Read, N.D.; Jaffe, D.; FitzHugh, W.; Ma, L.J.; Smirnov, S.; Purcell, S.; et al. The genome sequence of the filamentous fungus Neurospora crassa. Nature 2003, 422, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Ohm, R.A.; De Jong, J.F.; Lugones, L.G.; Aerts, A.; Kothe, E.; Stajich, J.E.; de Vries, R.P.; Record, E.; Levasseur, A.; Baker, S.E.; et al. Genome sequence of the model mushroom Schizophyllum commune. Nat. Biotechnol. 2010, 28, 957–963. [Google Scholar] [CrossRef] [Green Version]
- Li, W.C.; Huang, C.H.; Chen, C.L.; Chuang, Y.C.; Tung, S.Y.; Wang, T.F. Trichoderma reesei complete genome sequence, repeat-induced point mutation, and partitioning of CAZyme gene clusters. Biotechnol. Biofuels 2017, 10, 170. [Google Scholar] [CrossRef]
- Kämper, J.; Kahmann, R.; Bölker, M.; Ma, L.-J.; Brefort, T.; Saville, B.J.; Banuett, F.; Kronstad, J.W.; Gold, S.E.; Müller, O.; et al. Insights from the genome of the biotrophic fungal plant pathogen Ustilago maydis. Nature 2006, 444, 97–101. [Google Scholar] [CrossRef]
- Yin, Y.; Mao, X.; Yang, J.; Chen, X.; Mao, F.; Xu, Y. dbCAN: a web resource for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2012, 40, W445–W451. [Google Scholar] [CrossRef]
- Petersen, T.N.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 2011, 8, 785–786. [Google Scholar] [CrossRef]
- Breton, C.; Šnajdrová, L.; Jeanneau, C.; Koča, J.; Imberty, A. Structures and mechanisms of glycosyltransferases. Glycobiology 2006, 16, 29R–37R. [Google Scholar] [CrossRef]
- Lairson, L.L.; Henrissat, B.; Davies, G.J.; Withers, S.G. Glycosyltransferases: Structures, functions, and mechanisms. Annu. Rev. Biochem. 2008, 77, 521–555. [Google Scholar] [CrossRef] [Green Version]
- Coutinho, P.M.; Deleury, E.; Davies, G.J.; Henrissat, B. An evolving hierarchical family classification for glycosyltransferases. J. Mol. Biol. 2003, 328, 307–317. [Google Scholar] [CrossRef]
- Campbell, J.A.; Davies, G.J.; Bulone, V.; Henrissat, B. A classification of nucleotide-diphospho-sugar glycosyltransferases based on amino acid sequence similarities. Biochem. J. 1997, 326, 929–939. [Google Scholar] [CrossRef] [PubMed]
- Paulson, J.C.; Weinstein, J.; Ujita, E.L.; Riggs, K.J.; Lai, H. The membrane-binding domain of a rat liver Golgi sialyltransferase. Biochem. Soc. Trans. 1987, 15, 618–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wickner, W.T.; Lodish, H.F. Multiple mechanisms of protein insertion into and across membranes. Science 1985, 230, 400–407. [Google Scholar] [CrossRef]
- Mukai, Y.; Hirokawa, T.; Tomii, K.; Asai, K.; Akiyama, Y.; Suwa, M. Identification of glycosyltransferases focusing on Golgi transmembrane region. Trends Glycosci. Glycotechnol. 2008, 19, 41–47. [Google Scholar] [CrossRef] [Green Version]
- Bornscheuer, U.T. Microbial carboxyl esterases: Classification, properties and application in biocatalysis. FEMS Microbiol. Rev. 2002, 26, 73–81. [Google Scholar] [CrossRef]
- Jaeger, K.E.; Eggert, T. Lipases for biotechnology. Curr. Opin. Biotechnol. 2002, 13, 390–397. [Google Scholar] [CrossRef]
- Cantarel, B.L.; Coutinho, P.M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The Carbohydrate-Active EnZymes database (CAZy): An expert resource for glycogenomics. Nucleic Acids Res. 2009, 37, D233–D238. [Google Scholar] [CrossRef]
- Biely, P. Microbial carbohydrate esterases deacetylating plant polysaccharides. Biotechnol. Adv. 2012, 30, 1575–1588. [Google Scholar] [CrossRef]
