Developmental, Dietary, and Geographical Impacts on Gut Microbiota of Red Swamp Crayfish (Procambarus clarkii)

Abstract
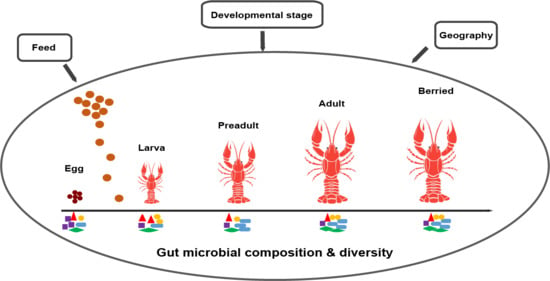
:
1. Introduction
2. Materials and Methods
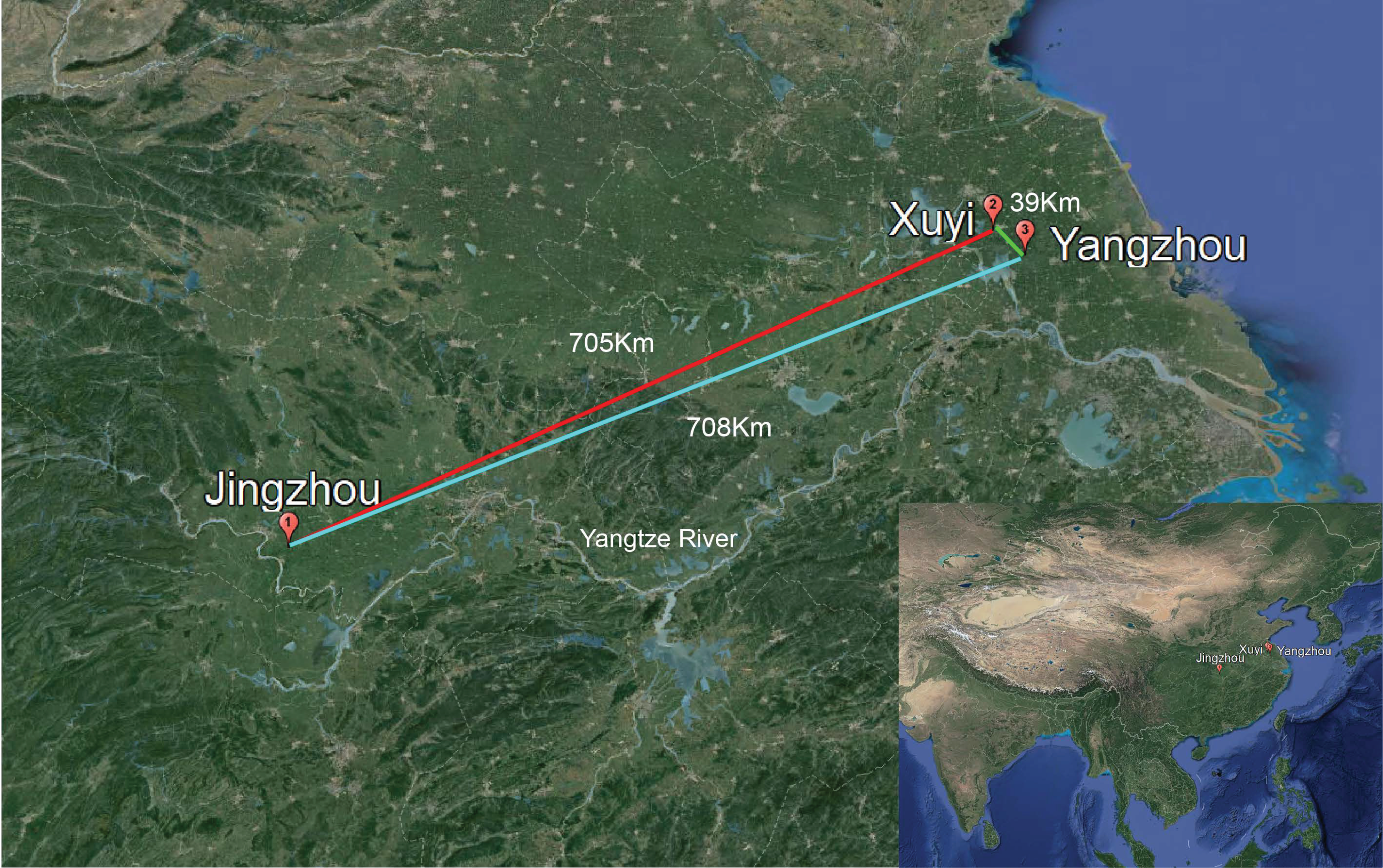
2.1. Sample Collection
2.2. DNA Extraction and 16S rRNA Gene Amplification
2.3. High-Throughput Sequencing and Data Analysis
3. Results
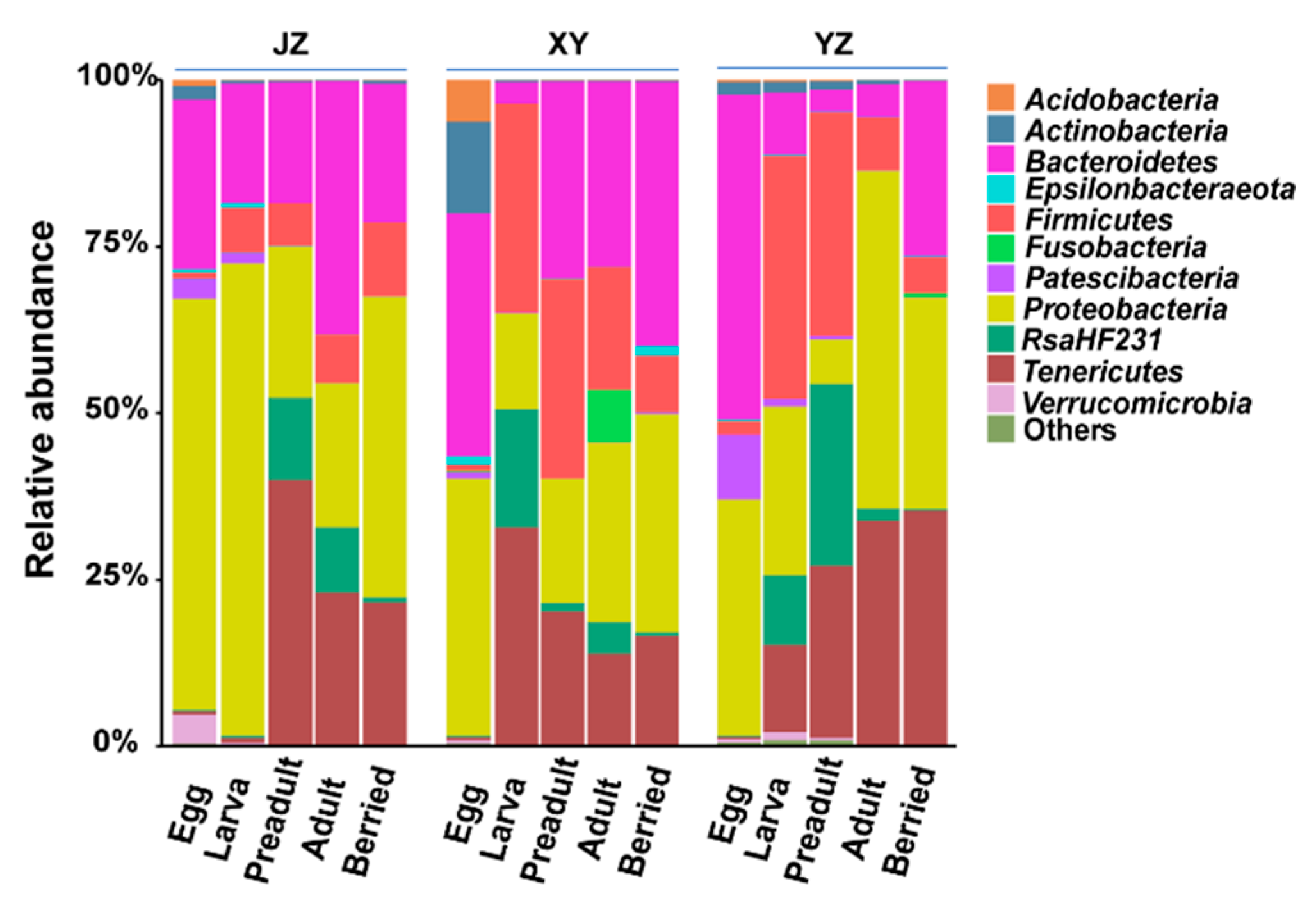
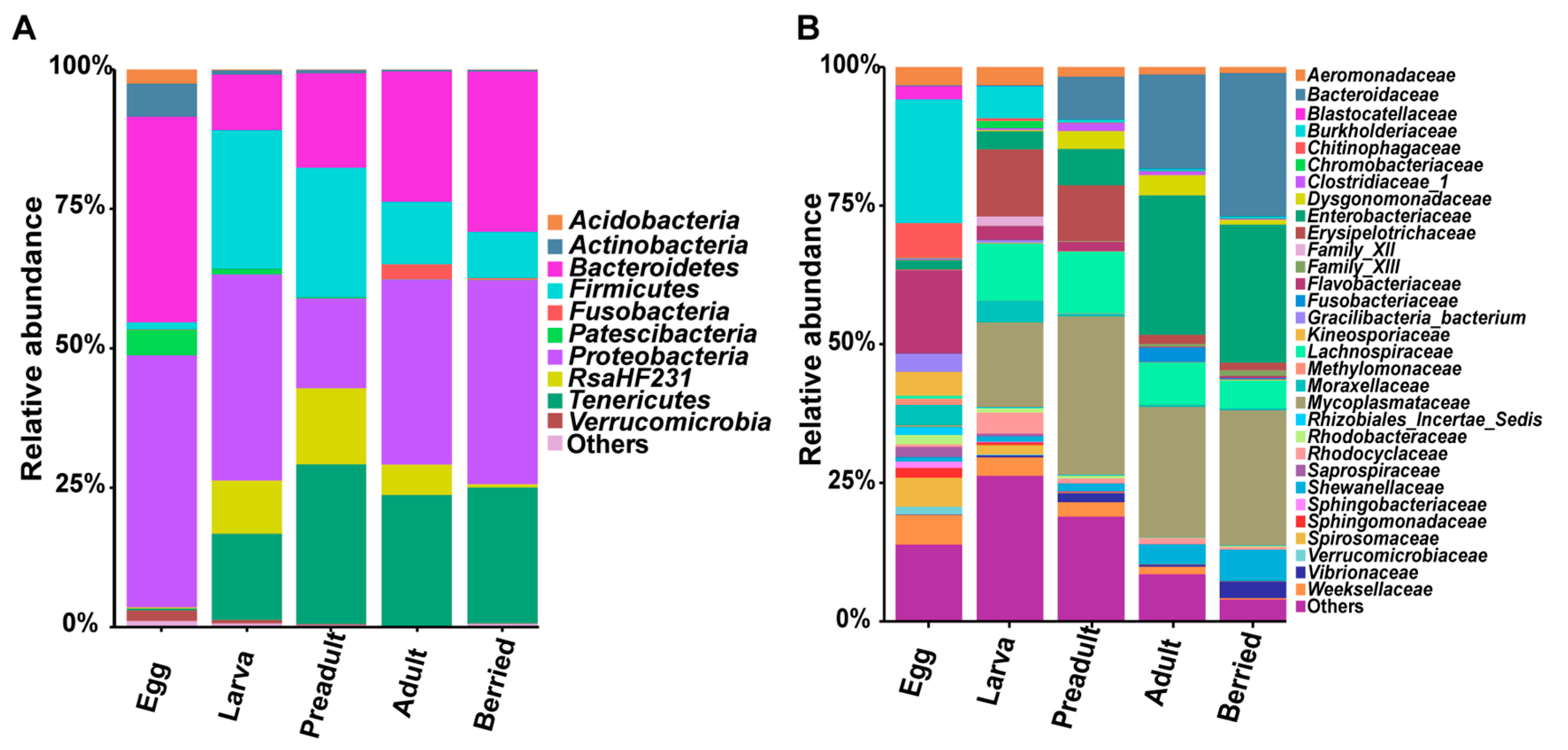
3.1. Dominant Bacteria at the Phyla Level
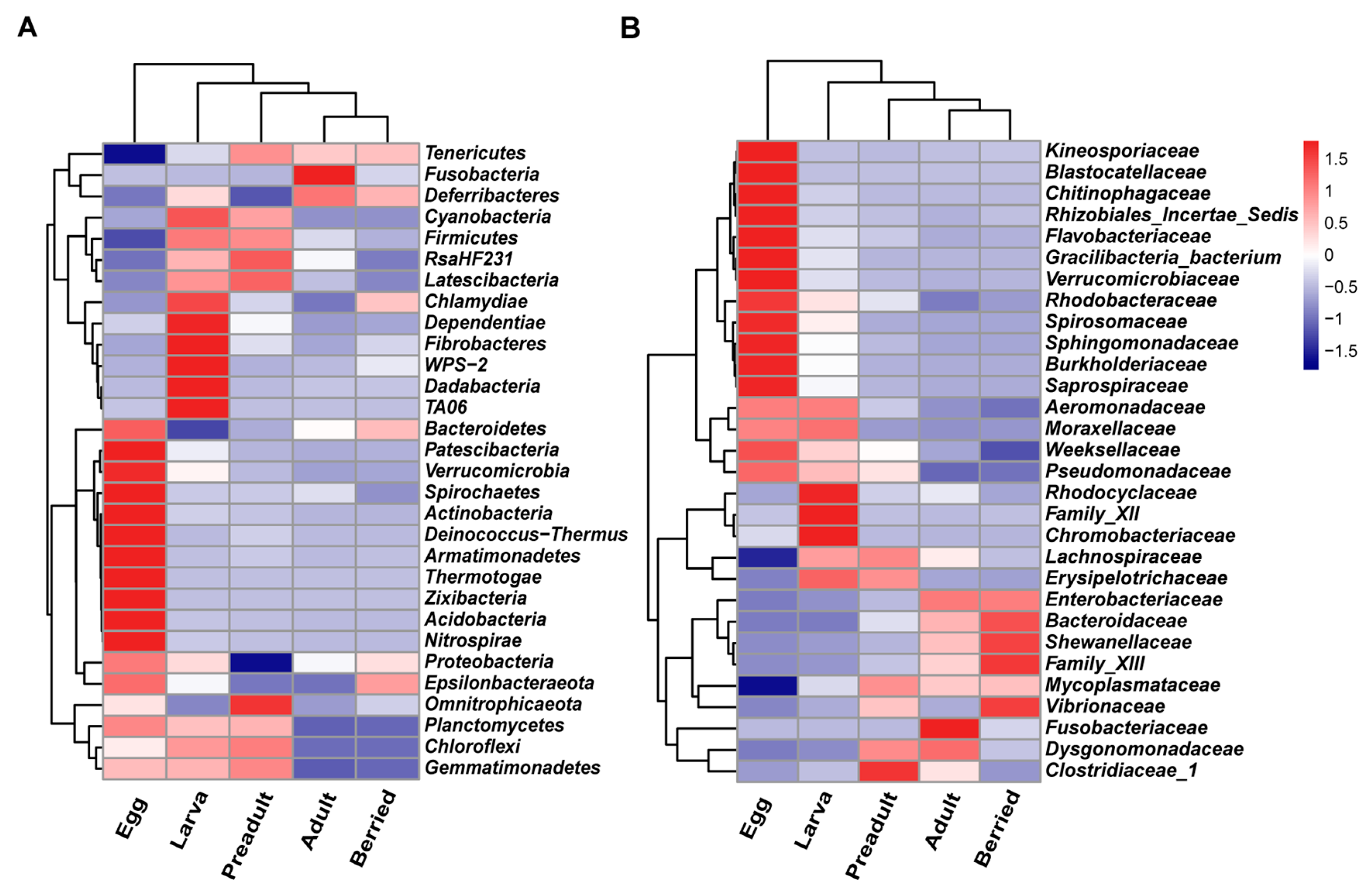
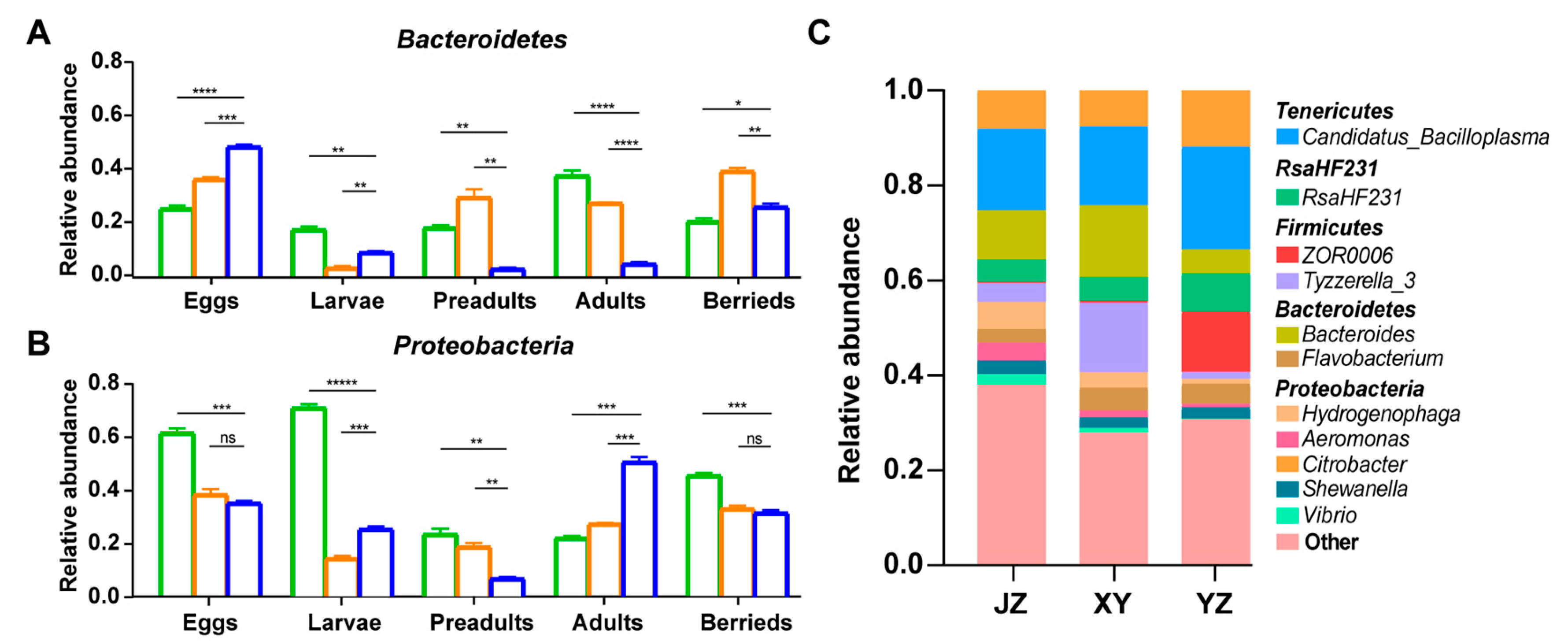
3.2. Composition and Dynamics of Gut Microbiota during Developmental Stages
3.3. Correlation between the Crayfish Gut Microbiota and Developmental Stages
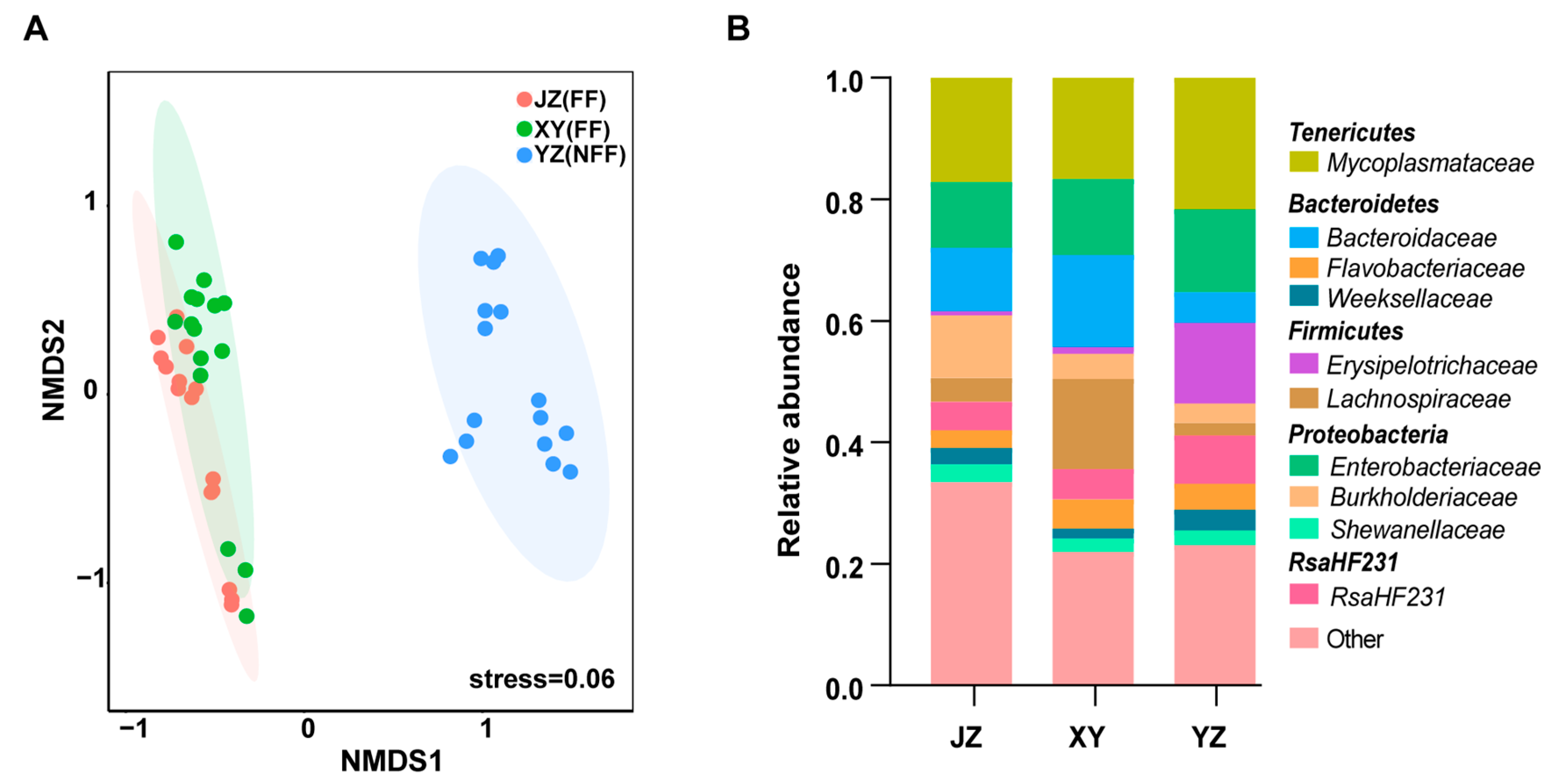
3.4. Effects of Diet and Geographical Location on Gut Microbiota
3.5. Fermented Feed Affects the Gut’s Microbial Composition
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Data Accessibility
References
- Ross, B.D.; Verster, A.J.; Radey, M.C.; Schmidtke, D.T.; Pope, C.E.; Hoffman, L.R.; Hajjar, A.M.; Peterson, S.B.; Borenstein, E.; Mougous, J.D. Human gut bacteria contain acquired interbacterial defence systems. Nature 2019, 575, 224–228. [Google Scholar] [CrossRef] [PubMed]
- Cho, I.; Blaser, M.J. The human microbiome: At the interface of health and disease. Nat. Rev. Genet. 2012, 13, 260–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hooper, L.V.; Littman, D.R.; Macpherson, A.J. Interactions between the microbiota and the immune system. Science 2012, 336, 1268–1273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piewngam, P.; Zheng, Y.; Nguyen, T.H.; Dickey, S.W.; Joo, H.S.; Villaruz, A.E.; Glose, K.A.; Fisher, E.L.; Hunt, R.L.; Li, B.; et al. Pathogen elimination by probiotic Bacillus via signalling interference. Nature 2018, 562, 532–537. [Google Scholar] [CrossRef] [PubMed]
- Tramontano, M.; Andrejev, S.; Pruteanu, M.; Klunemann, M.; Kuhn, M.; Galardini, M.; Jouhten, P.; Zelezniak, A.; Zeller, G.; Bork, P.; et al. Nutritional preferences of human gut bacteria reveal their metabolic idiosyncrasies. Nat. Microbiol. 2018, 3, 514–522. [Google Scholar] [CrossRef]
