In Silico Analysis of Genetic VapC Profiles from the Toxin-Antitoxin Type II VapBC Modules among Pathogenic, Intermediate, and Non-Pathogenic Leptospira

 ,
, 
Abstract
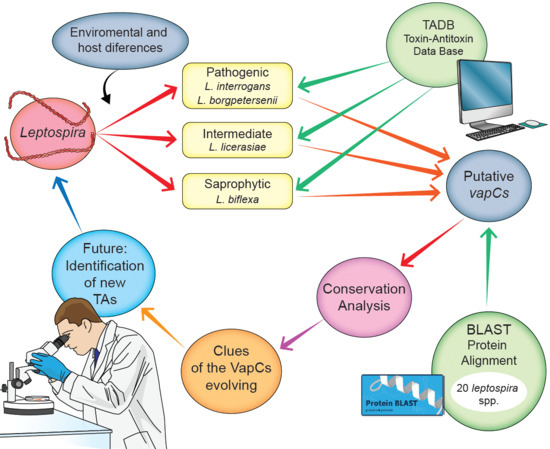
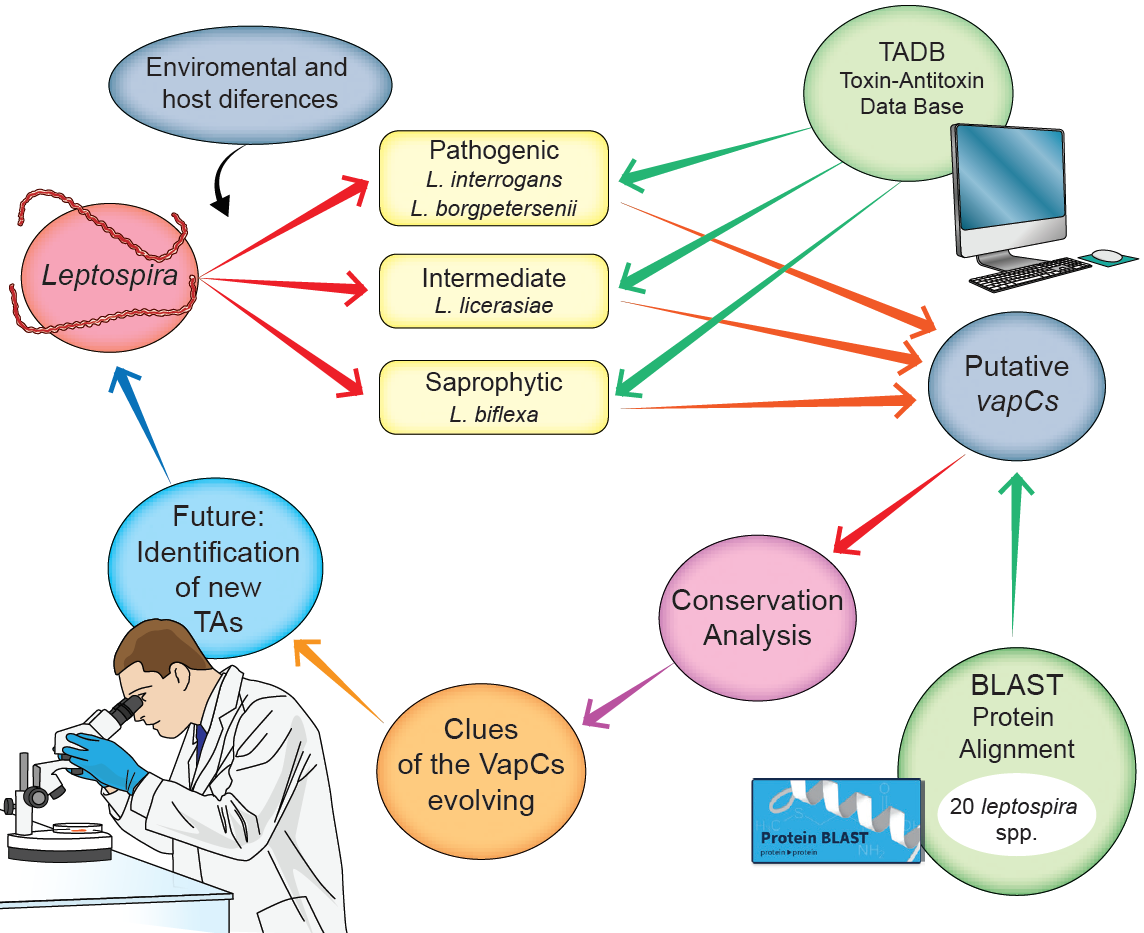
:
1. Introduction
2. Materials and Methods
2.1. Analysis of Type II TA Modules
2.2. Evaluation of the Presence of vapBC among Leptospira spp.
“Conservation Value” Index
2.3. Protein Alignment
3. Results
3.1. Analysis and Comparison of TA Profiles within Leptospira Species
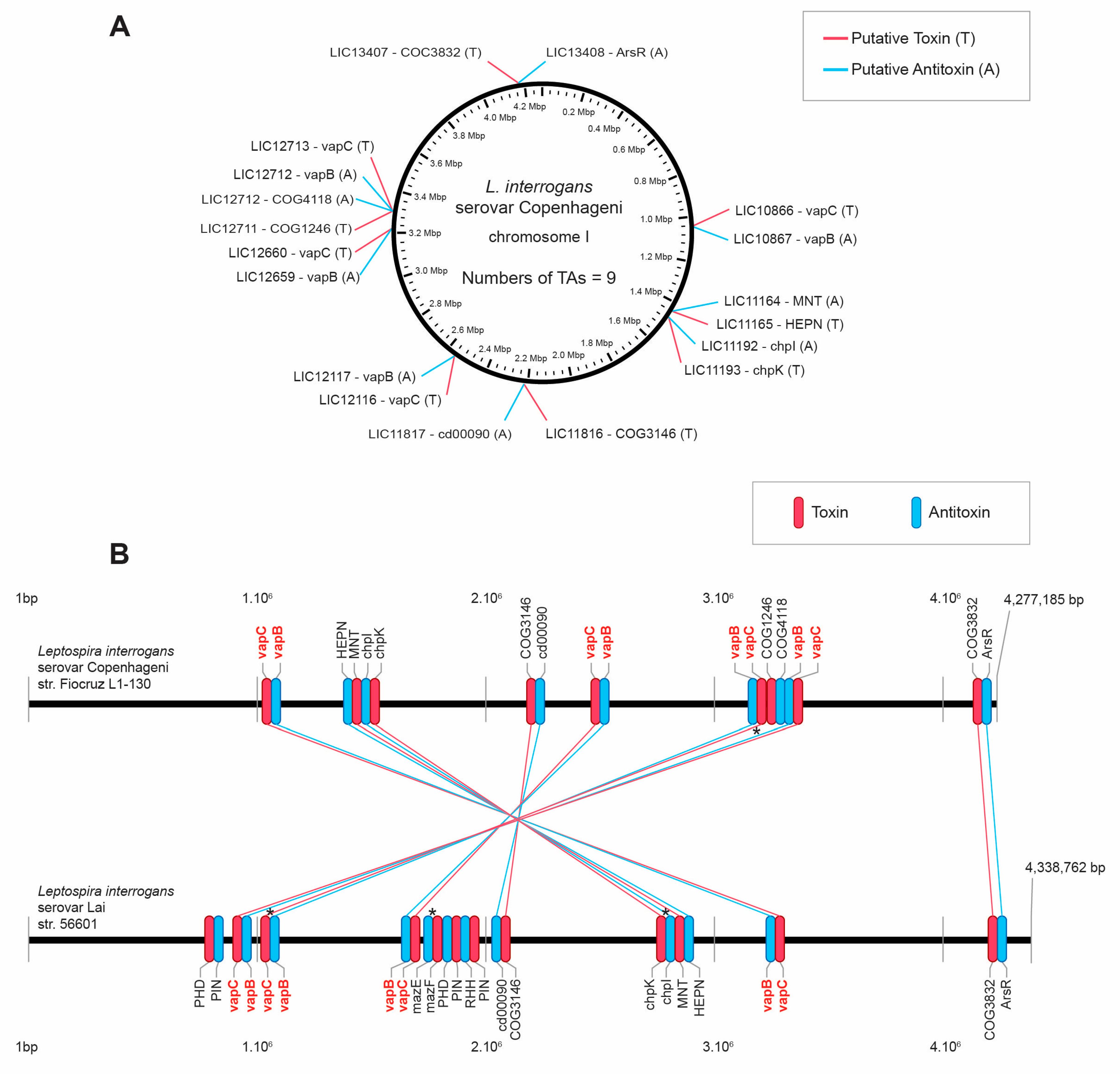
3.1.1. L. interrogans Serovar Copenhageni Fiocruz L1-130 and L. interrogans Serovar Lai Strain 56601
3.1.2. L. borgpetersenii Serovar Hardjo-bovis Strain JB197 and Strain L550
3.1.3. L. biflexa Serovar Patoc Strain Patoc 1 (Ames) and Strain Patoc 1 (Paris)
3.2. Variability of Amino Acids Sequences of VapCs within Leptospira Strains
3.3. Distribution of VapCs of Pathogenic, Intermediate, and Saprophytic Leptospira Strains
3.3.1. L. interrogans Serovar Copenhageni Fiocruz L1-130
3.3.2. L. borgpetersenii Serovar Hardjo-Bovis Strain JB197
3.3.3. L. licerasiae Serovar Varillal strain VAR010
3.3.4. L. biflexa Serovar Patoc 1 (Ames)
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Plank, R.; Dean, D. Overview of the epidemiology, microbiology, and pathogenesis of Leptospira spp. in humans. Microbes Infect. 2000, 2, 1265–1276. [Google Scholar] [CrossRef]
- Bharti, A.R.; Nally, J.E.; Ricaldi, J.N.; Matthias, M.A.; Diaz, M.M.; Lovett, M.A.; Levett, P.N.; Gilman, R.H.; Willig, M.R.; Gotuzzo, E.; et al. Leptospirosis: A zoonotic disease of global importance. Lancet Infect. Dis. 2003, 3, 757–771. [Google Scholar] [CrossRef]
- Levett, P.N. Leptospirosis. Clin. Microbiol. Rev. 2001, 14, 296–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehmann, J.S.; Matthias, M.A.; Vinetz, J.M.; Fouts, D.E. Leptospiral pathogenomics. Pathogens 2014, 3, 280–308. [Google Scholar] [CrossRef] [PubMed]
- Fouts, D.E.; Matthias, M.A.; Adhikarla, H.; Adler, B.; Amorim-Santos, L.; Berg, D.E.; Bulach, D.; Buschiazzo, A.; Chang, Y.F.; Galloway, R.L.; et al. What Makes a Bacterial Species Pathogenic?:Comparative Genomic Analysis of the Genus Leptospira. PLoS Negl. Trop. Dis. 2016, 10, e0004403. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, K.; Christensen, S.K.; Løbner-Olesen, A. Prokaryotic toxin-antitoxin stress response loci. Nat. Rev. Microbiol. 2005, 3, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Park, J.H.; Inouye, M. Toxin-antitoxin systems in bacteria and archaea. Annu. Rev. Genet. 2011, 45, 61–79. [Google Scholar] [CrossRef]
- Lioy, V.S.; Machon, C.; Tabone, M.; Gonzalez-Pastor, J.E.; Daugelavicius, R.; Ayora, S.; Alonso, J.C. The zeta toxin induces a set of protective responses and dormancy. PLoS ONE 2012, 7, e30282. [Google Scholar] [CrossRef]
- Komi, K.K.; Ge, Y.M.; Xin, X.Y.; Ojcius, D.M.; Sun, D.; Hu, W.L.; Zhao, X.; Lin, X.; Yan, J. ChpK and MazF of the toxin-antitoxin modules are involved in the virulence of Leptospira interrogans during infection. Microbes Infect. 2015, 17, 34–47. [Google Scholar] [CrossRef]