- Nakamura, A.M.; Nascimento, A.S.; Polikarpov, I. Structural diversity of carbohydrate esterases. Biotechnol. Res. Innov. 2017, 1, 35–51. [Google Scholar] [CrossRef]
- Adesioye, F.A.; Makhalanyane, T.P.; Biely, P.; Cowan, D.A. Phylogeny, classification and metagenomic bioprospecting of microbial acetyl xylan esterases. Enzym. Microb. Technol. 2016, 93, 79–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Y.; Schottel, J.L.; Derewenda, U.; Swenson, L.; Patkar, S.; Derewenda, Z.S. A novel variant of the catalytic triad in the Streptomyces scabies esterase. Nat. Struct. Biol. 1995, 2, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Petersen, E.I.; Valinger, G.; Sölkner, B.; Stubenrauch, G.; Schwab, H. A novel esterase from Burkholderia gladioli shows high deacetylation activity on cephalosporins is related to β-lactamases and dd-peptidases. J. Biotechnol. 2001, 89, 11–25. [Google Scholar] [CrossRef]
- Berlemont, R.; Martiny, A.C. Glycoside hydrolases across environmental microbial communities. PLoS Comput. Biol. 2016, 12, e1005300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henrissat, B. A classification of glycosyl hydrolases based on amino acid sequence similarities. Biochem. J. 1991, 280, 309–316. [Google Scholar] [CrossRef]
- Hahn, M.; Olsen, O.; Politz, O.; Borriss, R.; Heinemann, U. Crystal structure and site-directed mutagenesis of Bacillus macerans endo-1,3-1,4-β -glucanase. J. Biol. Chem. 1995, 270, 3081–3088. [Google Scholar] [CrossRef] [Green Version]
- Masuda, S.; Endo, K.; Koizumi, N.; Hayami, T.; Fukazawa, T.; Yatsunami, R.; Fukui, T.; Nakamura, S. Molecular identification of a novel β-1,3-glucanase from alkaliphilic Nocardiopsis sp. strain F96. Extremophiles 2006, 10, 251–255. [Google Scholar] [CrossRef]
- Kotake, T.; Hirata, N.; Degi, Y.; Ishiguro, M.; Kitazawa, K.; Takata, R.; Ichinose, H.; Kaneko, S.; Igarashi, K.; Samejima, M.; et al. Endo-β-1,3-galactanase from winter mushroom Flammulina velutipes. J. Biol. Chem. 2011, 286, 27848–27854. [Google Scholar] [CrossRef] [Green Version]
- Horton, P.; Park, K.J.; Obayashi, T.; Fujita, N.; Harada, H.; Adams-Collier, C.J.; Nakai, K. WoLF PSORT: Protein localization predictor. Nucleic Acids Res. 2007, 35, W585–W587. [Google Scholar] [CrossRef] [Green Version]
- Wilson, D.B. Microbial diversity of cellulose hydrolysis. Curr. Opin. Microbiol. 2011, 14, 259–263. [Google Scholar] [CrossRef]
- Berlemont, R.; Martiny, A.C. Phylogenetic distribution of potential cellulases in bacteria. Appl. Environ. Microbiol. 2013, 79, 1545–1554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berlemont, R.; Martiny, A.C. Genomic potential for polysaccharides deconstruction in bacteria. Appl. Environ. Microbiol. 2015, 81, 1513–1519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talamantes, D.; Biabini, N.; Dang, H.; Abdoun, K.; Berlemont, R. Natural diversity of cellulases, xylanases, and chitinases in bacteria. Biotechnol. Biofuels 2016, 9, 133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berlemont, R. Distribution and diversity of enzymes for polysaccharide degradation in fungi. Sci. Rep. 2017, 7, 222. [Google Scholar] [CrossRef] [Green Version]
- Eichlerová, I.; Homolka, L.; Žifčáková, L.; Lisá, L.; Dobiášová, P.; Baldrian, P. Enzymatic systems involved in decomposition reflects the ecology and taxonomy of saprotrophic fungi. Fungal Ecol. 2015, 13, 10–22. [Google Scholar] [CrossRef]