- Bascunan, P.; Nino-Garcia, J.P.; Galeano-Castaneda, Y.; Serre, D.; Correa, M.M. Factors shaping the gut bacterial community assembly in two main Colombian malaria vectors. Microbiome 2018, 6, 148. [Google Scholar] [CrossRef]
- Frese, S.A.; Parker, K.; Calvert, C.C.; Mills, D.A. Diet shapes the gut microbiome of pigs during nursing and weaning. Microbiome 2015, 3, 28. [Google Scholar] [CrossRef] [Green Version]
- Larry, J.L.; Scott, A.K.; Joseph, N.; Ryan, M.L.; Fangrui, M.; Kunjie, H.; Rohita, S.; Daniel, A.P.; Jens, W.; Andrew, K.B.; et al. Host genetics and diet, but not immunoglobulin A expression, converge to shape compositional features of the gut microbiome in an advanced intercross population of mice. Genomebiology 2014, 15, 552. [Google Scholar]
- Michl, S.C.; Beyer, M.; Ratten, J.M.; Hasler, M.; LaRoche, J.; Schulz, C. A diet-change modulates the previously established bacterial gut community in juvenile brown trout (Salmo trutta). Sci. Rep. 2019, 9, 2339. [Google Scholar] [CrossRef] [Green Version]
- Halmos, E.P.; Christophersen, C.T.; Bird, A.R.; Shepherd, S.J.; Gibson, P.R.; Muir, J.G. Diets that differ in their FODMAP content alter the colonic luminal microenvironment. Gut 2015, 64, 93–100. [Google Scholar] [CrossRef]
- Takagi, T.; Naito, Y.; Inoue, R.; Kashiwagi, S.; Uchiyama, K.; Mizushima, K.; Tsuchiya, S.; Dohi, O.; Yoshida, N.; Kamada, K.; et al. Differences in gut microbiota associated with age, sex, and stool consistency in healthy Japanese subjects. J. Gastroenterol. 2019, 54, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human gut microbiome viewed across age and geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Escalas, A.; Troussellier, M.; Yuan, T.; Bouvier, T.; Bouvier, C.; Mouchet, M.A.; Flores Hernandez, D.; Ramos Miranda, J.; Zhou, J.; Mouillot, D. Functional diversity and redundancy across fish gut, sediment and water bacterial communities. Environ. Microbiol. 2017, 19, 3268–3282. [Google Scholar] [CrossRef] [PubMed]
