Acanthamoeba castellanii as a Screening Tool for Mycobacterium avium Subspecies paratuberculosis Virulence Factors with Relevance in Macrophage Infection


Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and Cultures
2.2. Amoeba Culture
2.3. Mammalian Cell Culture
2.4. Construction of MAP Transposon Library
2.5. Transposon Mutant Sequencing
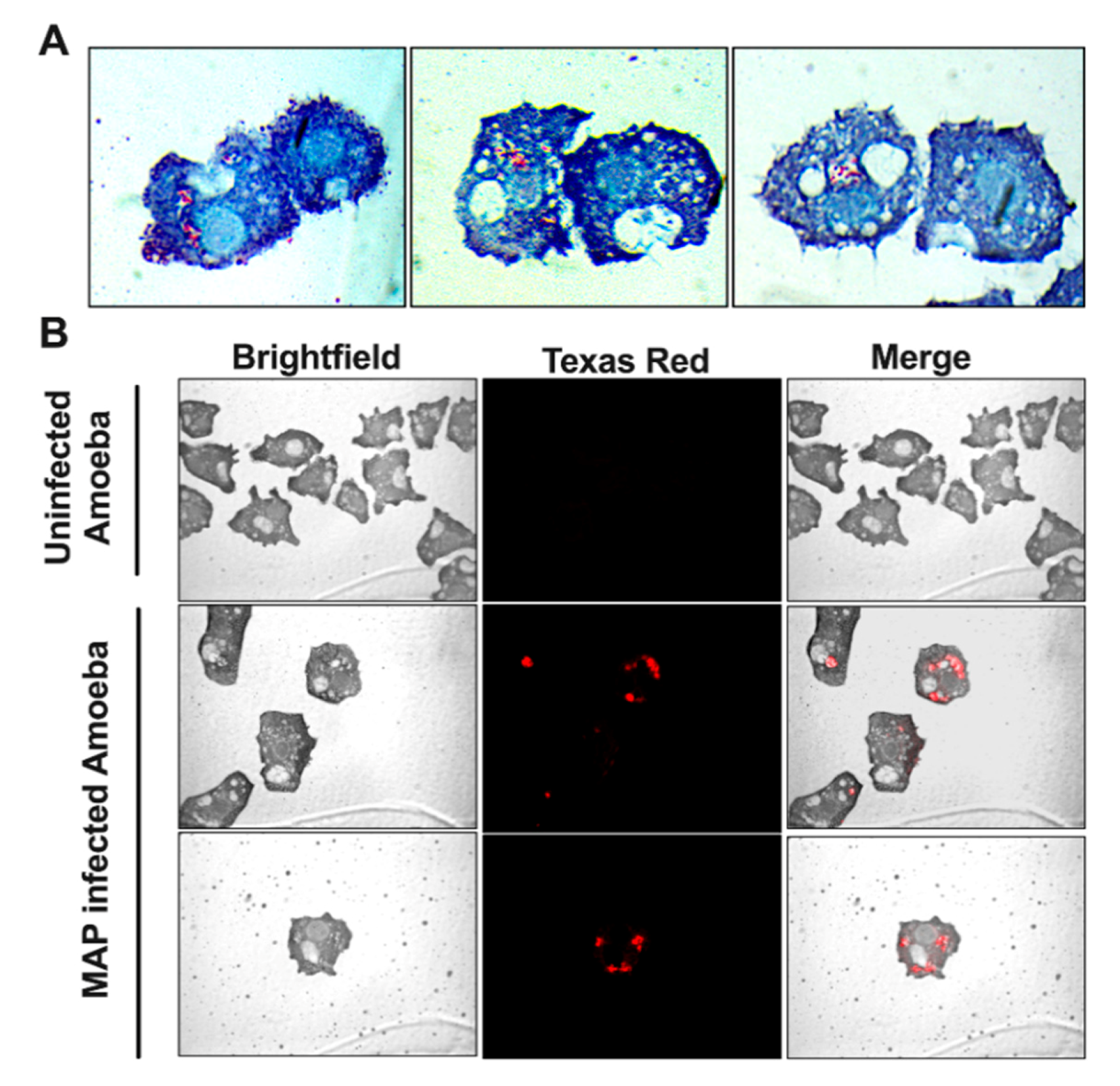
2.6. Visualization of Intracellular MAP within Amoeba
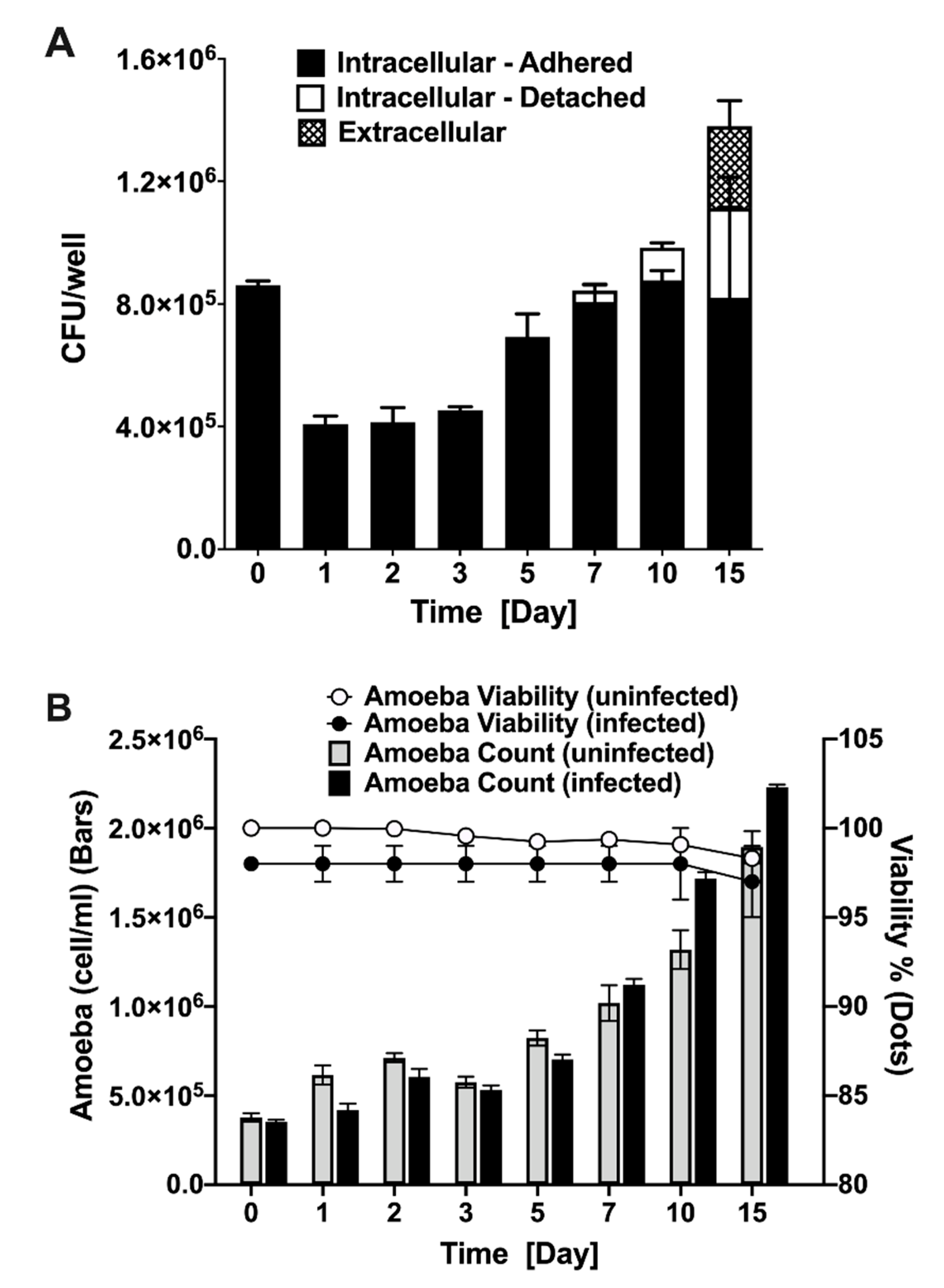
2.7. MAP Survival during Amoeba Infection and Amoeba Viability
2.8. Establishing Amoeba Metabolic Activity
2.9. Metabolic Screening of MAP Mutant Library Using Alamarblue
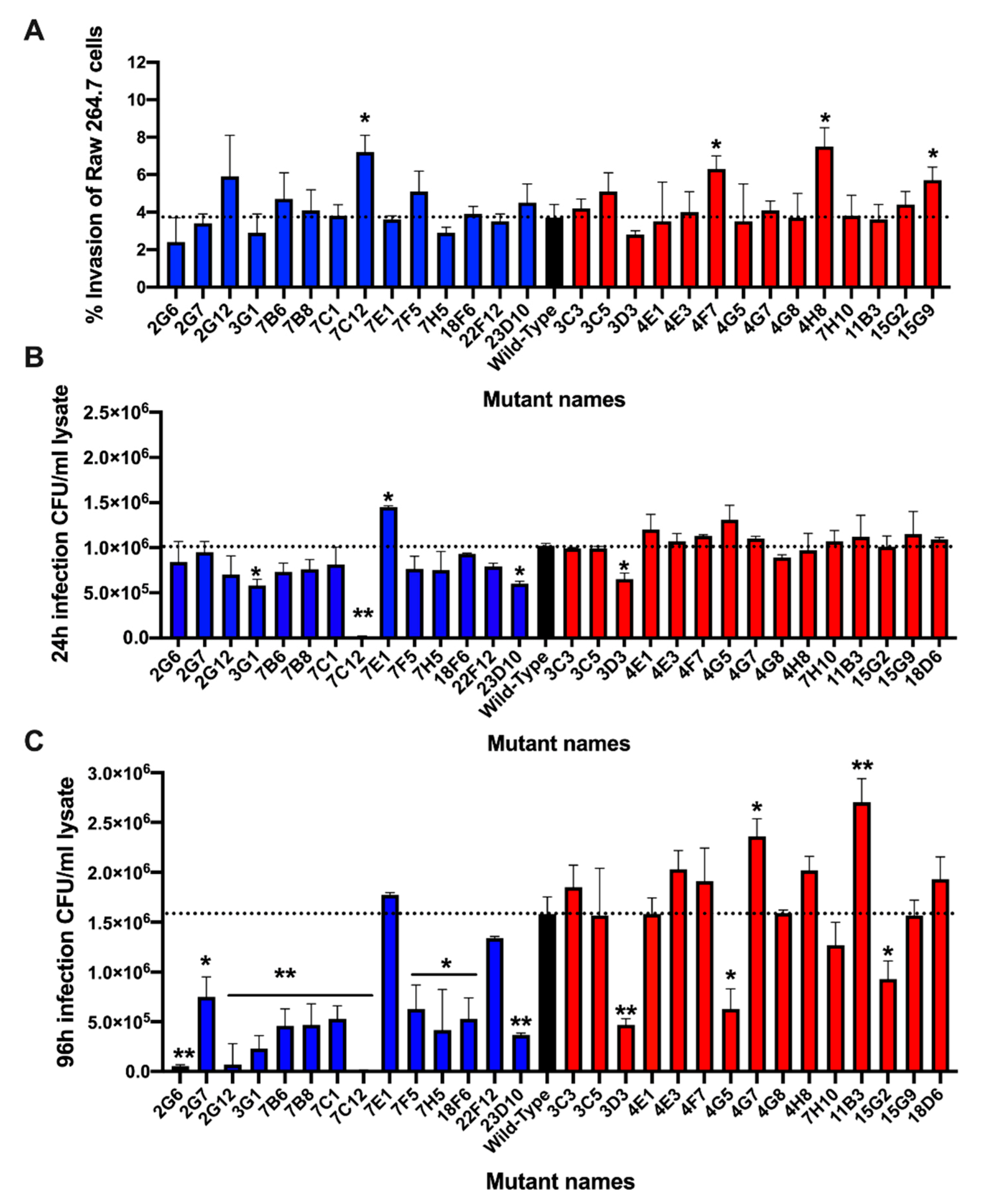
2.10. Macrophage Invasion and Survival Assays
2.11. Complementation of MAP Mutants
2.12. Statistics
3. Results
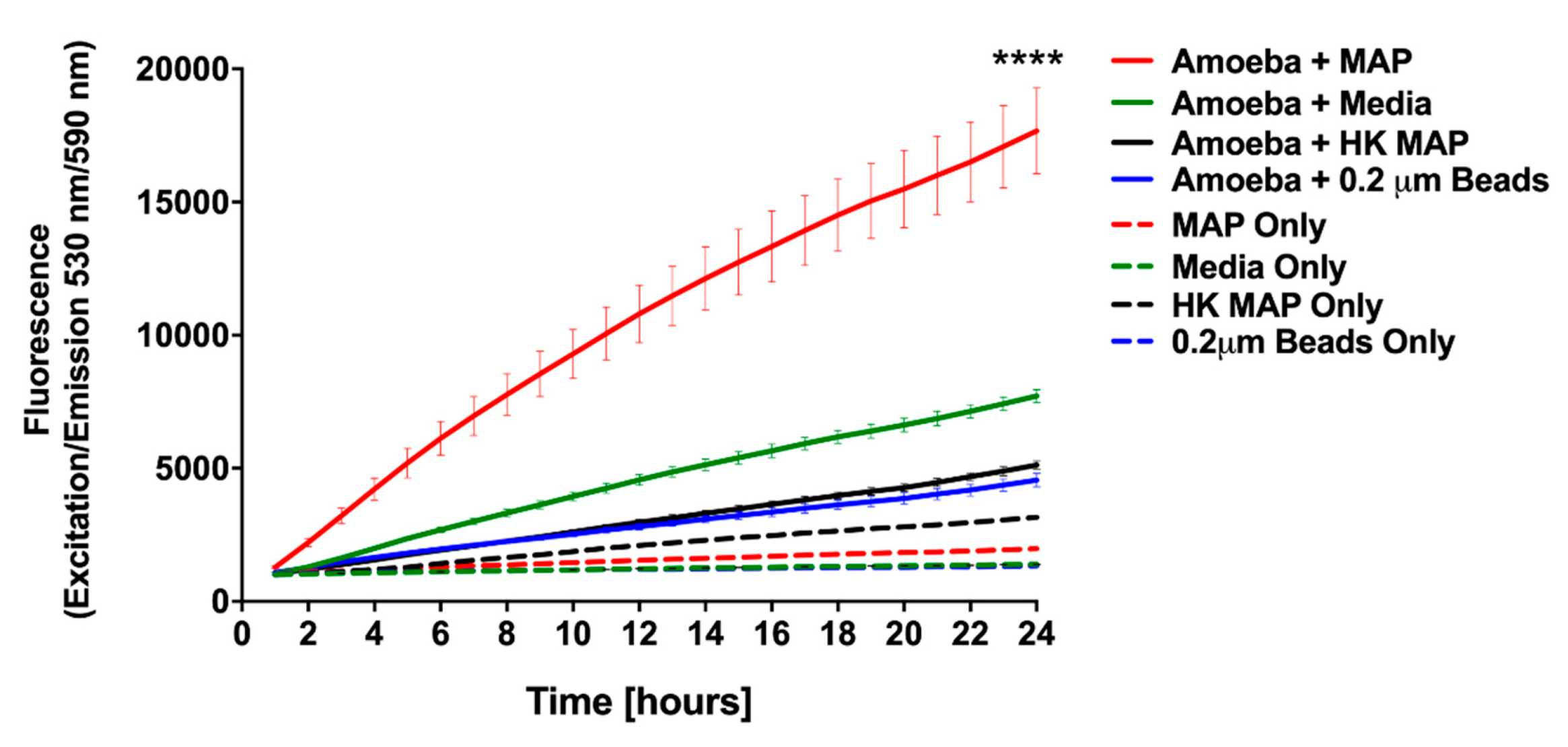
3.1. MAP is Readily Taken up by and Replicate within Acanthamoeba castellanii
3.2. Early Metabolic Response of Amoeba to MAP Infection
3.3. Late Metabolic Response of Amoeba to MAP Infection
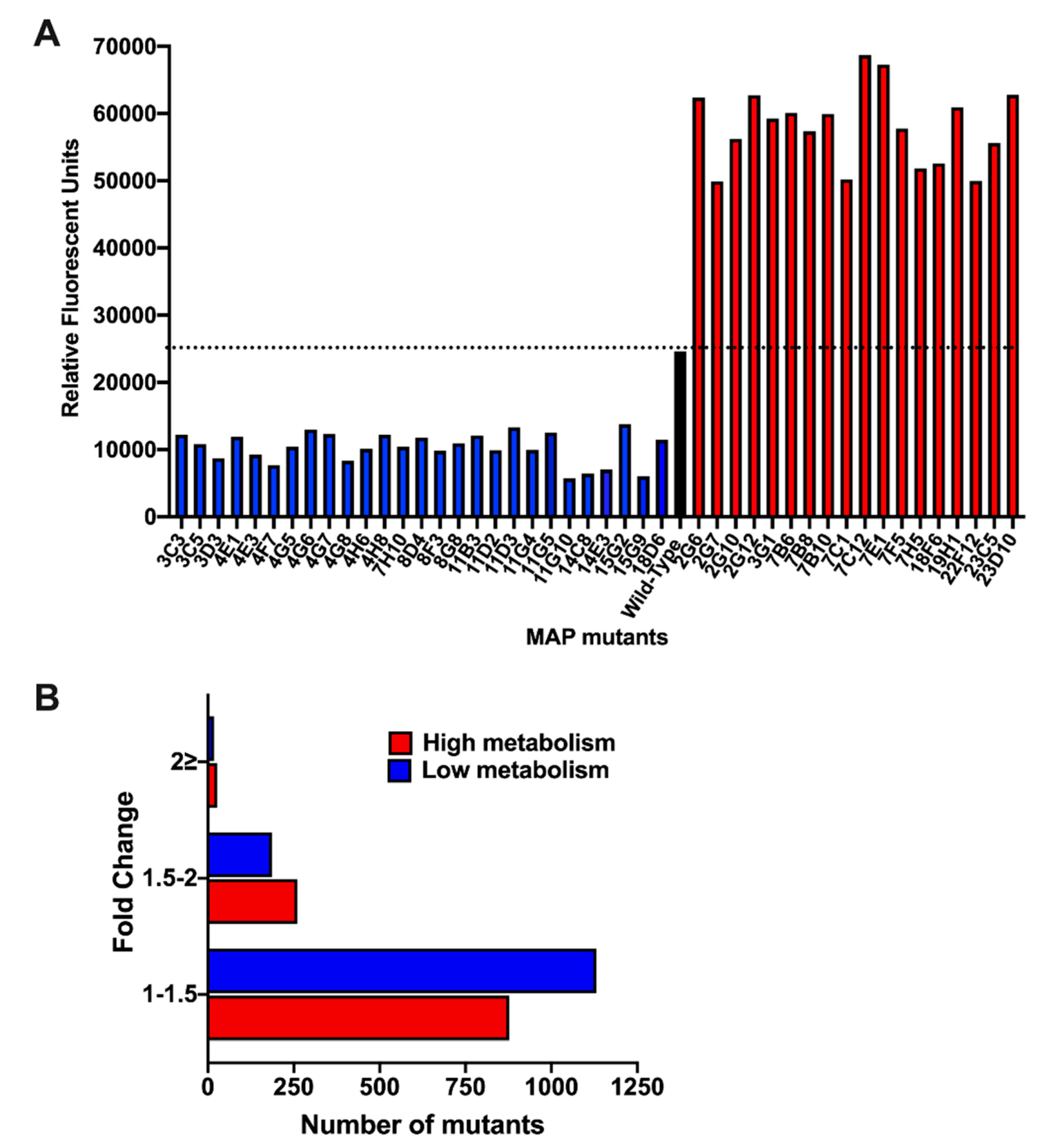
3.4. Stimulation of Amoeba Metabolism by MAP Gene Knockout Mutants
3.5. Validation of Metabolic Mutants in Macrophage Assay
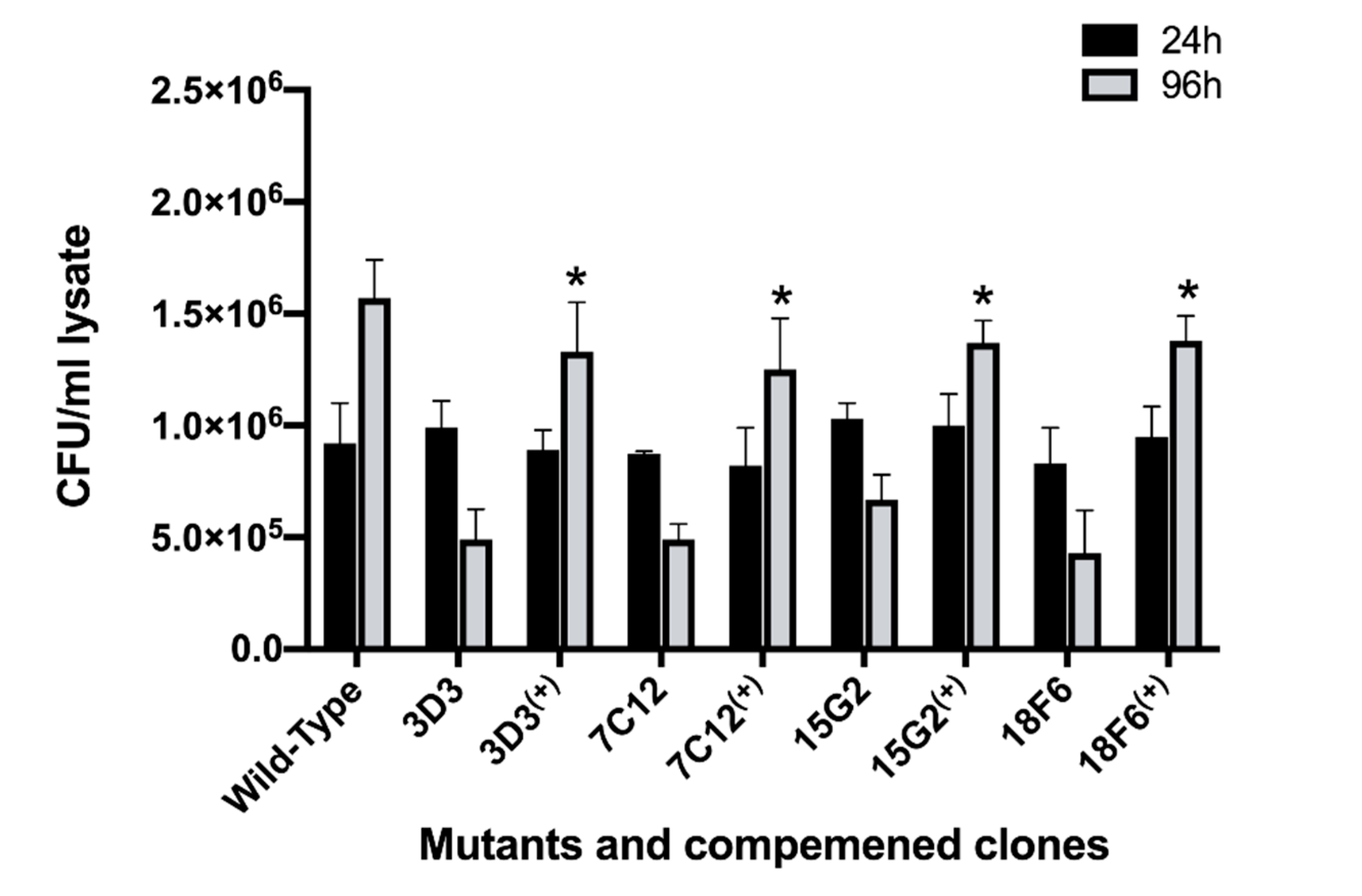
3.6. The Complementation Restores the Attenuation Phenotype in Mutants
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Li, L.; Katani, R.; Schilling, M.; Kapur, V. Molecular Epidemiology ofMycobacterium aviumsubsp.paratuberculosison Dairy Farms. Annu. Rev. Anim. Biosci. 2016, 4, 155–176. [Google Scholar] [CrossRef]
- Lombard, J.; Gardner, I.; Jafarzadeh, S.R.; Fossler, C.; Harris, B.; Capsel, R.; Wagner, B.; Johnson, W. Herd-level prevalence of Mycobacterium avium subsp. paratuberculosis infection in United States dairy herds in 2007. Prev. Veter Med. 2013, 108, 234–238. [Google Scholar] [CrossRef]
- Weiss, D.J.; Souza, C.D. Review Paper: Modulation of Mononuclear Phagocyte Function byMycobacterium aviumsubsp.paratuberculosis. Veter Pathol. 2008, 45, 829–841. [Google Scholar] [CrossRef] [PubMed]
- Arsenault, R.J.; Maattanen, P.; Daigle, J.; Potter, A.; Griebel, P.; Enapper, S. From mouth to macrophage: Mechanisms of innate immune subversion by Mycobacterium avium subsp. paratuberculosis. Veter Res. 2014, 45, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tessema, M.; Koets, A.; Rutten, V.; Gruys, E. Bacteriology: Review paratuberculosis: How doesmycobacterium aviumsubsp.Paratuberculosisresist intracellular degradation? Veter Q. 2001, 23, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Rathnaiah, G.; Zinniel, D.K.; Bannantine, J.P.; Stabel, J.R.; Gröhn, Y.T.; Collins, M.T.; Barletta, R.G. Pathogenesis, Molecular Genetics, and Genomics of Mycobacterium avium subsp. paratuberculosis, the Etiologic Agent of Johne’s Disease. Front. Veter Sci. 2017, 4, 187. [Google Scholar] [CrossRef] [Green Version]
- Abu Kwaik, Y.; Bumann, D. Host Delivery of Favorite Meals for Intracellular Pathogens. PLOS Pathog. 2015, 11, e1004866. [Google Scholar] [CrossRef] [Green Version]
- Hilbi, H.; Weber, S.S.; Ragaz, C.; Nyfeler, Y.; Urwyler, S. Environmental predators as models for bacterial pathogenesis. Environ. Microbiol. 2007, 9, 563–575. [Google Scholar] [CrossRef]