- Treseder, K.K.; Lennon, J.T. Fungal traits that drive ecosystem dynamics on land. Microbiol. Mol. Biol. Rev. 2015, 79, 243–262. [Google Scholar] [CrossRef] [Green Version]
- Yip, V.L.; Withers, S.G. Breakdown of oligosaccharides by the process of elimination. Curr. Opin. Chem. Biol. 2006, 10, 147–155. [Google Scholar] [CrossRef]
- Zhao, Z.; Liu, H.; Wang, C.; Xu, J.R. Correction: Comparative analysis of fungal genomes reveals different plant cell wall degrading capacity in fungi. BMC Genom. 2013, 14, 274. [Google Scholar] [CrossRef] [Green Version]
- Garron, M.L.; Cygler, M. Structural and mechanistic classification of uronic acid-containing polysaccharide lyases. Glycobiology 2010, 20, 1547–1573. [Google Scholar] [CrossRef] [Green Version]
- Linhardt, R.J.; Galliher, P.M.; Cooney, C.L. Polysaccharide lyases. Appl. Biochem. Biotechnol. 1987, 12, 135–176. [Google Scholar] [CrossRef]
- Xavier-Santos, S.; Carvalho, C.C.; Bonfá, M.; Silva, R.; Capelari, M.; Gomes, E. Screening for pectinolytic activity of wood-rotting basidiomycetes and characterization of the enzymes. Folia Microbiol. 2004, 49, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Levasseur, A.; Drula, E.; Lombard, V.; Coutinho, P.M.; Henrissat, B. Expansion of the enzymatic repertoire of the CAZy database to integrate auxiliary redox enzymes. Biotechnol. Biofuels 2013, 6, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiss, R.; Ihssen, J.; Richter, M.; Eichhorn, E.; Schilling, B.; Thöny-Meyer, L. Laccase versus laccase-like multi-copper oxidase: A comparative study of similar enzymes with diverse substrate spectra. PLoS ONE 2013, 8, e65633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez, I.S.; Ruiz-Duenas, F.J.; Santillana, E.; Ferreira, P.; Martinez, M.J.; Martinez, A.T.; Romero, A. Novel structural features in the GMC family of oxidoreductases revealed by the crystal structure of fungal aryl-alcohol oxidase. Acta Crystallogr D Biol Crystallogr. 2009, 65, 1196–1205. [Google Scholar] [CrossRef] [PubMed]
- Wierenga, R.K.; Drenth, J.; Schulz, G.E. Comparison of the 3-dimensional protein and nucleotide structure of the FAD-binding domain of parahydroxybenzoate hydroxylase with the FAD-binding as well as NADPH-binding domains of glutathionereductase. J. Mol. Biol. 1983, 167, 725–739. [Google Scholar] [CrossRef]
- Varela, E.; Martinet, M.J.; Martinez, A.T. Arylalcohol oxidase protein sequence: A comparison with glucose oxidase and other FAD oxidoreductases. Biochem. Biophys. Acta Protein Struct. Mol. Enzymol. 2000, 1481, 202–208. [Google Scholar] [CrossRef]
- Leonowicz, A.; Matuszewska, A.; Luterek, J.; Ziegenhagen, D.; Wojtas-Wasilewska, M.; Cho, N.S.; Hofrichter, M.; Rogalski, J. Biodegradation of lignin by white rot fungi. Fungal Genet. Biol. 1999, 27, 175–185. [Google Scholar] [CrossRef]
- Guillén, F.; Martínez, M.J.; Gutiérrez, A.; Del Rio, J.C. Biodegradation of lignocellulosics: Microbial, chemical, and enzymatic aspects of the fungal attack of lignin. Int. Microbiol. 2005, 8, 195–204. [Google Scholar]
- Boraston, A.B.; Bolam, D.N.; Gilbert, H.J.; Davies, G.J. Carbohydrate-binding modules: Fine-tuning polysaccharide recognition. Biochem. J. 2004, 382, 769–781. [Google Scholar] [CrossRef]
- Shoseyov, O.; Shani, Z.; Levy, I. Carbohydrate binding modules: Biochemical properties and novel applications. Microbiol. Mol. Biol. Rev. 2006, 70, 283–295. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Phylogenetic analysis of fungal species based on ortholog clustering using OrthoFinder.