- Goodrich, J.K.; Waters, J.L.; Poole, A.C.; Sutter, J.L.; Koren, O.; Blekhman, R.; Beaumont, M.; Van Treuren, W.; Knight, R.; Bell, J.T.; et al. Human genetics shape the gut microbiome. Cell 2014, 159, 789–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roeselers, G.; Mittge, E.K.; Stephens, W.Z.; Parichy, D.M.; Cavanaugh, C.M.; Guillemin, K.; Rawls, J.F. Evidence for a core gut microbiota in the zebrafish. ISME J. 2011, 5, 1595–1608. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Sun, Y.; Chen, K.; Yu, N.; Zhou, Z.; Chen, L.; Du, Z.; Li, E. Characterization of the intestinal microbiota in Pacific white shrimp, Litopenaeus vannamei, fed diets with different lipid sources. Aquaculture 2014, 434, 449–455. [Google Scholar] [CrossRef]
- Xiong, J.; Dai, W.; Li, C. Advances, challenges, and directions in shrimp disease control: The guidelines from an ecological perspective. Appl. Microbiol. Biotechnol. 2016, 100, 6947–6954. [Google Scholar] [CrossRef]
- Xiong, J.; Wang, K.; Wu, J.; Qiuqian, L.; Yang, K.; Qian, Y.; Zhang, D. Changes in intestinal bacterial communities are closely associated with shrimp disease severity. Appl. Microbiol. Biotechnol. 2015, 99, 6911–6919. [Google Scholar] [CrossRef]
- Xiong, J.; Zhu, J.; Dai, W.; Dong, C.; Qiu, Q.; Li, C. Integrating gut microbiota immaturity and disease-discriminatory taxa to diagnose the initiation and severity of shrimp disease. Environ. Microbiol. 2017, 19, 1490–1501. [Google Scholar] [CrossRef]
- Giatsis, C.; Sipkema, D.; Smidt, H.; Heilig, H.; Benvenuti, G.; Verreth, J.; Verdegem, M. The impact of rearing environment on the development of gut microbiota in tilapia larvae. Sci. Rep. 2015, 5, 18206. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Yang, R.; Hu, J.; Lin, S.; Gu, Z.; Ma, Z. The gut microbiota community and antioxidant enzymes activity of barramundi reared at seawater and freshwater. Fish Shellfish Immunol. 2019, 89, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Baldo, L.; Riera, J.L.; Tooming-Klunderud, A.; Alba, M.M.; Salzburger, W. Gut Microbiota Dynamics during Dietary Shift in Eastern African Cichlid Fishes. PLoS ONE 2015, 10, e0127462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chai, P.C.; Song, X.L.; Chen, G.F.; Xu, H.; Huang, J. Dietary supplementation of probiotic Bacillus PC465 isolated from the gut of Fenneropenaeus chinensis improves the health status and resistance of Litopenaeus vannamei against white spot syndrome virus. Fish Shellfish Immunol. 