- Thewes, S.; Soldati, T.; Eichinger, L. Editorial: Amoebae as Host Models to Study the Interaction with Pathogens. Front. Cell. Infect. Microbiol. 2019, 9, 47. [Google Scholar] [CrossRef]
- Cirillo, J.D.; Falkow, S.; Tompkins, L.S.; E Bermudez, L. Interaction of Mycobacterium avium with environmental amoebae enhances virulence. Infect. Immun. 1997, 65, 3759–3767. [Google Scholar] [CrossRef] [Green Version]
- Cirillo, J.D.; Cirillo, S.L.G.; Yan, L.; Bermudez, L.E.; Falkow, S.; Tompkins, L.S. Intracellular Growth in Acanthamoeba castellanii Affects Monocyte Entry Mechanisms and Enhances Virulence of Legionella pneumophila. Infect. Immun. 1999, 67, 4427–4434. [Google Scholar] [CrossRef] [Green Version]
- Samba-Louaka, A.; Robino, E.; Cochard, T.; Branger, M.; Delafont, V.; Aucher, W.; Wambeke, W.; Bannantine, J.P.; Biet, F.; Hechard, Y. Environmental Mycobacterium avium subsp. paratuberculosis Hosted by Free-Living Amoebae. Front. Cell. Infect. Microbiol. 2018, 8, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, V.; McDonnell, G. Relationship between mycobacteria and amoebae: Ecological and epidemiological concerns. Lett. Appl. Microbiol. 2007, 45, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Falkinham, J.O. Surrounded by mycobacteria: Nontuberculous mycobacteria in the human environment. J. Appl. Microbiol. 2009, 107, 356–367. [Google Scholar] [CrossRef] [PubMed]
- Drancourt, M.; Adekambi, T.; Raoult, D. Interactions between Mycobacterium xenopi, amoeba and human cells. J. Hosp. Infect. 2007, 65, 138–142. [Google Scholar] [CrossRef] [PubMed]
- Glaser, K.C.; Hetrick, N.D.; Molestina, R.E. Evidence for a Previously Unrecognized Mycobacterial Endosymbiont in Acanthamoeba castellanii Strain Ma (ATCC®50370™). J. Eukaryot. Microbiol. 2010, 58, 75–76. [Google Scholar] [CrossRef]
- Ben Salah, I.; Drancourt, M. Surviving within the amoebal exocyst: The Mycobacterium avium complex paradigm. BMC Microbiol. 2010, 10, 99. [Google Scholar] [CrossRef] [Green Version]
- Bozzaro, S. The past, present and future of Dictyostelium as a model system. Int. J. Dev. Biol. 2019, 63, 321–331. [Google Scholar] [CrossRef] [Green Version]
- Molmeret, M.; Horn, M.; Wagner, M.; Santic, M.; Abu Kwaik, Y. Amoebae as Training Grounds for Intracellular Bacterial Pathogens. Appl. Environ. Microbiol. 2005, 71, 20–28. [Google Scholar] [CrossRef] [Green Version]
- Boulais, J.; Trost, M.; Landry, C.R.; Dieckmann, R.; Levy, E.D.; Soldati, T.; Michnick, S.W.; Thibault, P.; Desjardins, M. Molecular characterization of the evolution of phagosomes. Mol. Syst. Biol. 2010, 6, 423. [Google Scholar] [CrossRef]
- Dunn, J.D.; Bosmani, C.; Barisch, C.; Raykov, L.; Lefrançois, L.H.; Cardenal-Muñoz, E.; López-Jiménez, A.T.; Soldati, T. Eat Prey, Live: Dictyostelium discoideum As a Model for Cell-Autonomous Defenses. Front. Immunol. 2018, 8, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tenant, R.; Bermudez, L.E. Mycobacterium avium Genes Upregulated Upon Infection of Acanthamoeba castellanii Demonstrate a Common Response to the Intracellular Environment. Curr. Microbiol. 