Figure 2.
Number of identification and annotation of carbohydrate-active enzyme genes in the Flammulina velutipes var. lupinicola genome based on three different databases including the HMMER (dbCAN CAZyme domain HMM database), DIAMOND (CAZy database), and Hotpep (short conserved motifs in the PRR library database) [29].
Figure 2.
Number of identification and annotation of carbohydrate-active enzyme genes in the Flammulina velutipes var. lupinicola genome based on three different databases including the HMMER (dbCAN CAZyme domain HMM database), DIAMOND (CAZy database), and Hotpep (short conserved motifs in the PRR library database) [29].

Figure 3.
Predicted and annotated glycosyltransferase (GT) families in Flammulina velutipes var. lupinicola from dbCAN, Hotpep, and CAZy databases.
Figure 3.
Predicted and annotated glycosyltransferase (GT) families in Flammulina velutipes var. lupinicola from dbCAN, Hotpep, and CAZy databases.

Figure 4.
Predicted and annotated carbohydrate esterase (CE) families in Flammulina velutipes var. lupinicola from dbCAN, Hotpep, and CAZy databases.
Figure 4.
Predicted and annotated carbohydrate esterase (CE) families in Flammulina velutipes var. lupinicola from dbCAN, Hotpep, and CAZy databases.

Figure 5.
Predicted and annotated glycoside hydrolase (GH) families in Flammulina velutipes var. lupinicola from dbCAN, Hotpep, and CAZy databases.
Figure 5.
Predicted and annotated glycoside hydrolase (GH) families in Flammulina velutipes var. lupinicola from dbCAN, Hotpep, and CAZy databases.

Figure 6.
Predicted and annotated polysaccharide lyase (PL) families in Flammulina velutipes var. lupinicola from dbCAN, Hotpep, and CAZy databases.
Figure 6.
Predicted and annotated polysaccharide lyase (PL) families in Flammulina velutipes var. lupinicola from dbCAN, Hotpep, and CAZy databases.

Figure 7.
Predicted and annotated auxiliary activities (AA) families in Flammulina velutipes var. lupinicola from dbCAN, Hotpep, and CAZy databases.
Figure 7.
Predicted and annotated auxiliary activities (AA) families in Flammulina velutipes var. lupinicola from dbCAN, Hotpep, and CAZy databases.

Figure 8.
Predicted and annotated carbohydrate-binding module (CBM) families in Flammulina velutipes var. lupinicola from dbCAN, Hotpep, and CAZy databases.
Figure 8.
Predicted and annotated carbohydrate-binding module (CBM) families in Flammulina velutipes var. lupinicola from dbCAN, Hotpep, and CAZy databases.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Flammulina velutipes var. lupinicola genome sequencing statistics.
| Hiseq 2000 NGS Analysis | Total Reads (100 bp) | 41,592,600 |
|---|---|---|
| Reads After Trimming (%), >Q 30 | 38,416,296 (92.36) | |
| De Novo Assembly | Optimized hash value (kmer) | 71 |
| Total number of contigs (Depth of coverage) | 2570 (110) | |
| Number of contigs (≥1 kb) | 1658 | |
| Contig N50 (bp) | 48,981 | |
| Length of longest contig (bp) | 814,722 | |
| Total bases in contigs (bp) | 33,223,357 | |
| Total bases in contigs (≥1 kb) | 32,579,599 | |
| GC content (%) | 49.53 | |
| Gene Prediction | Predicted gene | 15,337 |
| Average gene (bp) and protein (aa) length | 1122 and 486.40 | |
| Average exon per gene | 4.59 | |
| Average exon and intron size (bp) | 244.66 and 53.6 |
Table 2.
Comparison of the genome characteristics of Flammulina velutipes var. lupinicola and other basidiomycetes.
Table 2.