2016, 54, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Ooi, M.C.; Goulden, E.F.; Smith, G.G.; Nowak, B.F.; Bridle, A.R. Developmental and gut-related changes to microbiomes of the cultured juvenile spiny lobster Panulirus ornatus. FEMS Microbiol. Ecol. 2017, 93, fix159. [Google Scholar] [CrossRef] [Green Version]
- Stephens, W.Z.; Burns, A.R.; Stagaman, K.; Wong, S.; Rawls, J.F.; Guillemin, K.; Bohannan, B.J. The composition of the zebrafish intestinal microbial community varies across development. ISME J. 2016, 10, 644–654. [Google Scholar] [CrossRef] [Green Version]
- Gherardi, F. Crayfish invading Europe: The case study of Procambarus clarkii. Mar. Freshw. Behav. Physiol. 2006, 39, 175–191. [Google Scholar] [CrossRef]
- Piria, M.; Jaksic, G.; Jakovlic, I.; Treer, T. Dietary habits of invasive Ponto-Caspian gobies in the Croatian part of the Danube River basin and their potential impact on benthic fish communities. Sci. Total Environ. 2016, 540, 386–395. [Google Scholar] [CrossRef]
- Yi, S.; Li, Y.; Shi, L.; Zhang, L.; Li, Q.; Chen, J. Characterization of Population Genetic Structure of red swamp crayfish, Procambarus clarkii, in China. Sci. Rep. 2018, 8, 5586. [Google Scholar] [CrossRef] [Green Version]
- Holt, C.C.; van der Giezen, M.; Daniels, C.L.; Stentiford, G.D.; Bass, D. Spatial and temporal axes impact ecology of the gut microbiome in juvenile European lobster (Homarus gammarus). ISME J. 2020, 14, 531–543. [Google Scholar] [CrossRef] [Green Version]
- Meziti, A.; Kormas, K.A. Comparison of the Norway lobster (Nephrops norvegicus) gut bacterial communities using 16S rDNA clone libraries and pyrosequencing. Anaerobe 2013, 23, 9–11. [Google Scholar] [CrossRef]