2006, 52, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Bardarov, S.; Kriakov, J.; Carriere, C.; Yu, S.; Vaamonde, C.; McAdam, R.A.; Bloom, B.R.; Hatfull, G.F.; William, R.J., Jr. Conditionally replicating mycobacteriophages: A system for transposon delivery to Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 1997, 94, 10961–10966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose, S.J.; Bermudez, L.E. Identification of Bicarbonate as a Trigger and Genes Involved with Extracellular DNA Export in Mycobacterial Biofilms. mBio 2016, 7, e01597-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stover, C.K.; De La Cruz, V.F.; Fuerst, T.R.; Burlein, J.E.; Benson, L.A.; Bennett, L.T.; Bansal, G.P.; Young, J.F.; Lee, M.H.; Hatfull, G.F.; et al. New use of BCG for recombinant vaccines. Nat. Cell Biol. 1991, 351, 456–460. [Google Scholar] [CrossRef]
- Kumar, Y.; Valdivia, R.H. Leading a Sheltered Life: Intracellular Pathogens and Maintenance of Vacuolar Compartments. Cell Host Microbe 2009, 5, 593–601. [Google Scholar] [CrossRef] [Green Version]
- Cook, G.M.; Berney, M.; Gebhard, S.; Heinemann, M.; Cox, R.A.; Danilchanka, O.; Niederweis, M. Physiology of Mycobacteria. Adv. Bact. Pathog. Biol. 2009, 55, 81–319. [Google Scholar] [CrossRef] [Green Version]
- Berney, M.; Berney-Meyer, L. Mycobacterium tuberculosis in the Face of Host-Imposed Nutrient Limitation. Microbiol. Spectr. 2017, 5, 699–715. [Google Scholar] [CrossRef] [Green Version]
- Strassmann, J.E.; Shu, L. Ancient bacteria-amoeba relationships and pathogenic animal bacteria. PLoS Biol. 2017, 15, e2002460. [Google Scholar] [CrossRef]
- Dubois, V.; Pawlik, A.; Bories, A.; Le Moigne, V.; Sismeiro, O.; Legendre, R.; Varet, H.; Rodríguez-Ordóñez, M.D.P.; Gaillard, J.-L.; Coppée, J.-Y.; et al. Mycobacterium abscessus virulence traits unraveled by transcriptomic profiling in amoeba and macrophages. PLOS Pathog. 2019, 15, e1008069. [Google Scholar] [CrossRef] [Green Version]
- Bermudez, L.E.; Petrofsky, M.; Sangari, F.J. Intracellular phenotype of Mycobacterium avium enters macrophages primarily by a macropinocytosis-like mechanism and survives in a compartment that differs from that with extracellular phenotype. Cell Biol. Int. 2004, 28, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Early, J.; Bermudez, L.E. Mimicry of the Pathogenic Mycobacterium VacuoleIn VitroElicits the Bacterial Intracellular Phenotype, Including Early-Onset Macrophage Death. Infect. Immun. 2011, 79, 2412–2422. [Google Scholar] [CrossRef] [Green Version]
- Miltner, E.C.; Bermudez, L.E. Mycobacterium avium Grown inAcanthamoeba castellanii Is Protected from the Effects of Antimicrobials. Antimicrob. Agents Chemother. 2000, 44, 1990–1994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whan, L.; Grant, I.; Ball, H.; Scott, R.; Rowe, M. Bactericidal effect of chlorine on Mycobacterium paratuberculosis in drinking water. Lett. Appl. Microbiol. 2001, 33, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Hensel, M.; Hinsley, A.P.; Nikolaus, T.; Sawers, G.; Berks, B.C. The genetic basis of tetrathionate respiration in Salmonella typhimurium. Mol. Microbiol. 1999, 32, 275–287. [Google Scholar] [CrossRef]