Comparison of the genome characteristics of Flammulina velutipes var. lupinicola and other basidiomycetes.
| Fungal Species | F. Velutipes var. lupinicola | F. ononidis | F. fennae | F. elastica | F. velutipes | L. bicolar | C. cinerea | P. chrysosporium | U. maydis | S. commune | L. edodes |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Strain | ASI4195 | KACC 46186 | KACC 46185 | KACC 46182 | KACC 42780 | S238N-H82 | Okayama 7#130 | RP78 | 521 | H4-8 | W1-26 |
| Genome (Mb) | 33.22 | 34.5 | 32.4 | 35 | 35.6 | 60.71 | 36.19 | 35.15 | 19.6 | 38.67 | 48.3 |
| Genes | 15,337 | 12,269 | 11,591 | 12,536 | 12,218 | 23,132 | 13,393 | 13,602 | 6785 | 16,319 | 14,002 |
Table 3.
Ortholog analysis of Flammulina velutipes var. lupinicola and other fungal species.
| Fungal Species | Total Number of Genes | Number of Genes in Orthogroups Containing Species (%) | Number of Genes in Species-Specific Orthogroups (%) | Number of Genes Unassigned to Any Orthogroups (%) | Number of Orthogroups Containing Species (%) | Number of Species-Specific Orthogroups | |
|---|---|---|---|---|---|---|---|
| Basidiomycota | F. velutipes var. lupinicola ASI4195 | 15,337 | 14,563 (96.5) | 170 (1.1) | 526 (3.5) | 8431 (47.2) | 70 |
| Flammulina fennae KACC46185 | 11,591 | 11,318 (97.6) | 33 (0.3) | 273 (2.4) | 7631 (42.7) | 15 | |
| Flammulina ononidis KACC46186 | 12,269 | 11,948 (97.4) | 67 (0.5) | 321(2.6) | 7896 (44.2) | 29 | |
| Flammulina elastica KACC46182 | 12,536 | 12,079 (96) | 104 (0.8) | 457 (4) | 7856 (44.0) | 41 | |
| Flammulina velutipes KACC42780 | 12,218 | 10,957 (90) | 199 (1.6) | 1261 (10) | 7088 (39.7) | 81 | |
| Agaricus bisporus var. bisporus H97 | 10,438 | 9496 (91) | 1284 (12.3) | 942 (9) | 5960 (33.4) | 184 | |
| Coprinopsis cinerea okayama 7#130 | 13,393 | 11,646 (87) | 1945 (14.5) | 1747 (13) | 6821 (38.2) | 421 | |
| Cryptococcus neoformans var. grubii H99 | 6967 | 5931 (85) | 322 (4.6) | 1036 (15) | 4740 (26.5) | 83 | |
| Laccaria bicolor S238N-H82 | 23,132 | 20,555 (89) | 7400 (32) | 2577 (11) | 8202 (45.9) | 1478 | |
| Lentinula edodes W1-26 | 14,002 | 11,862 (85) | 947 (6.8) | 2140 (15) | 6688 (37.4) | 290 | |
| Phanerochaete chrysosporium RP78 | 13,602 | 11,236 (83) | 1480 (10.9) | 2366 (17) | 6650 (37.2) | 358 | |
| Schizophyllum commune H4-8 | 16,319 | 13,423 (82) | 2642 (16.2) | 2896 (18) | 7104 (39.8) | 597 | |
| Ustilago maydis 521 | 6785 | 5674 (84) | 227 (3.3) | 1111 (16) | 4765 (26.7) | 74 | |
| Ascomycota | Aspergillus nidulans FGSC-A4 | 10,680 | 1349 (87) | 268 (2.5) | 1349 (13) | 6383 (35.7) | 111 |
| Botrytis cinerea B05.10 | 16,447 | 6677 (59) | 587 (3.6) | 6677 (41) | 6533 (36.6) | 210 | |
| Cordyceps militaris CM01 | 9651 | 1162 (88) | 191 (2) | 1.162 (12) | 6323 (35.4) | 53 | |
| Neurospora crassa OR74A | 10,785 | 1809 (83) | 501 (4.6) | 1809 (17) | 6453 (36.1) | 183 | |
| Saccharomyces cerevisiae S288C | 6002 | 1790 (73) | 443 (6.7) | 1790 (27) | 3525 (19.7) | 136 | |
| Trichoderma reesei QM6a | 9115 | 1404 (86) | 167 (1.7) | 1404 (14) | 6580 (36.8) | 51 | |
Table 4.
