Profiling Bacterial Diversity and Potential Pathogens in Wastewater Treatment Plants Using High-Throughput Sequencing Analysis

 ,
,  ,
,
Abstract
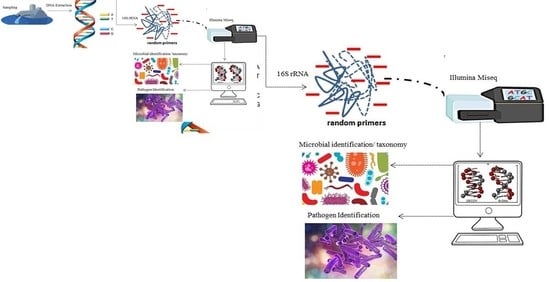
:
1. Introduction
2. Materials and Methods
2.1. Sample Collection and Field Measurements
2.2. Physicochemical Analysis of Water Samples
2.3. Nucleic Acid Extraction and 16S-rRNA-Based Amplicon Sequencing
2.4. Data Processing and Bioinformatics Analysis
3. Results
3.1. Physicochemical Profiles of the Wastewater Samples
3.2. Diversity Analysis for Bacterial Communities at the WWTPs
3.3. Taxonomic Composition of the WWTPs’ Microbial Communities
3.4. Significant Difference and Functions of Bacterial Communities in Influent and Effluent Samples
3.5. Detection of Potential Pathogenic Bacterial Members
4. Discussion
5. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Leclerc, H.; Schwartzbrod, L. Microbial Agents Associated with Waterborne Diseases. Crit. Rev. Microbiol. 2002, 28, 371–409. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Zhang, T. Detecting Human Bacterial Pathogens in Wastewater Treatment Plants by a High-Throughput Shotgun Sequencing Technique. Environ. Sci. Technol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Callejo, R.M.; Rodríguez-Naranjo, M.I.; Ubeda, C.; Hornedo-ortega, R.; Garcia-parrilla, M.C.; Troncoso, A.M. Reported Foodborne Outbreaks Due to Fresh Produce in the United States and European Union. Foodborne Pathog. Dis. 2015, 12, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Ramírez-Castillo, F.; Loera-Muro, A.; Jacques, M.; Garneau, P.; Avelar-González, F.; Harel, J.; Guerrero-Barrera, A. Waterborne Pathogens: Detection Methods and Challenges. Pathogens 2015, 4, 307–334. [Google Scholar] [CrossRef]
- Fong, T.-T.; Lipp, E.K. Enteric viruses of humans and animals in aquatic environments: Health risks, detection, and potential water quality assessment tools. Microbiol. Mol. Biol. Rev. 2005, 69, 357–371. [Google Scholar] [CrossRef]
- Okoh, A.I.; Odjadjare, E.E.; Igbinosa, E.O.; Osode, A.N. Wastewater treatment plants as a source of microbial pathogens in receiving watersheds. Afr. J. Biotechnol. 2007, 6, 2932–2944. [Google Scholar]
- Huang, K.; Zhao, F.; Zhang, X.-X.; Ye, L.; Ren, H.; Zhang, T.; Mao, Y.; Ju, F.; Wang, Y.; Li, B. Free-living bacteria and potential bacterial pathogens in sewage treatment plants. Appl. Microbiol. Biotechnol. 2018, 102, 2455–2464. [Google Scholar] [CrossRef]
- Cantalupo, P.G.; Calgua, B.; Zhao, G.; Hundesa, A.; Wier, A.D.; Katz, J.P.; Grabe, M.; Hendrix, R.W.; Girones, R.; Wang, D.; et al. Raw Sewage Harbors Diverse Viral Populations. MBio 2011. [Google Scholar] [CrossRef]
- Wang, X.; Hu, M.; Xia, Y.; Wen, X.; Ding, K. Pyrosequencing analysis of bacterial diversity in 14 wastewater treatment systems in china. Appl. Environ. Microbiol. 2012, 78, 7042–7047. [Google Scholar] [CrossRef]
- Jones, S.E.; Newton, R.J.; Mcmahon, K.D. Evidence for structuring of bacterial community composition by organic carbon source in temperate. Environ. Microbiol. 2009, 11, 2463–2472. [Google Scholar] [CrossRef]
- Cydzik-Kwiatkowska, A.; Zielińska, M. Bacterial communities in full-scale wastewater treatment systems. World J. Microbiol. Biotechnol. 2016, 32, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naidoo, S.; Olaniran, A.O. Treated wastewater effluent as a source of microbial pollution of surface water resources. Int. J. Environ. Res. Public Health 2013, 11, 249–270. [Google Scholar] [CrossRef] [PubMed]