- Cheung, M.K.; Yip, H.Y.; Nong, W.; Law, P.T.; Chu, K.H.; Kwan, H.S.; Hui, J.H. Rapid Change of Microbiota Diversity in the Gut but not the Hepatopancreas during Gonadal Development of the New Shrimp Model Neocaridina denticulata. Mar. Biotechnol. 2015, 17, 811–819. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Chen, L.; Mai, G.; Zhang, H.; Yang, J.; Deng, D.; Ma, Y. Dynamics of the gut microbiota in developmental stages of Litopenaeus vannamei reveal its association with body weight. Sci. Rep. 2019, 9, 734. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Zhu, J.; Dai, W.; Dong, C.; Qiu, Q.; Li, C. Integrating molecular and ecological approaches to identify potential polymicrobial pathogens over a shrimp disease progression. Appl. Microbiol. Biotechnol. 2018, 102, 3755–3764. [Google Scholar]
- Rungrassamee, W.; Klanchui, A.; Chaiyapechara, S.; Maibunkaew, S.; Tangphatsornruang, S.; Jiravanichpaisal, P.; Karoonuthaisiri, N. Bacterial population in intestines of the black tiger shrimp (Penaeus monodon) under different growth stages. PLoS ONE 2013, 8, e60802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, R.; Wang, Y.; Zhao, S.; Yin, M.; Fang, W. The composition of the microbial community associated with Macrobrachium rosenbergii zoeae varies throughout larval development. J. Fish Dis. 2020, 43, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magoc, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2471. [Google Scholar] [CrossRef] [Green Version]
- McHugh, M.L. Multiple comparison analysis testing in ANOVA. Biochem. Med. 2011, 21, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2-approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Anderson, M.J. A new method for non-parametric multivariate analysis of variance. Austral Ecol. 2011, 26, 32–46. [Google Scholar]
- Catalan, N.; Villasante, A.; Wacyk, J.; Ramirez, C.; Romero, J. Fermented Soybean Meal Increases Lactic Acid Bacteria in Gut Microbiota of Atlantic Salmon (Salmo salar). Probiotics Antimicrob. Proteins 2018, 10, 566–576. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Ran, C.; Wang, Y.; Zhang, Z.; Ding, Q.; Yang, Y.; Olsen, R.E.; Ringo, E.; Bindelle, J.; Zhou, Z. Use of probiotics in aquaculture of China-a review of the past decade. Fish Shellfish Immunol. 2019, 86, 734–755. [Google Scholar] [CrossRef] [PubMed]