- Ramage, H.R.; Connolly, L.E.; Cox, J.S. Comprehensive functional analysis of Mycobacterium tuberculosis toxin-antitoxin systems: Implications for pathogenesis, stress responses, and evolution. PLoS Genet. 2009, 5, e1000767. [Google Scholar] [CrossRef]
- Fernández-García, L.; Blasco, L.; Lopez, M.; Bou, G.; García-Contreras, R.; Wood, T.; Tomas, M. Toxin-Antitoxin Systems in Clinical Pathogens. Toxins 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Norton, J.P.; Mulvey, M.A. Toxin-antitoxin systems are important for niche-specific colonization and stress resistance of uropathogenic Escherichia coli. PLoS Pathog. 2012, 8, e1002954. [Google Scholar] [CrossRef] [PubMed]
- Ren, D.; Walker, A.N.; Daines, D.A. Toxin-antitoxin loci vapBC-1 and vapXD contribute to survival and virulence in nontypeable Haemophilus influenzae. BMC Microbiol. 2012, 12, 263. [Google Scholar] [CrossRef] [PubMed]
- De la Cruz, M.A.; Zhao, W.; Farenc, C.; Gimenez, G.; Raoult, D.; Cambillau, C.; Gorvel, J.P.; Méresse, S. A toxin-antitoxin module of Salmonella promotes virulence in mice. PLoS Pathog. 2013, 9, e1003827. [Google Scholar] [CrossRef] [PubMed]
- Sharma, B.; Brown, A.V.; Matluck, N.E.; Hu, L.T.; Lewis, K. Borrelia burgdorferi, the Causative Agent of Lyme Disease, Forms Drug-Tolerant Persister Cells. Antimicrob. Agents Chemother. 2015, 59, 4616–4624. [Google Scholar] [CrossRef] [PubMed]
- Pandey, D.P.; Gerdes, K. Toxin-antitoxin loci are highly abundant in free-living but lost from host-associated prokaryotes. Nucleic Acids Res. 2005, 33, 966–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Díaz-Orejas, R.; Espinosa, M.; Yeo, C.C. The Importance of the Expendable: Toxin-Antitoxin Genes in Plasmids and Chromosomes. Front. Microbiol. 2017, 8, 1479. [Google Scholar] [CrossRef]
- Kang, S.M.; Kim, D.H.; Jin, C.; Lee, B.J. A Systematic Overview of Type II and III Toxin-Antitoxin Systems with a Focus on Druggability. Toxins 2018, 10. [Google Scholar] [CrossRef]
- Kędzierska, B.; Hayes, F. Emerging Roles of Toxin-Antitoxin Modules in Bacterial Pathogenesis. Molecules 2016, 21. [Google Scholar] [CrossRef]
- Gerdes, K.; Maisonneuve, E.; Gottesman, S.; Harwood, C.; Schneewind, O. Bacterial Persistence and Toxin-Antitoxin Loci. Annu. Rev. Microbiol. 2012, 66, 103–123. [Google Scholar] [CrossRef]