- Stoffels, L.; Krehenbrink, M.; Berks, B.C.; Unden, G. Thiosulfate Reduction in Salmonella enterica Is Driven by the Proton Motive Force. J. Bacteriol. 2011, 194, 475–485. [Google Scholar] [CrossRef] [Green Version]
- Kentner, D.; Martano, G.; Callon, M.; Chiquet, P.; Brodmann, M.; Burton, O.; Wahlander, A.; Nanni, P.; Delmotte, N.; Grossmann, J.; et al. Shigellareroutes host cell central metabolism to obtain high-flux nutrient supply for vigorous intracellular growth. Proc. Natl. Acad. Sci. USA 2014, 111, 9929–9934. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.; VanderVen, B.C.; Fahey, R.J.; Russell, D.G. Intracellular Mycobacterium tuberculosis Exploits Host-derived Fatty Acids to Limit Metabolic Stress. J. Biol. Chem. 2013, 288, 6788–6800. [Google Scholar] [CrossRef] [Green Version]
- Olive, A.J.; Sassetti, C.M. Metabolic crosstalk between host and pathogen: Sensing, adapting and competing. Nat. Rev. Genet. 2016, 14, 221–234. [Google Scholar] [CrossRef]
- Abu Kwaik, Y.; Bumann, D. Microbial quest for foodin vivo: ‘Nutritional virulence’ as an emerging paradigm. Cell. Microbiol. 2013, 15, 882–890. [Google Scholar] [CrossRef]
- Kedia-Mehta, N.; Finlay, D.K. Competition for nutrients and its role in controlling immune responses. Nat. Commun. 2019, 10, 2123. [Google Scholar] [CrossRef]
- Christian, J.G.; Heymann, J.; Paschen, S.A.; Vier, J.; Schauenburg, L.; Rupp, J.; Meyer, T.F.; Häcker, G.; Heuer, D. Targeting of a Chlamydial Protease Impedes Intracellular Bacterial Growth. PLoS Pathog. 2011, 7, e1002283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elwell, C.A.; Jiang, S.; Kim, J.H.; Lee, A.; Wittmann, T.; Hanada, K.; Melançon, P.; Engel, J.N. Chlamydia trachomatis Co-opts GBF1 and CERT to Acquire Host Sphingomyelin for Distinct Roles during Intracellular Development. PLoS Pathog. 2011, 7, e1002198. [Google Scholar] [CrossRef] [Green Version]
- Price, C.T.D.; Al-Quadan, T.; Santic, M.; Rosenshine, I.; Abu Kwaik, Y. Host Proteasomal Degradation Generates Amino Acids Essential for Intracellular Bacterial Growth. Science 2011, 334, 1553–1557. [Google Scholar] [CrossRef]
- Lamont, E.A.; Xu, W.; Sreevatsan, S. Host-Mycobacterium avium subsp. paratuberculosis interactome reveals a novel iron assimilation mechanism linked to nitric oxide stress during early infection. BMC Genom. 2013, 14, 694. [Google Scholar] [CrossRef] [Green Version]
- Weigoldt, M.; Meens, J.; Doll, K.; Fritsch, I.; Möbius, P.; Egoethe, R.; Gerlach, G.F. Differential proteome analysis of Mycobacterium avium subsp. paratuberculosis grown in vitro and isolated from cases of clinical Johne’s disease. Microbiology 2011, 157, 557–565. [Google Scholar] [CrossRef] [Green Version]
- Walburger, A.; Koul, A.; Ferrari, G.; Nguyen, L.; Prescianotto-Baschong, C.; Huygen, K.; Klebl, B.; Thompson, C.; Bacher, G.; Pieters, J. Protein Kinase G from Pathogenic Mycobacteria Promotes Survival Within Macrophages. Science 2004, 304, 1800–1804. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.; Walburger, A.; Houben, E.N.G.; Koul, A.; Muller, S.; Morbitzer, M.; Klebl, B.; Ferrari, G.; Epieters, J. Role of Protein Kinase G in Growth and Glutamine Metabolism of Mycobacterium bovis BCG. J. Bacteriol. 2005, 187, 5852–5856. [Google Scholar] [CrossRef] [Green Version]