- Shui, Y.; Guan, Z.B.; Liu, G.F.; Fan, L.M. Gut microbiota of red swamp crayfish Procambarus clarkii in integrated crayfish-rice cultivation model. AMB Express. 2020, 10, 5. [Google Scholar] [CrossRef]
- Holt, C.C.; Bass, D.; Stentiford, G.D.; van der Giezen, M. Understanding the role of the shrimp gut microbiome in health and disease. J. Invertebr. Pathol. 2020, 107387. [Google Scholar] [CrossRef]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [Green Version]
- Simpson, H.L.; Campbell, B.J. Review article: Dietary fibre-microbiota interactions. Aliment. Pharmacol. Ther. 2015, 42, 158–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Youssef, N.H.; Couger, M.B.; McCully, A.L.; Criado, A.E.; Elshahed, M.S. Assessing the global phylum level diversity within the bacterial domain: A review. J. Adv. Res. 2015, 6, 269–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duguma, D.; Hall, M.W.; Rugman-Jones, P.; Stouthamer, R.; Terenius, O.; Neufeld, J.D.; Walton, W.E. Developmental succession of the microbiome of Culex mosquitoes. BMC Microbiol. 2015, 15, 140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, C.L.; Jin, W.Z.; Tao, X.H.; Zhang, Q.; Zhu, J.; Feng, S.Y.; Xu, X.H.; Li, H.Y.; Wang, Z.H.; Zhang, Z.J. Black soldier fly larvae (Hermetia illucens) strengthen the metabolic function of food waste biodegradation by gut microbiome. Microb. Biotechnol. 2019, 12, 528–543. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Bruni, L.; Jaramillo-Torres, A.; Gajardo, K.; Kortner, T.M.; Krogdahl, Å. Differential Response of Digesta- and Mucosa-Associated Intestinal Microbiota to Dietary Black Soldier Fly (Hermetia illucens) Larvae Meal in Seawater Phase Atlantic Salmon (Salmo salar). bioRxiv 2020. [Google Scholar] [CrossRef]
- Kostanjsek, R.; Strus, J.; Avgustin, G. “Candidatus Bacilloplasma,” a novel lineage of Mollicutes associated with the hindgut wall of the terrestrial isopod Porcellio scaber (Crustacea: Isopoda). Appl. Environ. Microbiol. 2007, 73, 5566–5573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burns, A.R.; Stephens, W.Z.; Stagaman, K.; Wong, S.; Rawls, J.F.; Guillemin, K.; Bohannan, B.J. Contribution of neutral processes to the assembly of gut microbial communities in the zebrafish over host development. ISME J. 2016, 10, 655–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyde, C.J.; Elizur, A.; Ventura, T. The Crustacean Ecdysone Cassette: A Gatekeeper for Molt and Metamorphosis. J. Steroid Biochem. Mol. Biol. 2019, 185, 172–183. [Google Scholar] [CrossRef]