- Ramisetty, B.C.M.; Santhosh, R.S. Endoribonuclease type II toxin-antitoxin systems: Functional or selfish? Microbiology 2017, 163, 931–939. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Harrison, E.M.; Bi, D.; Tai, C.; He, X.; Ou, H.Y.; Rajakumar, K.; Deng, Z. TADB: A web-based resource for Type 2 toxin-antitoxin loci in bacteria and archaea. Nucleic Acids Res. 2011, 39, D606–D611. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Wei, Y.; Shen, Y.; Li, X.; Zhou, H.; Tai, C.; Deng, Z.; Ou, H.Y. TADB 2.0: An updated database of bacterial type II toxin-antitoxin loci. Nucleic Acids Res. 2018, 46, D749–D753. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, J.; Hoeflich, K.; Ikura, M.; Qing, G.; Inouye, M. MazF cleaves cellular mRNAs specifically at ACA to block protein synthesis in Escherichia coli. Mol. Cell 2003, 12, 913–923. [Google Scholar] [CrossRef]
- Pedersen, K.; Zavialov, A.V.; Pavlov, M.Y.; Elf, J.; Gerdes, K.; Ehrenberg, M. The bacterial toxin RelE displays codon-specific cleavage of mRNAs in the ribosomal A site. Cell 2003, 112, 131–140. [Google Scholar] [CrossRef]
- Jørgensen, M.G.; Pandey, D.P.; Jaskolska, M.; Gerdes, K. HicA of Escherichia coli defines a novel family of translation-independent mRNA interferases in bacteria and archaea. J. Bacteriol. 2009, 191, 1191–1199. [Google Scholar] [CrossRef] [PubMed]
- Schifano, J.M.; Edifor, R.; Sharp, J.D.; Ouyang, M.; Konkimalla, A.; Husson, R.N.; Woychik, N.A. Mycobacterial toxin MazF-mt6 inhibits translation through cleavage of 23S rRNA at the ribosomal A site. Proc. Natl. Acad. Sci. USA 2013, 110, 8501–8506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schifano, J.M.; Cruz, J.W.; Vvedenskaya, I.O.; Edifor, R.; Ouyang, M.; Husson, R.N.; Nickels, B.E.; Woychik, N.A. tRNA is a new target for cleavage by a MazF toxin. Nucleic Acids Res. 2016, 44, 1256–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopes, A.P.Y.; Lopes, L.M.; Fraga, T.R.; Chura-Chambi, R.M.; Sanson, A.L.; Cheng, E.; Nakajima, E.; Morganti, L.; Martins, E.A.L. VapC from the Leptospiral VapBC Toxin-Antitoxin Module Displays Ribonuclease Activity on the Initiator tRNA. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Winther, K.S.; Gerdes, K. Enteric virulence associated protein VapC inhibits translation by cleavage of initiator tRNA. Proc. Natl. Acad. Sci. USA 2011, 108, 7403–7407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walling, L.R.; Butler, J.S. Homologous VapC Toxins Inhibit Translation and Cell Growth by Sequence-Specific Cleavage of tRNA. J. Bacteriol. 2018, 200. [Google Scholar] [CrossRef] [PubMed]
- Winther, K.; Tree, J.J.; Tollervey, D.; Gerdes, K. VapCs of Mycobacterium tuberculosis cleave RNAs essential for translation. Nucleic Acids Res. 2016, 44, 9860–9871. [Google Scholar] [CrossRef] [PubMed]
- Winther, K.S.; Brodersen, D.E.; Brown, A.K.; Gerdes, K. VapC20 of Mycobacterium tuberculosis cleaves the Sarcin-Ricin loop of 23S rRNA. Nat. Commun. 2013, 4, 2796. [Google Scholar] [CrossRef] [PubMed]
- Kositanont, U.; Saetun, P.; Krittanai, C.; Doungchawee, G.; Tribuddharat, C.; Thongboonkerd, V. Application of immunoproteomics to leptospirosis: Towards clinical diagnostics and vaccine discovery. Proteom. Clin. Appl. 2007, 1, 400–409. [Google Scholar] [CrossRef] [PubMed]
