The Core and Distinction of the Gut Microbiota in Chinese Populations across Geography and Ethnicity

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Groups
2.2. Analysis of Fecal Microbiota by 16S rRNA Sequencing
2.3. Feature Selection
2.4. The α-Diversity and β-Distance
2.5. Enterotypes and Bacterial Biomarkers Analysis
3. Results
3.1. Overall Gut Microbiota Distribution in Chinese Populations
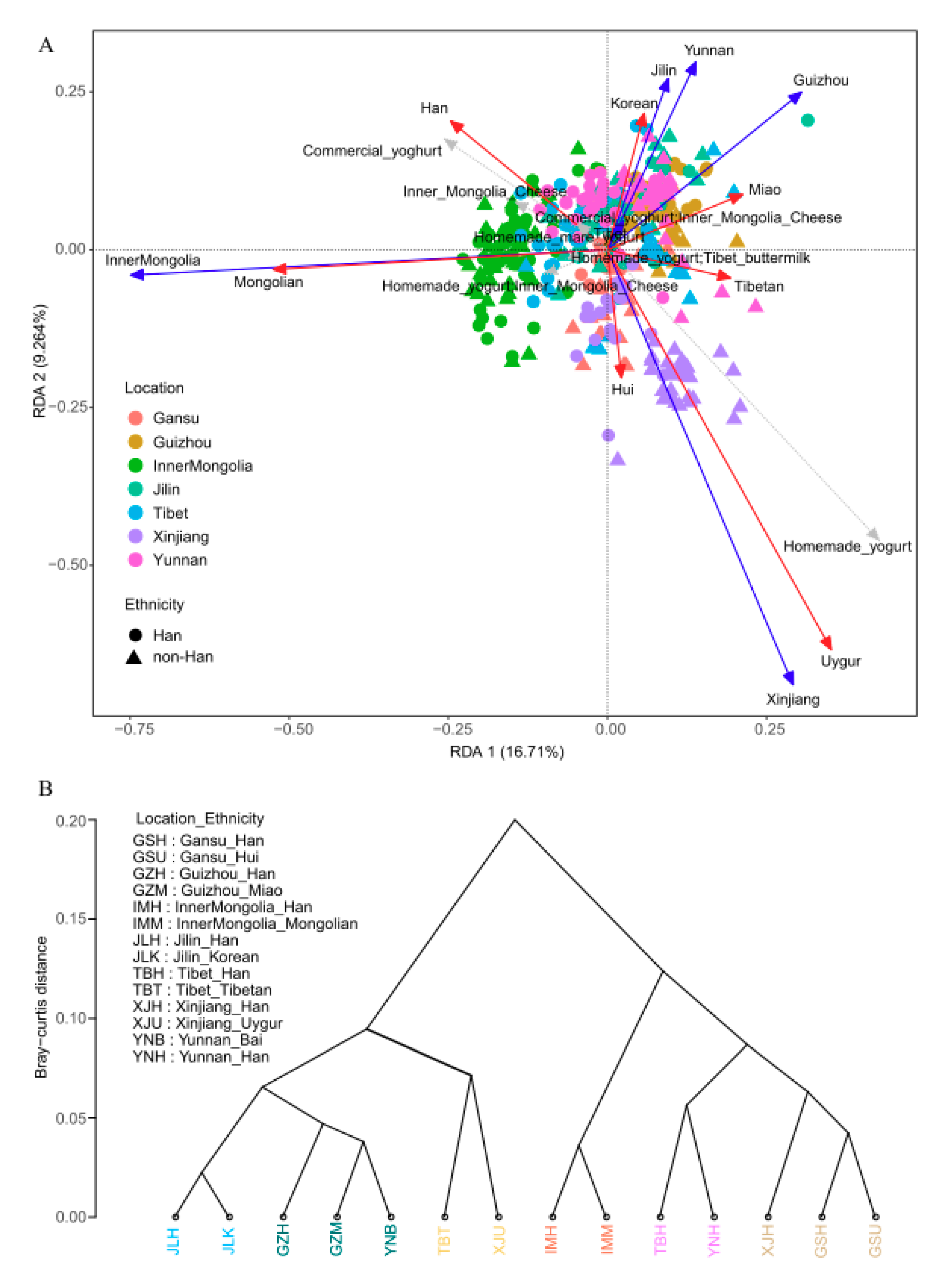
3.2. Geographical Location and Ethnicity Were Major Factors Influencing Composition of the Gut Microbiota
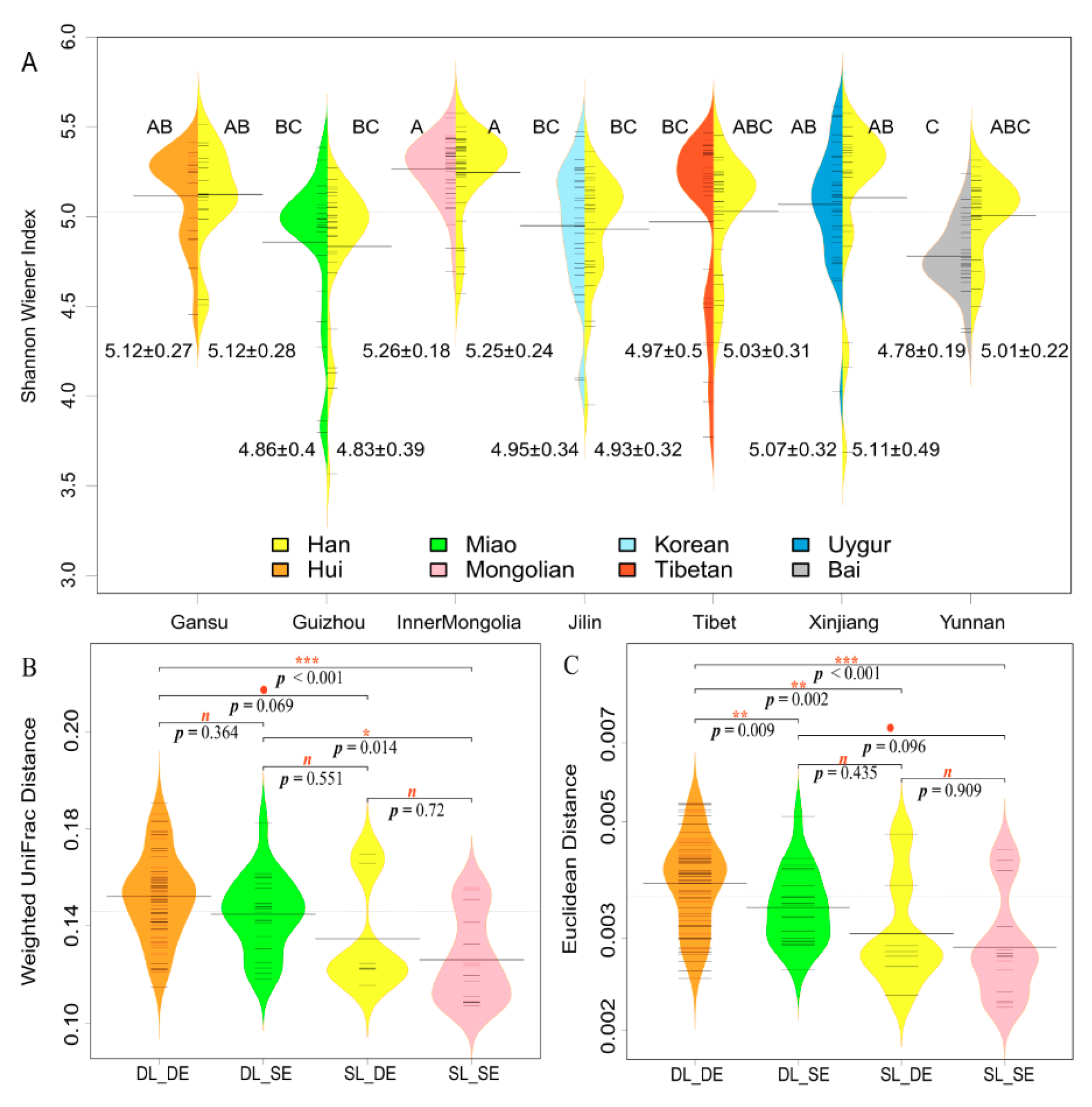
3.3. Geographical Location Has a Greater Influence than Ethnicity on α-Diversity and β-Distance
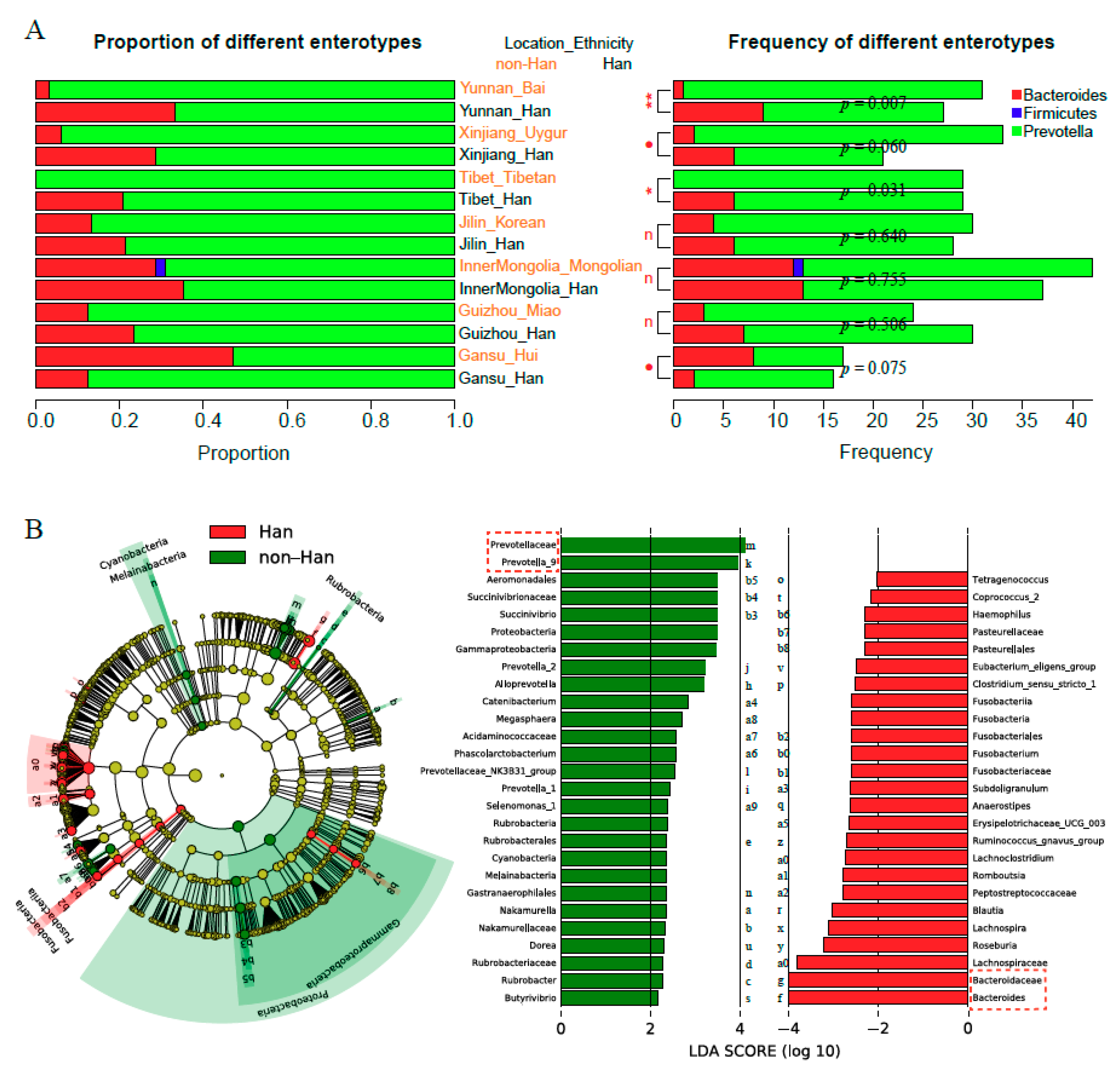
3.4. Prevotella and Bacteroidetes Contribute to Differences of Enterotype, Community Composition and Diversity of Gut Microbiota
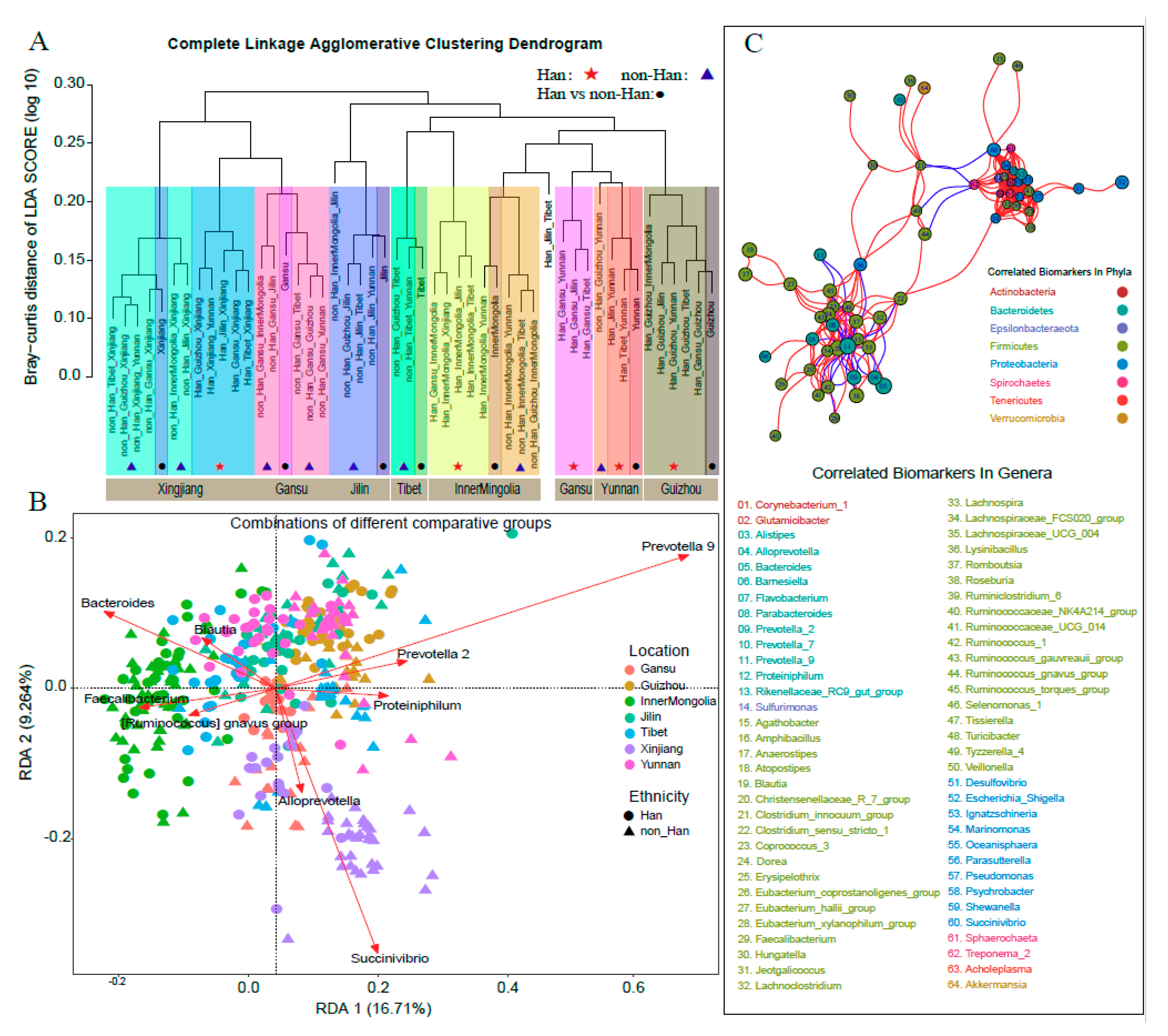
3.5. Bacterial Biomarkers of Various Ethnicity in Different Geographical Location
3.6. Bacterial Biomarkers Were Correlated with Each Other in Each Communities