CAZymes of Flammulina velutipes var. lupinicola and other fungal species.
| Taxon | Species | CAZymes | No. of CAZyme (Annotation DB) | Reference | |||||
|---|---|---|---|---|---|---|---|---|---|
| AA | GH | GT | CE | CBM | PL | ||||
| Basidiomycota | F. velutipes var. lupinicola | 95 | 188 | 71 | 59 | 7 | 19 | 439 (Hmmer dbCAN) | This study |
| 57 | 145 | 55 | 18 | 67 | 18 | 360 (Hotpep) | |||
| 54 | 157 | 73 | 13 | 33 | 6 | 336 (CAZy database) | |||
| F. fennae | 86 | 220 | 85 | 57 | 45 | 20 | 513 | [9] | |
| F. ononidis | 87 | 228 | 87 | 61 | 40 | 21 | 524 | [10] | |
| F. elastica | 82 | 218 | 89 | 59 | 42 | 18 | 508 | [8] | |
| F. velutipes | 85 | 239 | 84 | 63 | 44 | 25 | 540 | [8] | |
| A. bisporus | 81 | 174 | 54 | 33 | 44 | 9 | 395 | JGI database | |
| C. cinerea | 111 | 195 | 83 | 60 | 105 | 16 | 570 | [8] | |
| L. bicolor | 55 | 170 | 96 | 18 | 31 | 7 | 377 | JGI database | |
| L. edodes | 82 | 254 | 85 | 44 | 61 | 11 | 537 | [8] | |
| P. chrysosporium | 85 | 175 | 65 | 16 | 62 | 4 | 407 | JGI database | |
| S. commune | 78 | 241 | 85 | 57 | 37 | 18 | 516 | [8] | |
| U. maydis | 28 | 113 | 61 | 29 | 10 | 2 | 243 | ||
| C. neoformans | 14 | 97 | 70 | 5 | 12 | 4 | 202 | CAZy database | |
| Ascomycota | C. militaris | 54 | 165 | 91 | 34 | 39 | 5 | 388 | [8] |
| T. reesei | 59 | 210 | 90 | 32 | 44 | 5 | 440 | [8] | |
| S. cerevisiae | 5 | 57 | 68 | 2 | 12 | 0 | 144 | CAZy database | |
| A. nudulans | 33 | 267 | 91 | 30 | 46 | 23 | 490 | CAZy database | |
| N. crassa | 35 | 177 | 80 | 21 | 42 | 4 | 359 | CAZy database | |
| B. cinerea | 77 | 287 | 119 | 37 | 89 | 10 | 619 | CAZy database | |
Table 5.
Conserved motifs of CE families in Flammulina velutipes var. lupinicola.
| CE Family | Gene | Motifs |
|---|---|---|
| CE1, 4, 5 | GXSXG, SHD, FDDGP * | |
| CE1 | g5266 | GDSLG |
| g5267 | GDSLG | |
| g8160 | GDSLG, SHD | |
| CE4 | g374 | GDSDG, FDDGP |
| g2000 | FDDGP | |
| g4109 | FDDGP | |
| g7304 | FDDGP | |
| g7333 | GTSEG, FDDGP | |
| g8346 | FDDGP | |
| g10068 | GPSFG, FDDGP | |
| g10591 | GDSNG | |
| g13055 | FDDGP | |
| g13320 | GSSSG, FDDGP | |
| g14347 | GDSAG, FDDGP | |
| g14354 | GDSAG, FDDGP | |
| CE5 | g4988 | GWSQG |
| g15044 | GWSQG | |
| CE3, 16 | GDS(L) | |
| CE3 | g4116 | GDS |
| CE16 | g5620 | GDS |
| g5623 | GDS | |
| g5624 | GDS | |
| g8182 | GDS | |
| g15048 | GDS |
* Asterisk indicates CE family 4 specific motif.
Table 6.