- Jover-Smet, M.; Martín-Pascual, J.; Trapote, A. Model of suspended solids removal in the primary sedimentation tanks for the treatment of urban wastewater. Water 2017, 9, 448. [Google Scholar] [CrossRef]
- Lu, X.; Zhang, X.; Wang, Z.; Huang, K.; Wang, Y.; Liang, W. Bacterial Pathogens and Community Composition in Advanced Sewage Treatment Systems Revealed by Metagenomics Analysis Based on High-Throughput Sequencing. PLoS ONE 2015, 5, e0125549. [Google Scholar] [CrossRef] [PubMed]
- Rajasulochana, P. Comparison on efficiency of various techniques in treatment of waste and sewage water – A comprehensive review. Res.-Effic. Technol. 2016, 2, 175–184. [Google Scholar] [CrossRef]
- Narmadha, D.; Mary Selvam Kavitha, V.J. Treatment of domestic wastewater using natural flocculants. Int. J. Life Sci. Biotechnol. Pharm. Res. 2012, 3, 206–213. [Google Scholar]
- Rizzo, L.; Malato, S.; Antakyali, D.; Beretsou, V.G.; Dolic, M.B.; Gernjak, W.; Heath, E.; Ivancev-Tumbas, I.; Karaolia, P.; Lado Ribeiro, A.R.; et al. Consolidated vs new advanced treatment methods for the removal of contaminants of emerging concern from urban wastewater. Sci. Total Environ. 2018, 655, 986–1008. [Google Scholar] [CrossRef]
- Sonune, A.; Ghate, R. Developments in wastewater treatment methods. Desalination 2004, 167, 55–63. [Google Scholar] [CrossRef]
- Collivignarelli, M.C.; Abbà, A.; Benigna, I.; Sorlini, S.; Torretta, V. Overview of the Main Disinfection Processes for Wastewater and Drinking Water Treatment Plants. Sustainability 2018, 10, 86. [Google Scholar] [CrossRef]
- Lucena, F.; Duran, A.E.; Morón, A.; Calderón, E.; Campos, C.; Gantzer, C.; Skraber, S.; Jofre, J. Reduction of bacterial indicators and bacteriophages infecting faecal bacteria in primary and secondary wastewater treatments. J. Appl. Microbiol. 2004, 97, 1069–1076. [Google Scholar] [CrossRef]
- José Figueras, M.; Borrego, J.J. New perspectives in monitoring drinking water microbial quality. Int. J. Environ. Res. Public Health 2010, 7, 4179–4202. [Google Scholar] [CrossRef] [PubMed]
- Gholizadeh, M.H.; Melesse, A.M.; Reddi, L. A comprehensive review on water quality parameters estimation using remote sensing techniques. Sensors 2016, 16, 1298. [Google Scholar] [CrossRef] [PubMed]
- Vartoukian, S.R.; Palmer, R.M.; Wade, W.G. Strategies for culture of “unculturable” bacteria. FEMS Microbiol. Lett. 2010, 309, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Schmeisser, C.; Steele, H.; Streit, W.R. Metagenomics, biotechnology with non-culturable microbes. Appl. Microbiol. Biotechnol. 2007, 75, 955–962. [Google Scholar] [CrossRef] [PubMed]
- Girones, R.; Ferrús, M.A.; Alonso, J.L.; Rodriguez-Manzano, J.; Calgua, B.; de Abreu Corrêa, A.; Hundesa, A.; Carratala, A.; Bofill-Mas, S. Molecular detection of pathogens in water - The pros and cons of molecular techniques. Water Res. 2010, 44, 4325–4339. [Google Scholar] [CrossRef] [PubMed]
- Gugliandolo, C.; Lentini, V.; Spanò, A.; Maugeri, T.L. Conventional and molecular methods to detect bacterial pathogens in mussels. Lett. Appl. Microbiol. 2011, 52, 15–21. [Google Scholar] [CrossRef]
- Botes, M.; De Kwaadsteniet, M.; Cloete, T.E. Application of quantitative PCR for the detection of microorganisms in water. Anal. Bioanal. Chem. 2013, 405, 91–108. [Google Scholar] [CrossRef]