- Lewin, G.R.; Carlos, C.; Chevrette, M.G.; Horn, H.A.; McDonald, B.R.; Stankey, R.J.; Fox, B.G.; Currie, C.R. Evolution and Ecology of Actinobacteria and Their Bioenergy Applications. Annu. Rev. Microbiol. 2016, 70, 235–254. [Google Scholar] [CrossRef] [Green Version]
- Martens, E.C.; Chiang, H.C.; Gordon, J. Mucosal Glycan Foraging Enhances Fitness and Transmission of a Saccharolytic Human Gut Bacterial Symbiont. Cell Host Microbe 2008, 4, 447–457. [Google Scholar] [CrossRef] [Green Version]
- Magnúsdóttir, S.; Heinken, A.; Kutt, L.; Ravcheev, D.A.; Bauer, E.; Noronha, A.; Greenhalgh, K.; Jäger, C.; Baginska, J.; Wilmes, P.; et al. Generation of genome-scale metabolic reconstructions for 773 members of the human gut microbiota. Nat. Biotechnol. 2017, 35, 81–89. [Google Scholar] [CrossRef]
- Tong, L.C.; Wang, Y.; Wang, Z.B.; Liu, W.Y.; Sun, S.; Li, L.; Su, D.F.; Zhang, L.C. Propionate Ameliorates Dextran Sodium Sulfate-Induced Colitis by Improving Intestinal Barrier Function and Reducing Inflammation and Oxidative Stress. Front. Pharmacol. 2016, 7, 253. [Google Scholar] [CrossRef]
- Rahimnejad, S.; Lu, K.; Wang, L.; Song, K.; Mai, K.; Davis, D.A.; Zhang, C. Replacement of fish meal with Bacillus pumillus SE5 and Pseudozyma aphidis ZR1 fermented soybean meal in diets for Japanese seabass (Lateolabrax japonicus). Fish Shellfish Immunol. 2019, 84, 987–997. [Google Scholar] [CrossRef]
- Rimoldi, S.; Gini, E.; Koch, J.F.A.; Iannini, F.; Brambilla, F.; Terova, G. Effects of hydrolyzed fish protein and autolyzed yeast as substitutes of fishmeal in the gilthead sea bream (Sparus aurata) diet, on fish intestinal microbiome. BMC Vet. Res. 2020, 16, 118. [Google Scholar]