Conserved motifs of GH16 family in Flammulina velutipes var. lupinicola.
| GH Family | Gene | Motifs |
|---|---|---|
| EXDX[X]E | ||
| GH16 | g2766 | EIDLE, EIDLIE |
| g9518, g12441 | EIDVE | |
| g11166 | EIDWE | |
| g14715 | EADDE | |
| g311, g4184, g7248, g7679, g8220, g9061, g10939 | EIDIIE | |
| g3620, g9596 | EIDVFE | |
| g4175 | EVDIGE | |
| g4746, g5694, g7094, g12591, g14715 | EIDIFE | |
| g4760 | EIDIME | |
| g5774, g11509, g12838, g14181 | EIDILE | |
| g7259 | EIDVLE | |
| g11952 | EVDILE | |
| g11992 | EIDIVE |
Table 7.
Conserved motifs of GH families in Flammulina velutipes var. lupinicola.
| AA Family | Gene | Motifs | |||
|---|---|---|---|---|---|
| AA1 | HXHG | HXH | HXXHXH | HCHXXXHXXXXM/L/F | |
| g658 | HWHG | HSH | HPFHLH | HCHIDWHLEAGL | |
| g2787 | HHHG | HGH | HPFHFH | HCHIEWHLEVGL | |
| g4337 | HWHG | HSH | HPFHLH | HCHIDWHIEAGL | |
| g4797 | HWHG | HSH | HPFHLH | HCHVDWHMEAGL | |
| g8250 | HWHG | HSH | HPFHLH | HCHIDWHLEIGL | |
| g8252 | HWHG | HSH | HPFHLH | HCHIDWHLDIGL | |
| g10087 | HWHG | HSH | HPFHLH | HCHIDWHLELGL | |
| g12086 | HGHG | HAH | HPIHKH | HCHVSQHAAGGM | |
| g12686 | HHHG | HAH | HPFHLH | HCHIEWHLEVGL | |
| AA3 | GXGXXGX18E | ||||
| g1272 | GGGTAGLALAARLSE-DSNTTVLVLE | ||||
| g2268 | GAGLAGTTVAARLAE-DAGVSVLLIE | ||||
| g2484 | GSGSAGSIIATRLAE-DPNVSVCLLE | ||||
| g7746 | GGGTAGLVVAARLSE-DPNTSVLVLE | ||||
| g8286 | GGGTAGLILGARLSE-DSDTTVLVLE | ||||
| g8482 | GAGPGGSTVANRLTE-DPSLSVLLVE | ||||
| g9880 | GGGTAGVTLATRLAE-DGTHTVGVIE | ||||
| g10382 | GGGIGGAVVANRLTE-TSSVNVLLLE | ||||
| g11534 | GAGTAGSVVANRLTE-DRNVTVLVLE | ||||
| g11997 | GAGSAGMVVATRLAE-NPDASVCIIE | ||||
| g12094 | GAGTAGLVLARRLSE-KTSLKVGVIE | ||||
| g13184 | GGGAAGAVIANRLTE-IDTFSVLILE | ||||
| g13185 | GGGAAGAVVANRLTE-IDRFSVLVLE | ||||
| g13756 | GAGTAGNVVAARLSE-NRNMSVLVIE | ||||
| g13888 | GGGTAGLIIAARLSE-NADTTVLVLE | ||||
| g14144 | GGGTAGLIIAARLSE-NADTTVLVLE | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Yu, H.-W.; Im, J.-H.; Kong, W.-S.; Park, Y.-J. Comparative Analysis of Carbohydrate Active Enzymes in the Flammulina velutipes var. lupinicola Genome. Microorganisms 2021, 9, 20. https://doi.org/10.3390/microorganisms9010020
AMA Style
Yu H-W, Im J-H, Kong W-S, Park Y-J. Comparative Analysis of Carbohydrate Active Enzymes in the Flammulina velutipes var. lupinicola Genome. Microorganisms. 2021; 9(1):20. https://doi.org/10.3390/microorganisms9010020
Chicago/Turabian StyleYu, Hye-Won, Ji-Hoon Im, Won-Sik Kong, and Young-Jin Park. 2021. "Comparative Analysis of Carbohydrate Active Enzymes in the Flammulina velutipes var. lupinicola Genome" Microorganisms 9, no. 1: 20. https://doi.org/10.3390/microorganisms9010020
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.