- Toze, S. PCR and the Detection of Microbial Pathogens in Water and Wastewater. Water Res. 1999, 33, 3545–3556. [Google Scholar] [CrossRef]
- Ricchi, M.; Bertasio, C.; Boniotti, M.B.; Vicari, N.; Russo, S.; Tilola, M.; Bellotti, M.A.; Bertasi, B. Comparison among the quantification of bacterial pathogens by qPCR, dPCR, and cultural methods. Front. Microbiol. 2017, 8, 1–15. [Google Scholar] [CrossRef]
- Järvinen, A.K.; Laakso, S.; Piiparinen, P.; Aittakorpi, A.; Lindfors, M.; Huopaniemi, L.; Piiparinen, H.; Mäki, M. Rapid identification of bacterial pathogens using a PCR- and microarray-based assay. BMC Microbiol. 2009, 9, 161. [Google Scholar]
- Lee, D.-Y.; Lauder, H.; Cruwys, H.; Falletta, P.; Beaudette, L.A. Development and application of an oligonucleotide microarray and real-time quantitative PCR for detection of wastewater bacterial pathogens. Sci. Total Environ. 2008, 398, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Osunmakinde, C.O.; Selvarajan, R.; Sibanda, T.; Mamba, B.B.; Msagati, T.A.M. Overview of trends in the application of metagenomic techniques in the analysis of human enteric viral diversity in Africa’s environmental regimes. Viruses 2018, 10, 429. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.R.; Montoya, V.; Gardy, J.L.; Patrick, D.M.; Tang, P. Metagenomics for pathogen detection in public health. Genome Med. 2013, 5, 81. [Google Scholar] [CrossRef] [PubMed]
- Breitbart, M.; Hoare, A.; Nitti, A.; Siefert, J.; Haynes, M.; Dinsdale, E.; Edwards, R.; Souza, V.; Rohwer, F.; Hollander, D. Metagenomic and stable isotopic analyses of modern freshwater microbialites in Cuatro Ciénegas, Mexico. Environ. Microbiol. 2009, 11, 16–34. [Google Scholar] [CrossRef] [PubMed]
- Thomas, T.; Gilbert, J.; Meyer, F. Metagenomics - a guide from sampling to data analysis. Microb. Inform. Exp. 2012, 2, 3. [Google Scholar] [CrossRef] [PubMed]
- Sharpton, T.J. An introduction to the analysis of shotgun metagenomic data. Front. Plant. Sci. 2014, 5, 1–14. [Google Scholar] [CrossRef]
- Ramganesh, S.; Timothy, S.; Memory, T. Thermophilic bacterial communities inhabiting the microbial mats of ‘indifferent’ and chalybeate (iron-rich) thermal springs: Diversity and biotechnological analysis. Microbiologyopen 2018, 7, 1–12. [Google Scholar]
- Sibanda, T.; Selvarajan, R.; Msagati, T.; Venkatachalam, S.; Meddows-Taylor, S. Defunct gold mine tailings are natural reservoir for unique bacterial communities revealed by high-throughput sequencing analysis. Sci. Total Environ. 2019, 650, 2199–2209. [Google Scholar] [CrossRef]
- Sibanda, T.; Selvarajan, R.; Tekere, M. Targeted 16S rRNA amplicon analysis reveals the diversity of bacterial communities in carwash effluents. Int. Microbiol. 2018, 22, 181–189. [Google Scholar] [CrossRef]
- Zhang, T.; Shao, M.F.; Ye, L. 454 Pyrosequencing reveals bacterial diversity of activated sludge from 14 sewage treatment plants. ISME J. 2012, 6, 1137–1147. [Google Scholar] [CrossRef]
- Ahmed, W.; Staley, C.; Sidhu, J.; Sadowsky, M.; Toze, S. Amplicon-based profiling of bacteria in raw and secondary treated wastewater from treatment plants across Australia. Appl. Microbiol. Biotechnol. 2017, 101, 1253–1266. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed]
- Aronesty, E. Comparison of Sequencing Utility Programs. Open Bioinform. J. 2013, 7, 1–8. [Google Scholar] [CrossRef]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Glo, F.O.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, 590–596. [Google Scholar] [CrossRef]