- Leonard, C.J.; Aravind, L.; Koonin, E.V. Novel Families of Putative Protein Kinases in Bacteria and Archaea: Evolution of the “Eukaryotic” Protein Kinase Superfamily. Genome Res. 1998, 8, 1038–1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolff, K.A.; Nguyen, H.T.; Cartabuke, R.H.; Singh, A.; Ogwang, S.; Nguyen, L. Protein Kinase G Is Required for Intrinsic Antibiotic Resistance in Mycobacteria. Antimicrob. Agents Chemother. 2009, 53, 3515–3519. [Google Scholar] [CrossRef] [Green Version]
- Wolff, K.A.; De La Peña, A.H.; Nguyen, H.T.; Pham, T.H.; Amzel, L.M.; Gabelli, S.B.; Nguyen, L. A Redox Regulatory System Critical for Mycobacterial Survival in Macrophages and Biofilm Development. PLoS Pathog. 2015, 11, e1004839. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, G.; Shrivastva, R.; Mukhopadhyay, S. Mycobacterial PknG Targets the Rab7l1 Signaling Pathway To Inhibit Phagosome–Lysosome Fusion. J. Immunol. 2018, 201, 1421–1433. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.Z.; Bhaskar, A.; Upadhyay, S.; Kumari, P.; Rajmani, R.S.; Jain, P.; Singh, A.; Kumar, D.; Bhavesh, N.S.; Nandicoori, V.K. Protein kinase G confers survival advantage toMycobacterium tuberculosisduring latency-like conditions. J. Biol. Chem. 2017, 292, 16093–16108. [Google Scholar] [CrossRef] [Green Version]
- Rieck, B.; Degiacomi, G.; Zimmermann, M.; Cascioferro, A.; Boldrin, F.; Adler, N.R.L.; Bottrill, A.R.; Le Chevalier, F.; Frigui, W.; Bellinzoni, M.; et al. PknG senses amino acid availability to control metabolism and virulence of Mycobacterium tuberculosis. PLoS Pathog. 2017, 13, e1006399. [Google Scholar] [CrossRef]
- Scherr, N.; Honnappa, S.; Kunz, G.; Mueller, P.; Jayachandran, R.; Winkler, F.; Epieters, J.; Steinmetz, M.O. Structural basis for the specific inhibition of protein kinase G, a virulence factor of Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2007, 104, 12151–12156. [Google Scholar] [CrossRef] [Green Version]
- Purcell, E.B.; Tamayo, R. Cyclic diguanylate signaling in Gram-positive bacteria. FEMS Microbiol. Rev. 2016, 40, 753–773. [Google Scholar] [CrossRef]
- Freitas, T.A.K.; Saito, J.A.; Wan, X.; Hou, S.; Alam, M. Protoglobin and Globin-coupled Sensors. In The Smallest Biomolecules: Diatomics and their Interactions with Heme Proteins; Elsevier: Amsterdam, The Netherlands, 2008; pp. 175–202. [Google Scholar]
- Römling, U.; Galperin, M.Y.; Gomelsky, M. Cyclic di-GMP: The First 25 Years of a Universal Bacterial Second Messenger. Microbiol. Mol. Biol. Rev. 2013, 77, 1–52. [Google Scholar] [CrossRef] [Green Version]
- Valentini, M.; Filloux, A. Biofilms and Cyclic di-GMP (c-di-GMP) Signaling: Lessons from Pseudomonas aeruginosa and Other Bacteria. J. Biol. Chem. 2016, 291, 12547–12555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simm, R.; Morr, M.; Kader, A.; Nimtz, M.; Römling, U. GGDEF and EAL domains inversely regulate cyclic di-GMP levels and transition from sessility to motility. Mol. Microbiol. 2004, 53, 1123–1134. [Google Scholar] [CrossRef]
- Valentini, M.; Filloux, A. Multiple Roles of c-di-GMP Signaling in Bacterial Pathogenesis. Annu. Rev. Microbiol. 2019, 73, 387–406. [Google Scholar] [CrossRef]