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Clemente, J.C.; Ursell, L.K.; Parfrey, L.W.; Knight, R. The impact of the gut microbiota on human health: An integrative view. Cell 2012, 148, 1258–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flint, H.J.; Scott, K.P.; Louis, P.; Duncan, S.H. The role of the gut microbiota in nutrition and health. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Gill, S.R.; Pop, M.; Deboy, R.T.; Eckburg, P.B.; Turnbaugh, P.J.; Samuel, B.S.; Gordon, J.I.; Relman, D.A.; Fraser-Liggett, C.M.; Nelson, K.E. Metagenomic analysis of the human distal gut microbiome. Science 2006, 312, 1355–1359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huttenhower, C.; Gevers, D.; Knight, R.; Abubucker, S.; Badger, J.H.; Chinwalla, A.T.; Creasy, H.H.; Earl, A.M.; FitzGerald, M.G.; Fulton, R.S.; et al. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [Google Scholar] [CrossRef] [Green Version]
- Costello, E.K.; Stagaman, K.; Dethlefsen, L.; Bohannan, B.J.; Relman, D.A. The application of ecological theory toward an understanding of the human microbiome. Science 2012, 336, 1255–1262. [Google Scholar] [CrossRef] [Green Version]
- Li, E.; Hamm, C.M.; Gulati, A.S.; Sartor, R.B.; Chen, H.; Wu, X.; Zhang, T.; Rohlf, F.J.; Zhu, W.; Gu, C.; et al. Inflammatory bowel diseases phenotype, C. difficile and NOD2 genotype are associated with shifts in human ileum associated microbial composition. PLoS ONE 2012, 7, e26284. [Google Scholar] [CrossRef] [Green Version]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D.; et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef]
- Fei, N.; Zhao, L. An opportunistic pathogen isolated from the gut of an obese human causes obesity in germfree mice. ISME J. 2013, 7, 880–884. [Google Scholar] [CrossRef]
- Hall, A.B.; Tolonen, A.C.; Xavier, R.J. Human genetic variation and the gut microbiome in disease. Nat. Rev. Genet. 2017, 18, 690–699. [Google Scholar] [CrossRef]
- Gilbert, J.A.; Quinn, R.A.; Debelius, J.; Xu, Z.Z.; Morton, J.; Garg, N.; Jansson, J.K.; Dorrestein, P.C.; Knight, R. Microbiome-wide association studies link dynamic microbial consortia to disease. Nature 2016, 535, 94–103. [Google Scholar] [CrossRef]