- Pruesse, E.; Quast, C.; Knittel, K.; Fuchs, B.M.; Ludwig, W.; Peplies, J.; Glöckner, F.O. SILVA: A comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007, 35, 7188–7196. [Google Scholar] [CrossRef]
- Arndt, D.; Xia, J.; Liu, Y.; Zhou, Y.; Guo, A.C.; Cruz, J.A.; Sinelnikov, I.; Budwill, K.; Nesbø, C.L.; Wishart, D.S. METAGENassist: A comprehensive web server for comparative metagenomics. Nucleic Acids Res. 2012, 40, 88–95. [Google Scholar] [CrossRef]
- Hammer, Ø.; Harper, D.; Ryan, P.D. Paleontological statistics software package for education and data analysis. Palaeontol. Electron. 2001, 4, 9–18. [Google Scholar]
- Segata, N.; Huttenhower, C. Toward an efficient method of identifying core genes for evolutionary and functional microbial phylogenies. PLoS ONE 2011. [Google Scholar] [CrossRef]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; Mcdonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Thurber, R.L.V.; Knight, R.; et al. Analysis Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Native Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Li, S.; Yang, L.; Huang, P.; Li, W.; Wang, S.; Zhao, G.; Zhang, M.; Pang, X.; Yan, Z.; et al. Structural modulation of gut microbiota in life-long calorie-restricted mice. Nat. Commun. 2013, 4, 2163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Xia, Y.; Wen, X.; Yang, Y.; Zhou, J. Microbial community functional structures in wastewater treatment plants as characterized by GeoChip. PLoS ONE 2014, 9, e93422. [Google Scholar] [CrossRef] [PubMed]
- National Water Act 36 of 1998 Wastewater Limit Values Applicable to the Irrigation of Any Land or Property up to 50 Cubic Metres. Gov. Gaz. No. 19182 2013, Notice No., 17–18. Available online: https://cer.org.za/wp-content/uploads/2014/02/Revision-of-General-Authorisations-2013.pdf (accessed on 8 March 2018).
- WHO Guidelines for Drinking-water Quality. Available online: https://www.who.int/water_sanitation_health/dwq/GDWQ2004web.pdf (accessed on 8 May 2018).
- Tanyol, M.; Demir, V. Correlations between some operation parameters and efficiency evaluation of domestic wastewater treatment plant in Tunceli (Turkey). Desalin. Water Treat. 2016, 57, 28115–28121. [Google Scholar] [CrossRef]
- WHO Guidelines for Drinking-water Quality Second Addendum to Third Edition WHO Library Cataloguing-in-Publication Data. 2008, pp. 17–19. Available online: https://www.who.int/water_sanitation_health/dwq/fulltext.pdf (accessed on 7 July 2018).
- Ma, Y.; Egodawatta, P.; McGree, J.; Liu, A.; Goonetilleke, A. Human health risk assessment of heavy metals in urban stormwater. Sci. Total Environ. 2016, 557–558, 764–772. [Google Scholar] [CrossRef] [PubMed]
- Duan, B.; Zhang, W.; Zheng, H.; Wu, C.; Zhang, Q.; Bu, Y. Comparison of health risk assessments of heavy metals and as in sewage sludge from wastewater treatment plants (WWTPs) for adults and children in the urban district of Taiyuan, China. Int. J. Environ. Res. Public Health 2017, 14, 1194. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Chen, X.; Jia, L.; Liu, Q.; Chen, X.; Han, D.; Cheng, J. Effluent concentration and removal efficiency of nine heavy metals in secondary treatment plants in Shanghai, China. Environ. Sci. Pollut. Res. 2018, 25, 17058–17065. [Google Scholar] [CrossRef]