- Nascimento, A.L.; Verjovski-Almeida, S.; Van Sluys, M.A.; Monteiro-Vitorello, C.B.; Camargo, L.E.; Digiampietri, L.A.; Harstkeerl, R.A.; Ho, P.L.; Marques, M.V.; Oliveira, M.C.; et al. Genome features of Leptospira interrogans serovar Copenhageni. Braz. J. Med. Biol. Res. 2004, 37, 459–477. [Google Scholar] [CrossRef] [PubMed]
- Adler, B.; Lo, M.; Seemann, T.; Murray, G.L. Pathogenesis of leptospirosis: The influence of genomics. Vet. Microbiol. 2011, 153, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.X.; Fu, G.; Jiang, X.G.; Zeng, R.; Miao, Y.G.; Xu, H.; Zhang, Y.X.; Xiong, H.; Lu, G.; Lu, L.F.; et al. Unique physiological and pathogenic features of Leptospira interrogans revealed by whole-genome sequencing. Nature 2003, 422, 888–893. [Google Scholar] [CrossRef]
- Gamberini, M.; Gómez, R.M.; Atzingen, M.V.; Martins, E.A.; Vasconcellos, S.A.; Romero, E.C.; Leite, L.C.; Ho, P.L.; Nascimento, A.L. Whole-genome analysis of Leptospira interrogans to identify potential vaccine candidates against leptospirosis. FEMS Microbiol. Lett. 2005, 244, 305–313. [Google Scholar] [CrossRef]
- Ricaldi, J.N.; Fouts, D.E.; Selengut, J.D.; Harkins, D.M.; Patra, K.P.; Moreno, A.; Lehmann, J.S.; Purushe, J.; Sanka, R.; Torres, M.; et al. Whole genome analysis of Leptospira licerasiae provides insight into leptospiral evolution and pathogenicity. PLoS Negl. Trop. Dis. 2012, 6, e1853. [Google Scholar] [CrossRef]
- Llanes, A.; Restrepo, C.M.; Rajeev, S. Whole Genome Sequencing Allows Better Understanding of the Evolutionary History of Leptospira interrogans Serovar Hardjo. PLoS ONE 2016, 11, e0159387. [Google Scholar] [CrossRef]
- Nascimento Filho, E.G.; Vieira, M.L.; Teixeira, A.F.; Santos, J.C.; Fernandes, L.G.V.; Passalia, F.J.; Daroz, B.B.; Rossini, A.; Kochi, L.T.; Cavenague, M.F.; et al. Proteomics as a tool to understand Leptospira physiology and virulence: Recent advances, challenges and clinical implications. J. Proteom. 2018, 180, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Bulach, D.M.; Zuerner, R.L.; Wilson, P.; Seemann, T.; McGrath, A.; Cullen, P.A.; Davis, J.; Johnson, M.; Kuczek, E.; Alt, D.P.; et al. Genome reduction in Leptospira borgpetersenii reflects limited transmission potential. Proc. Natl. Acad. Sci. USA 2006, 103, 14560–14565. [Google Scholar] [CrossRef]
- Picardeau, M.; Bulach, D.M.; Bouchier, C.; Zuerner, R.L.; Zidane, N.; Wilson, P.J.; Creno, S.; Kuczek, E.S.; Bommezzadri, S.; Davis, J.C.; et al. Genome sequence of the saprophyte Leptospira biflexa provides insights into the evolution of Leptospira and the pathogenesis of leptospirosis. PLoS ONE 2008, 3, e1607. [Google Scholar] [CrossRef] [PubMed]