- Gilbert, J.A.; Blaser, M.J.; Caporaso, J.G.; Jansson, J.K.; Lynch, S.V.; Knight, R. Current understanding of the human microbiome. Nat. Med. 2018, 24, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Guo, Z.; Xue, Z.; Sun, Z.; Zhang, M.; Wang, L.; Wang, G.; Wang, F.; Xu, J.; Cao, H.; et al. A phylo-functional core of gut microbiota in healthy young Chinese cohorts across lifestyles, geography and ethnicities. ISME J. 2015, 9, 1979–1990. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Hamady, M.; Yatsunenko, T.; Cantarel, B.L.; Duncan, A.; Ley, R.E.; Sogin, M.L.; Jones, W.J.; Roe, B.A.; Affourtit, J.P.; et al. A core gut microbiome in obese and lean twins. Nature 2009, 457, 480–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothschild, D.; Weissbrod, O.; Barkan, E.; Kurilshikov, A.; Korem, T.; Zeevi, D.; Costea, P.I.; Godneva, A.; Kalka, I.N.; Bar, N.; et al. Environment dominates over host genetics in shaping human gut microbiota. Nature 2018, 555, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human gut microbiome viewed across age and geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Deschasaux, M.; Bouter, K.E.; Prodan, A.; Levin, E.; Groen, A.K.; Herrema, H.; Tremaroli, V.; Bakker, G.J.; Attaye, I.; Pinto-Sietsma, S.J.; et