- Xu, S.; Yao, J.; Ainiwaer, M.; Hong, Y.; Zhang, Y. Analysis of Bacterial Community Structure of Activated Sludge from Wastewater Treatment Plants in Winter. Biomed. Res. Int. 2018, 2018, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Numberger, D.; Ganzert, L.; Zoccarato, L.; Mühldorfer, K.; Sauer, S.; Grossart, H.-P.; Greenwood, A.D. Characterization of bacterial communities in wastewater with enhanced taxonomic resolution by full-length 16S rRNA sequencing. Sci. Rep. 2019, 9, 9673. [Google Scholar] [CrossRef]
- Tian, H.; Liu, J.; Feng, T.; Li, H.; Wu, X.; Li, B. Assessing the performance and microbial structure of biofilms adhering on aerated membranes for domestic saline sewage treatment. RSC Adv. 2017, 7, 27198–27205. [Google Scholar] [CrossRef] [Green Version]
- Naik, M.; Duraphe, M.D. Review paper on-Parameters affecting bioremediation. Int. J. Life Sci. 2012, 2, L77–L80. [Google Scholar]
- Bernard, E.I.; Stanley, I.R.; Okoduwa, G.O.; Ebere, P.A.; Abraham, O.A.; Ibe, K.E. Toxicity and Bioremediation of Heavy Metals Contaminated Ecosystem from Tannery Wastewater: A Review. J. Toxicol. 2018, 2018, 2568038. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Joia, J.; Sood, A.; Sood, R.; Sidhu, C.; Kaur, G. Microbes as Potential Tool for Remediation of Heavy Metals: A Review. J. Microb. Biochem. Technol. 2016, 8, 364–372. [Google Scholar] [CrossRef] [Green Version]
- Malla, M.A.; Dubey, A.; Yadav, S.; Kumar, A.; Hashem, A.; Abd-Allah, E.F. Understanding and designing the strategies for the microbe-mediated remediation of environmental contaminants using omics approaches. Front. Microbiol. 2018, 9, 1132. [Google Scholar] [CrossRef]
- Ojuederie, O.B.; Babalola, O.O. Microbial and plant-assisted bioremediation of heavy metal polluted environments: A review. Int. J. Environ. Res. Public Health 2017, 14, 1504. [Google Scholar] [CrossRef]
- Kreth, J.; Lengeler, J.W.; Jahreis, K. Characterization of Pyruvate Uptake in Escherichia coli K-12. PLoS ONE 2013, 8, 6–12. [Google Scholar] [CrossRef]
- Trotsenko, V.A.; Khmelenina, V.N. Aerobic methanotrophic bacteria of cold ecosystems. FEMS Microbiol. Ecol. 2015, 53, 15–26. [Google Scholar] [CrossRef]
- Semrau, J.D.; Dispirito, A.A.; Yoon, S. Methanotrophs and copper. FEMS Microbiol. Rev. 2010, 34, 496–531. [Google Scholar] [CrossRef] [Green Version]
- Pandey, P.K.; Kass, P.H.; Soupir, M.L.; Biswas, S.; Singh, V.P. Contamination of water resources by pathogenic bacteria. AMB Express 2014, 4, 1–16. [Google Scholar] [CrossRef]
- Kim, Y.K.; Moon, J.S.; Song, K.E.; Lee, W.K. Two cases of bacteremia due to roseomonas mucosa. Ann. Lab. Med. 2016, 36, 367–370. [Google Scholar] [CrossRef]
- Romano-Bertrand, S.; Bourdier, A.; Aujoulat, F.; Michon, A.L.; Masnou, A.; Parer, S.; Marchandin, H.; Jumas-Bilak, E. Skin microbiota is the main reservoir of Roseomonas mucosa, an emerging opportunistic pathogen so far assumed to be environmental. Clin. Microbiol. Infect. 2016, 22, 737.e1–737.e7. [Google Scholar] [CrossRef] [PubMed]
- Diesendorf, N.; Köhler, S.; Geißdörfer, W.; Grobecker-Karl, T.; Karl, M.; Burkovski, A. Characterisation of Roseomonas mucosa isolated from the root canal of an infected tooth. BMC Res. Notes 2017, 1, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Igbinosa, I.H.; Igumbor, E.U.; Aghdasi, F.; Tom, M.; Okoh, A.I. Emerging Aeromonas species infections and their significance in public health. Sci. World, J. 2012, 2012, 1326. [Google Scholar] [CrossRef] [PubMed]