- Nascimento, A.L.; Ko, A.I.; Martins, E.A.; Monteiro-Vitorello, C.B.; Ho, P.L.; Haake, D.A.; Verjovski-Almeida, S.; Hartskeerl, R.A.; Marques, M.V.; Oliveira, M.C.; et al. Comparative genomics of two Leptospira interrogans serovars reveals novel insights into physiology and pathogenesis. J. Bacteriol. 2004, 186, 2164–2172. [Google Scholar] [CrossRef] [PubMed]
- Arcus, V.L.; McKenzie, J.L.; Robson, J.; Cook, G.M. The PIN-domain ribonucleases and the prokaryotic VapBC toxin-antitoxin array. Protein Eng. Des. Sel. 2011, 24, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.X.; Li, J.; Guo, X.K.; Wu, C.; Bi, B.; Ren, S.X.; Wu, C.F.; Zhao, G.P. Characterization of a novel toxin-antitoxin module, VapBC, encoded by Leptospira interrogans chromosome. Cell Res. 2004, 14, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Picardeau, M.; Le Dantec, C.; Richard, G.F.; Saint Girons, I. The spirochetal chpK-chromosomal toxin-antitoxin locus induces growth inhibition of yeast and mycobacteria. FEMS Microbiol. Lett. 2003, 229, 277–281. [Google Scholar] [CrossRef]
- Gerdes, K. Toxin-antitoxin modules may regulate synthesis of macromolecules during nutritional stress. J. Bacteriol. 2000, 182, 561–572. [Google Scholar] [CrossRef] [PubMed]
- Masuda, Y.; Miyakawa, K.; Nishimura, Y.; Ohtsubo, E. chpA and chpB, Escherichia coli chromosomal homologs of the pem locus responsible for stable maintenance of plasmid R100. J. Bacteriol. 1993, 175, 6850–6856. [Google Scholar] [CrossRef] [PubMed]
- Harms, A.; Brodersen, D.E.; Mitarai, N.; Gerdes, K. Toxins, Targets, and Triggers: An Overview of Toxin-Antitoxin Biology. Mol. Cell 2018, 70, 768–784. [Google Scholar] [CrossRef]
- Sierra, R.; Viollier, P.; Renzoni, A. Linking toxin-antitoxin systems with phenotypes: A Staphylococcus aureus viewpoint. Biochim. Biophys. Acta Gene Regul. Mech. 2018. [Google Scholar] [CrossRef] [PubMed]
- Winther, K.; Gerdes, K. Regulation of Enteric vapBC Transcription: Induction by VapC Toxin Dimer-Breaking. Nucleic Acids Res. 2012, 40, 4347–4357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lobato-Márquez, D.; Moreno-Córdoba, I.; Figueroa, V.; Díaz-Orejas, R.; García-del Portillo, F. Distinct type I and type II toxin-antitoxin modules control Salmonella lifestyle inside eukaryotic cells. Sci. Rep. 2015, 5, 9374. [Google Scholar] [CrossRef] [PubMed]
- Cooper, C.R.; Daugherty, A.J.; Tachdjian, S.; Blum, P.H.; Kelly, R.M. Role of vapBC toxin-antitoxin loci in the thermal stress response of Sulfolobus solfataricus. Biochem. Soc. Trans. 2009, 37, 123–126. [Google Scholar] [CrossRef]
- Nikolic, N. Autoregulation of bacterial gene expression: Lessons from the MazEF toxin-antitoxin system. Curr. Genet. 2019, 65, 133–138. [Google Scholar] [CrossRef]