al. Depicting the composition of gut microbiota in a population with varied ethnic origins but shared geography. Nat. Med. 2018, 24, 1526–1531. [Google Scholar] [CrossRef]
- De Filippo, C.; Cavalieri, D.; Di Paola, M.; Ramazzotti, M.; Poullet, J.B.; Massart, S.; Collini, S.; Pieraccini, G.; Lionetti, P. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl. Acad. Sci. USA 2010, 107, 14691–14696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, V.K.; Paul, S.; Dutta, C. Geography, Ethnicity or Subsistence-Specific Variations in Human Microbiome Composition and Diversity. Front. Microbiol. 2017, 8, 1162. [Google Scholar] [CrossRef] [Green Version]
- Dehingia, M.; Devi, K.T.; Talukdar, N.C.; Talukdar, R.; Reddy, N.; Mande, S.S.; Deka, M.; Khan, M.R. Gut bacterial diversity of the tribes of India and comparison with the worldwide data. Sci. Rep. 2015, 5, 18563. [Google Scholar] [CrossRef] [Green Version]
- O’Donnell, L.J.; Virjee, J.; Heaton, K.W. Detection of pseudodiarrhoea by simple clinical assessment of intestinal transit rate. BMJ 1990, 300, 439–440. [Google Scholar] [CrossRef] [Green Version]
- Hijmans, R.J.; Cameron, S.E.; Parra, J.L.; Jones, P.G.; Jarvis, A. Very high resolution interpolated climate surfaces for global land areas. Int. J. Clim. 2005, 25, 1965–1978. [Google Scholar] [CrossRef]
- Kriticos, D.J.; Webber, B.L.; Leriche, A.; Ota, N.; Macadam, I.; Bathols, J.; Scott, J.K. CliMond: Global high-resolution historical and future scenario climate surfaces for bioclimatic modelling. Methods Ecol. Evol. 2012, 3, 53–64. [Google Scholar] [CrossRef]
- Armougom, F. Exploring Microbial Diversity Using 16S rRNA High-Throughput Methods. J. Comput. Sci. Syst. Biol. 2009, 2. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glockner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Asshauer, K.P.; Wemheuer, B.; Daniel, R.; Meinicke, P. Tax4Fun: Predicting functional profiles from metagenomic 16S rRNA data. Bioinformatics 2015, 31, 2882–2884. [Google Scholar] [CrossRef]
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; Szoecs, E.; Stevens, M.H.; et al. Vegan: Community Ecology Package. In R Package Version 2.5-6; 2019; Available online: https://CRAN.R-project.org/package=vegan (accessed on 1 September 2019).