- Oyeka, M.; Antony, S. Citrobacter braakii Bacteremia: Case Report and Review of the Literature. J. Infect. Disord. Drug Targets 2017, 17, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Baldissera, M.D.; Souza, C.F.; Junior, G.B.; Moreira, K.L.S.; da Veiga, M.L.; da Rocha, M.; Baldisserotto, B. Citrobacter freundii impairs the phosphoryl transfer network in the gills of Rhamdia quelen: Impairment of bioenergetics homeostasis. Microb. Pathog. 2018, 117, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Austin, B.; Austin, D.A. Bacterial Fish Pathogens: Diseases of Farmed and Wild Fish; Springer: Berlin, Germany, 2007. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Stage | Egg | Larvae | Preadult | Adult | Berried | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Egg | - | - | ||||||||
| Larvae | 0.725 | 0.002 | - | - | ||||||
| Preadult | 1.000 | 0.001 | 0.147 | 0.057 | - | - | ||||
| Adult | 1.000 | 0.001 | 0.420 | 0.001 | 0.189 | 0.034 | - | - | ||
| Berried | 1.000 | 0.001 | 0.632 | 0.001 | 0.439 | 0.001 | 0.004 | 0.411 | - | - |
| Group | Jingzhou | Yangzhou | Xuyi | |||
|---|---|---|---|---|---|---|
| Jingzhou | - | - | ||||
| Yangzhou | 0.126 | 0.028 | - | - | ||
| Xuyi | 0.069 | 0.101 | 0.204 | 0.002 | - | - |
| Factor | N | R | p |
|---|---|---|---|
| Age | 5 | 0.561 | 0.001 |
| Diet | 2 | 0.204 | 0.002 |
| Geography | 3 | 0.144 | 0.001 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Z.; Liu, J.; Jin, X.; Liu, C.; Fan, C.; Guo, L.; Liang, Y.; Zheng, J.; Peng, N. Developmental, Dietary, and Geographical Impacts on Gut Microbiota of Red Swamp Crayfish (Procambarus clarkii). Microorganisms 2020, 8, 1376. https://doi.org/10.3390/microorganisms8091376
Zhang Z, Liu J, Jin X, Liu C, Fan C, Guo L, Liang Y, Zheng J, Peng N. Developmental, Dietary, and Geographical Impacts on Gut Microbiota of Red Swamp Crayfish (Procambarus clarkii). Microorganisms. 2020; 8(9):1376. https://doi.org/10.3390/microorganisms8091376
Chicago/Turabian StyleZhang, Zhenting, Jiali Liu, Xuexia Jin, Chao Liu, Chenwei Fan, Li Guo, Yunxiang Liang, Jinshui Zheng, and Nan Peng. 2020. "Developmental, Dietary, and Geographical Impacts on Gut Microbiota of Red Swamp Crayfish (Procambarus clarkii)" Microorganisms 8, no. 9: 1376. https://doi.org/10.3390/microorganisms8091376