- Kalia, D.; Merey, G.; Nakayama, S.; Zheng, Y.; Zhou, J.; Luo, Y.; Guo, M.; Roembke, B.T.; Sintim, H.O. Nucleotide, c-di-GMP, c-di-AMP, cGMP, cAMP, (p)ppGpp signaling in bacteria and implications in pathogenesis. Chem. Soc. Rev. 2013, 42, 305–341. [Google Scholar] [CrossRef] [PubMed]
- Hall, C.L.; Lee, V.T. Cyclic-di-GMP regulation of virulence in bacterial pathogens. Wiley Interdiscip. Rev. RNA 2017, 9, e1454. [Google Scholar] [CrossRef]
- Johnson, R.M.; McDonough, K.A. Cyclic nucleotide signaling in Mycobacterium tuberculosis: An expanding repertoire. Pathog. Dis. 2018, 76. [Google Scholar] [CrossRef] [Green Version]
- Hong, Y.; Zhou, X.; Fang, H.; Yu, D.; Li, C.; Sun, B. Cyclic di-GMP mediates Mycobacterium tuberculosis dormancy and pathogenecity. Tuberculosis 2013, 93, 625–634. [Google Scholar] [CrossRef]
- Pathania, R.; Navani, N.K.; Rajamohan, G.; Dikshit, K.L. Mycobacterium tuberculosisHemoglobin HbO Associates with Membranes and Stimulates Cellular Respiration of RecombinantEscherichia coli. J. Biol. Chem. 2002, 277, 15293–15302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hade, M.D.; Sethi, D.; Datta, H.; Singh, S.; Thakur, N.; Chhaya, A.; Dikshit, K.L. Truncated Hemoglobin O Carries an Autokinase Activity and Facilitates Adaptation of Mycobacterium tuberculosis Under Hypoxia. Antioxid. Redox Signal. 2019, 32, 351–362. [Google Scholar] [CrossRef] [PubMed]
- Bustamante, J.P.; Radusky, L.; Boechi, L.; Estrin, D.A.; Have, A.T.; Salierno, M. Evolutionary and Functional Relationships in the Truncated Hemoglobin Family. PLoS Comput. Biol. 2016, 12, e1004701. [Google Scholar] [CrossRef]
- Fabozzi, G.; Ascenzi, P.; Di Renzi, S.; Visca, P. Truncated hemoglobin GlbO from Mycobacterium leprae alleviates nitric oxide toxicity. Microb. Pathog. 2006, 40, 211–220. [Google Scholar] [CrossRef]
- Visca, P.; Fabozzi, G.; Petrucca, A.; Ciaccio, C.; Coletta, M.; De Sanctis, G.; Bolognesi, M.; Milani, M.; Ascenzi, P. The truncated hemoglobin from Mycobacterium leprae. Biochem. Biophys. Res. Commun. 2002, 294, 1064–1070. [Google Scholar] [CrossRef]
- Visca, P.; Fabozzi, G.; Milani, M.; Bolognesi, M.; Ascenzi, P. Nitric Oxide and Mycobacterium leprae Pathogenicity. IUBMB Life 2002, 54, 95–99. [Google Scholar] [CrossRef]
- Ascenzi, P.; Milani, M.; Visca, P. Peroxynitrite scavenging by ferrous truncated hemoglobin GlbO from Mycobacterium leprae. Biochem. Biophys. Res. Commun. 2006, 351, 528–533. [Google Scholar] [CrossRef]
- Pawaria, S.; Lama, A.; Raje, M.; Dikshit, K.L. Responses of Mycobacterium tuberculosis Hemoglobin Promoters to In Vitro and In Vivo Growth Conditions. Appl. Environ. Microbiol. 2008, 74, 3512–3522. [Google Scholar] [CrossRef] [Green Version]
- Mainardi, J.-L.; Villet, R.; Bugg, T.D.; Mayer, C.; Arthur, M. Evolution of peptidoglycan biosynthesis under the selective pressure of antibiotics in Gram-positive bacteria. FEMS Microbiol. Rev. 2008, 32, 386–408. [Google Scholar] [CrossRef] [Green Version]
- Schoonmaker, M.K.; Bishai, W.R.; Lamichhane, G. Nonclassical Transpeptidases of Mycobacterium tuberculosis Alter Cell Size, Morphology, the Cytosolic Matrix, Protein Localization, Virulence, and Resistance to -Lactams. J. Bacteriol. 2014, 196, 1394–1402. [Google Scholar] [CrossRef] [Green Version]