- Chen, J.; Bittinger, K.; Charlson, E.S.; Hoffmann, C.; Lewis, J.; Wu, G.D.; Collman, R.G.; Bushman, F.D.; Li, H. Associating microbiome composition with environmental covariates using generalized UniFrac distances. Bioinformatics 2012, 28, 2106–2113. [Google Scholar] [CrossRef]
- Costea, P.I.; Hildebrand, F.; Arumugam, M.; Backhed, F.; Blaser, M.J.; Bushman, F.D.; de Vos, W.M.; Ehrlich, S.D.; Fraser, C.M.; Hattori, M.; et al. Enterotypes in the landscape of gut microbial community composition. Nat. Microbiol. 2018, 3, 8–16. [Google Scholar] [CrossRef] [Green Version]
- Arumugam, M.; Raes, J.; Pelletier, E.; Le Paslier, D.; Yamada, T.; Mende, D.R.; Fernandes, G.R.; Tap, J.; Bruls, T.; Batto, J.M.; et al. Enterotypes of the human gut microbiome. Nature 2011, 473, 174–180. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [Green Version]
- Tyakht, A.V.; Kostryukova, E.S.; Popenko, A.S.; Belenikin, M.S.; Pavlenko, A.V.; Larin, A.K.; Karpova, I.Y.; Selezneva, O.V.; Semashko, T.A.; Ospanova, E.A.; et al. Human gut microbiota community structures in urban and rural populations in Russia. Nat. Commun. 2013, 4, 2469. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Wu, W.; Zheng, H.M.; Li, P.; McDonald, D.; Sheng, H.F.; Chen, M.X.; Chen, Z.H.; Ji, G.Y.; Zheng, Z.D.; et al. Regional variation limits applications of healthy gut microbiome reference ranges and disease models. Nat. Med. 2018, 24, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Fan, B.; Liu, W.; Ren, R.; Chen, H.; Bai, S.; Zhu, L.; Sun, G.; Yang, Y.; Wang, X. Investigation into the stability and culturability of Chinese enterotypes. Sci. Rep. 2017, 7, 7947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandberg, J.; Kovatcheva-Datchary, P.; Bjorck, I.; Backhed, F.; Nilsson, A. Abundance of gut Prevotella at baseline and metabolic response to barley prebiotics. Eur. J. Nutr. 2019, 58, 2365–2376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mejia-Leon, M.E.; Barca, A.M. Diet, Microbiota and Immune System in Type 1 Diabetes Development and Evolution. Nutrients 2015, 7, 9171–9184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.C.; Fang, F.S.; Gong, Y.P.; Yang, G.; Li, C.L. Characteristics of repaglinide and its mechanism of action on insulin secretion in patients with newly diagnosed type-2 diabetes mellitus. Medicine 2018, 97, e12476. [Google Scholar] [CrossRef] [PubMed]
- Roth, C.; Petricevic, M.; John, A.; Goddard-Borger, E.D.; Davies, G.J.; Williams, S.J. Structural and mechanistic insights into a Bacteroides vulgatus retaining N-acetyl-beta-galactosaminidase that uses neighbouring group participation. Chem. Commun. 2016, 52, 11096–11099. [Google Scholar] [CrossRef] [Green Version]
- Shmagel, A.; Demmer, R.; Knights, D.; Butler, M.; Langsetmo, L.; Lane, N.E.; Ensrud, K. The Effects of Glucosamine and Chondroitin Sulfate on Gut Microbial Composition: A Systematic Review of Evidence from Animal and Human Studies. Nutrients 2019, 11, 294. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Zhao, X. Comparative analyses of fecal microbiota in Tibetan and Chinese Han living at low or high altitude by barcoded 454 pyrosequencing. Sci. Rep. 2015, 5, 14682. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Dan, Z.; Gesang, L.; Wang, H.; Zhou, Y.; Du, Y.; Ren, Y.; Shi, Y.; Nie, Y. Comparative Analysis of Gut Microbiota of Native Tibetan and Han Populations Living at Different Altitudes. PLoS ONE 2016, 11, e0155863. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, D.; Wang, R.; Luo, J.; Ren, F.; Gu, Z.; Zhao, Y.; Zhao, L. The Core and Distinction of the Gut Microbiota in Chinese Populations across Geography and Ethnicity. Microorganisms 2020, 8, 1579. https://doi.org/10.3390/microorganisms8101579
Lin D, Wang R, Luo J, Ren F, Gu Z, Zhao Y, Zhao L. The Core and Distinction of the Gut Microbiota in Chinese Populations across Geography and Ethnicity. Microorganisms. 2020; 8(10):1579. https://doi.org/10.3390/microorganisms8101579
Chicago/Turabian StyleLin, Deng, Ran Wang, Junjie Luo, Fazheng Ren, Zhenglong Gu, Yiqiang Zhao, and Liang Zhao. 2020. "The Core and Distinction of the Gut Microbiota in Chinese Populations across Geography and Ethnicity" Microorganisms 8, no. 10: 1579. https://doi.org/10.3390/microorganisms8101579