- Jin, D.; Kong, X.; Cui, B.; Jin, S.; Xie, Y.; Wang, X.; Deng, Y. Bacterial communities and potential waterborne pathogens within the typical urban surface waters. Sci. Rep. 2018, 8, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Janda, J.M.; Abbott, S.L. The genus Aeromonas: Taxonomy, pathogenicity, and infection. Clin. Microbiol. Rev. 2010, 23, 35–73. [Google Scholar] [CrossRef]
- Ibekwe, A.M.; Leddy, M.; Murinda, S.E. Potential human pathogenic bacteria in a mixed urban watershed as revealed by pyrosequencing. PLoS ONE 2013, 8, e79490. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Influent | Effluent | ||||||
|---|---|---|---|---|---|---|---|---|
| DI | FHI | PSI | p-Value | DE | FHE | PSE | p-Value | |
| Temp °C | 21.2 | 15.9 | 18.4 | <0.0001 | 19.9 | 15.6 | 18.5 | 0.0002 |
| pH | 6.64 ± 0.01 | 7.8 ± 0.01 | 7.2 ± 0.01 | 0.0028 | 7.05 ± 0.03 | 8.12 ± 0.06 | 7.2 ± 0.01 | 0.144 |
| DO mg/L | 0.44 ± 0.6 | 0.67 ± 0.1 | 0.48 ± 0.4 | <0.0001 | 3.45 ± 0.4 | 3.04 ± 0.5 | 1.89 ± 0.8 | 0.0001 |
| EC µScm−1 | 840.5 ± 2.1 | 902 ± 5.6 | 685.5 ± 6.4 | <0.0001 | 506 ± 0.01 | 774 ± 2.8 | 1016 ± 7 | 0.0085 |
| Salinity µg/L | 0.42 ± 0.01 | 0.45 ± 0.01 | 0.34 ± 0.01 | <0.0001 | 0.25 ± 0.01 | 0.38 ± 0.01 | 0.51 ± 0.01 | <0.0001 |
| NH3–N | 0.04 ± 0.01 | 0.17 ± 0.04 | ND | 0.0001 | 0.06 ± 0.01 | 0.13 ± 0.04 | 0.04 ± 0.01 | <0.0001 |
| TDS mg/L | 420.5 ± 0.7 | 451.5 ± 4 | 513.3 ± 18 | 0.0003 | 253.0 ± 0.1 | 387.5 ± 2 | 508.0 ± 4.2 | <0.0001 |
| DOC mg/L | 20.39 ± 0.62 | 55.13 ± 0.8 | 154.93 ± 2.5 | 0.0001 | 5.59 ± 0.7 | 11.73 ± 0.7 | 18.16 ± 0.9 | 0.033 |
| Cl− mg/L | 28.64 ± 4.75 | 21.54 ± 0.6 | 60.57 ± 0.01 | <0.0001 | 41.31 ± 0.34 | 19.34 ± 1.0 | 44.08 ± 2.0 | <0.0001 |
| F− mg/L | 0.23 ± 0.09 | 0.13 ± 0.01 | 16.97 ± 0.5 | 0.0003 | ND | 0.20 ± 0.01 | 0.20 ± 0.01 | <0.0001 |
| Br− mg/L | 0.54 ± 0.1 | 0.91 ± 0.1 | ND | 0.0003 | ND | ND | ND | - |
| SO4 mg/L | 6.68 ± 1.0 | 26.98 ± 0.1 | 3.51 ± 0.1 | <0.0001 | 40.49 ± 0.5 | 22.91 ± 1.3 | 58.11 ± 1.4 | 0.0002 |
| PO4 mg/L | 5.07 ± 0.6 | 12.72 ± 0.1 | 23.29 ± 0.1 | <0.0001 | ND | 0.71 ± 0.03 | 4.33 ± 0.1 | 0.0085 |
| Parameters | DI | DE | FHI | FHE | PSI | PSE |
|---|---|---|---|---|---|---|
| Quality reads | 25,287 | 23,066 | 32,176 | 32,469 | 36,742 | 46,924 |
| Average read length | 518 | 491 | 471 | 469 | 471 | 473 |
| OTUs | 519 | 159 | 229 | 104 | 144 | 351 |
| Good’s coverage % | 99.90 | 99.40 | 99.90 | 99.90 | 100 | 99.90 |
| ACE | 659.6 | 179.8 | 257.5 | 116.5 | 150.1 | 369.2 |
| Chao1 | 620.7 | 170 | 240.2 | 110.1 | 146.2 | 357.3 |
| Shannon_H | 1.304 | 1.524 | 1.523 | 1.04 | 1.198 | 1.883 |
| Simpson_1-D | 0.663 | 0.347 | 0.484 | 0.568 | 0.51 | 0.383 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oluseyi Osunmakinde, C.; Selvarajan, R.; Mamba, B.B.; Msagati, T.A.M. Profiling Bacterial Diversity and Potential Pathogens in Wastewater Treatment Plants Using High-Throughput Sequencing Analysis. Microorganisms 2019, 7, 506. https://doi.org/10.3390/microorganisms7110506
Oluseyi Osunmakinde C, Selvarajan R, Mamba BB, Msagati TAM. Profiling Bacterial Diversity and Potential Pathogens in Wastewater Treatment Plants Using High-Throughput Sequencing Analysis. Microorganisms. 2019; 7(11):506. https://doi.org/10.3390/microorganisms7110506
Chicago/Turabian StyleOluseyi Osunmakinde, Cecilia, Ramganesh Selvarajan, Bhekie B. Mamba, and Titus A.M. Msagati. 2019. "Profiling Bacterial Diversity and Potential Pathogens in Wastewater Treatment Plants Using High-Throughput Sequencing Analysis" Microorganisms 7, no. 11: 506. https://doi.org/10.3390/microorganisms7110506