- Sabe, V.T.; Tolufashe, G.F.; Ibeji, C.U.; Maseko, S.B.; Govender, T.; Maguire, G.E.M.; Lamichhane, G.; Honarparvar, B.; Kruger, H.G. Identification of potent L,D-transpeptidase 5 inhibitors for Mycobacterium tuberculosis as potential anti-TB leads: Virtual screening and molecular dynamics simulations. J. Mol. Model. 2019, 25, 328. [Google Scholar] [CrossRef]
- Dubée, V.; Triboulet, S.; Mainardi, J.-L.; Ethève-Quelquejeu, M.; Gutmann, L.; Marie, A.; Dubost, L.; Hugonnet, J.-E.; Arthur, M. Inactivation of Mycobacterium tuberculosis l,d-Transpeptidase LdtMt1by Carbapenems and Cephalosporins. Antimicrob. Agents Chemother. 2012, 56, 4189–4195. [Google Scholar] [CrossRef] [Green Version]
- Kumar, P.; Chauhan, V.; Silva, J.R.A.; Lameira, J.; D’Andrea, F.; Li, S.-G.; Ginell, S.L.; Freundlich, J.S.; Alves, C.N.; Bailey, S.; et al. Mycobacterium abscessus l,d-Transpeptidases Are Susceptible to Inactivation by Carbapenems and Cephalosporins but Not Penicillins. Antimicrob. Agents Chemother. 2017, 61, e00866-17. [Google Scholar] [CrossRef] [Green Version]
- Kumar, P.; Kaushik, A.; Lloyd, E.P.; Li, S.-G.; Mattoo, R.; Ammerman, N.C.; Bell, D.T.; Perryman, A.L.; Zandi, T.A.; Ekins, S.; et al. Non-classical transpeptidases yield insight into new antibacterials. Nat. Chem. Biol. 2016, 13, 54–61. [Google Scholar] [CrossRef] [Green Version]
- Geiger, T.; Pazos, M.; Lara-Tejero, M.; Vollmer, W.; Galán, J.E. Peptidoglycan editing by a specific ld-transpeptidase controls the muramidase-dependent secretion of typhoid toxin. Nat. Microbiol. 2018, 3, 1243–1254. [Google Scholar] [CrossRef]
- Juan, C.; Torrens, G.; Barceló, I.M.; Oliver, A. Interplay between Peptidoglycan Biology and Virulence in Gram-Negative Pathogens. Microbiol. Mol. Biol. Rev. 2018, 82, e00033-18. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name Mutant | Sequence |
|---|---|
| MAP_0949 7C12 | Forward (HindIII) 5′-TTTTTAAGCTTGTGCCACGCAGCCTGGAC-3′ Reverse (NotI) 5′-TTTTTGCGGCCGCCTAGATCGAGCCCATC-3′ |
| MAP_2291 18F6 | Forward (HindIII) 5′-TTTTTAAGCTTATGGATCAGGTGAGCTTC-3′ Reverse (NotI) 5′-TTTTTGCGGCCGCTCACAACGGAGAATTCAC-3′ |
| MAP_3634 15G2 | Forward (HindIII) 5′-TTTTTAAGCTTATGTCGGGCTGGACGAG-3′ Reverse (NotI) 5′-TTTTTGCGGCCGCCTAGTTCATCCAGTCG-3′ |
| MAP_3893c 3D3 | Forward (BamHI) 5′-TTTTTGGATCCATGGCCGAGCCGGA -3′ Reverse (EcoRI) 5′-TTTTTGAATTCTCAGAACGTGCTGGTGGG -3′ |
| Mutant | Gene Name | Amoeba Metabolism | Attenuated in Macrophages | Function/Domains/Notes |
|---|---|---|---|---|
| 15G9 | MAP_0800c | Low | No | Helicase_C_3 domain-containing protein |
| 2G7 | MAP_0122 | High | Yes | PE (proline-glutamate) family protein |
| 4E3 | MAP_0294c/pca | Low | No | Pyruvate carboxylase |
| 18D6 | MAP_0338c | Low | No | DUF772 domain-containing protein |
| 3C5 | MAP_0824/aurF | Low | No | Metalloenzyme P-aminobenzoate N-oxygenase |
| 4F7 | MAP_0847 | Low | No | DUF4185 domain-containing protein |
| 7C12 | MAP_0949 | High | Yes | Hypothetical protein with EAL domain/Diguanylate cyclase domain |
| 7E1 | MAP_1024/cysM2 | High | No | Cystathionine beta-synthase |
| 14C8 | MAP_1076 | Low | N/A | Hypothetical protein |
| 2G12 | MAP_1133 | High | Yes | Methionyl-tRNA formyltransferase |
| 7B10 | MAP_1221 | High | N/A | DNA-binding response regulator, OmpR family, contains REC and winged-helix (wHTH) domain |
| 4H8 | MAP_1301/chaA | Low | No | Ca2+/H+ antiporter |
| 14E3 | MAP_1320c | Low | No | Thiolase_N domain-containing protein; Lipid-transfer protein |
| 23D10 | MAP_1423 | High | Yes | Major Facilitator Superfamily (MFS) domain-containing protein; Benzoate transport |
| 4G7 | MAP_1450c | Low | No | Flavoprotein CzcO associated with the cation diffusion facilitator CzcD |
| 7B8 | MAP_1591 | High | Yes | Methylmuconolactone methyl-isomerase; EthD domain-containing protein |
| 7B6 | MAP_1592 | High | Yes | Putative_PNPOx domain-containing protein |
| 11D3 | MAP_1642 | Low | N/A | Lactamase_B domain-containing protein |
| 11D2 | MAP_1824c | Low | N/A | Integrase catalytic domain-containing protein |
| 22F12 | MAP_1842c | High | No | tRNA (adenine(58)-N(1))-methyltransferase TrmI |
| 2G6 | MAP_2127 | High | Yes | Flavin-utilizing monoxygenases; Bac_luciferase domain-containing protein |
| 3C3 | MAP_2228 | Low | No | Pimeloyl-CoA dehydrogenase |
| 18F6 | MAP_2291/glbO | High | Yes | Truncated hemoglobin, group 2 (O) |
| 7H5 | MAP_2324c | High | Yes | YdfJ uncharacterized membrane protein/MmpL |
| 2G10 | MAP_2363c | High | N/A | Acyl-CoA dehydrogenase related to the alkylation response protein AidB |
| 11B3 | MAP_2535 | Low | No | Hypothetical protein |
| 7F5 | MAP_2843c | High | Yes | Hypothetical protein |
| 3G1 | MAP_2973/llpW | High | Yes | Penicillin binding protein transpeptidase domain-containing protein; Lipid transport |
| 15G2 | MAP_3634 | Low | Yes | IgD-like repeat domain of mycobacterial L,D-transpeptidases |
| 4G8 | MAP_3717c | Low | No | Dipeptidyl aminopeptidase/acylaminoacyl peptidase domain |
| 4H6 | MAP_3761c | Low | N/A | Transmembrane sulfolipid-1 addressing protein SAP |
| 4G5 | MAP_3832c | Low | Yes | Molecular chaperone DnaK |
| 3D3 | MAP_3893c/pknG | Low | Yes | Serine/Threonine kinase |
| 7C1 | MAP_3947/mmpl4_7 | High | Yes | Uncharacterized membrane protein mmpL |
| 7H10 | MAP_4350c | Low | No | 50s ribosomal protein L34 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Phillips, I.L.; Everman, J.L.; Bermudez, L.E.; Danelishvili, L. Acanthamoeba castellanii as a Screening Tool for Mycobacterium avium Subspecies paratuberculosis Virulence Factors with Relevance in Macrophage Infection. Microorganisms 2020, 8, 1571. https://doi.org/10.3390/microorganisms8101571
Phillips IL, Everman JL, Bermudez LE, Danelishvili L. Acanthamoeba castellanii as a Screening Tool for Mycobacterium avium Subspecies paratuberculosis Virulence Factors with Relevance in Macrophage Infection. Microorganisms. 2020; 8(10):1571. https://doi.org/10.3390/microorganisms8101571
Chicago/Turabian StylePhillips, Ida L., Jamie L. Everman, Luiz E. Bermudez, and Lia Danelishvili. 2020. "Acanthamoeba castellanii as a Screening Tool for Mycobacterium avium Subspecies paratuberculosis Virulence Factors with Relevance in Macrophage Infection" Microorganisms 8, no. 10: 1571. https://doi.org/10.3390/microorganisms8101571