- Victoria, B.; Ahmed, A.; Zuerner, R.L.; Ahmed, N.; Bulach, D.M.; Quinteiro, J.; Hartskeerl, R.A. Conservation of the S10-spc-alpha locus within otherwise highly plastic genomes provides phylogenetic insight into the genus Leptospira. PLoS ONE 2008, 3, e2752. [Google Scholar] [CrossRef]
- Chan, W.T.; Yeo, C.C.; Sadowy, E.; Espinosa, M. Functional validation of putative toxin-antitoxin genes from the Gram-positive pathogen Streptococcus pneumoniae: Phd-doc is the fourth bona-fide operon. Front. Microbiol. 2014, 5, 677. [Google Scholar] [CrossRef]
- Ahidjo, B.A.; Kuhnert, D.; McKenzie, J.L.; Machowski, E.E.; Gordhan, B.G.; Arcus, V.; Abrahams, G.L.; Mizrahi, V. VapC toxins from Mycobacterium tuberculosis are ribonucleases that differentially inhibit growth and are neutralized by cognate VapB antitoxins. PLoS ONE 2011, 6, e21738. [Google Scholar] [CrossRef]
- Malmström, J.; Beck, M.; Schmidt, A.; Lange, V.; Deutsch, E.W.; Aebersold, R. Proteome-wide cellular protein concentrations of the human pathogen Leptospira interrogans. Nature 2009, 460, 762–765. [Google Scholar] [CrossRef]
- Ramisetty, B.C.; Santhosh, R.S. Horizontal gene transfer of chromosomal Type II toxin-antitoxin systems of Escherichia coli. FEMS Microbiol. Lett. 2016, 363. [Google Scholar] [CrossRef]
- Fiedoruk, K.; Daniluk, T.; Swiecicka, I.; Sciepuk, M.; Leszczynska, K. Type II toxin-antitoxin systems are unevenly distributed among Escherichia coli phylogroups. Microbiology 2015, 161, 158–167. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| A—L. interrogans Serovar Copenhageni str. Fiocruz L1-130—Identity (%) | B—L. biflexa Serovar Patoc Strain Patoc 1 (Ames)—Identity (%) | ||||||||||||||||
| VapC-1 | VapC-2 | VapC-3 | VapC-4 | VapC-1 | VapC-2 | VapC-3 | VapC-4 | VapC-5 | VapC-6 | VapC-7 | VapC-8 | ||||||
| VapC-1 | 100 | 19 | 23 | 18 | VapC-1 | 100 | 17 | 13 | 24 | 19 | 19 | 27 | 21 | ||||
| VapC-2 | 100 | 19 | 17 | VapC-2 | 100 | 18 | 23 | 19 | 22 | 20 | 17 | ||||||
| VapC-3 | 100 | 26 | VapC-3 | 100 | 27 | 22 | 16 | 16 | 19 | ||||||||
| VapC-4 | 100 | VapC-4 | 100 | 22 | 22 | 22 | 28 | ||||||||||
| VapC-5 | 100 | 36 | 19 | 46 | |||||||||||||
| VapC-6 | 100 | 17 | 38 | ||||||||||||||
| VapC-7 | 100 | 22 | |||||||||||||||
| VapC-8 | 100 | ||||||||||||||||
| C—L. licersiae Serovar Varillal str. VAR 010—Identity (%) | |||||||||||||||||
| VapC-1 | VapC-2 | VapC-3 | VapC-4 | VapC-5 | VapC-6 | VapC-7 | VapC-8 | VapC-9 | VapC-10 | VapC-11 | VapC-12 | VapC-13 | VapC-14 | VapC-15 | |||
| VapC-1 | 100 | 26 | 18 | 19 | 28 | 17 | 22 | 14 | 24 | 18 | 17 | 14 | 23 | 21 | 14 | ||
| VapC-2 | 100 | 13 | 19 | 31 | 21 | 18 | 17 | 19 | 16 | 19 | 17 | 20 | 21 | 17 | |||
| VapC-3 | 100 | 21 | 14 | 17 | 19 | 17 | 20 | 17 | 24 | 17 | 22 | 23 | 17 | ||||
| VapC-4 | 100 | 18 | 21 | 28 | 25 | 21 | 18 | 22 | 25 | 20 | 31 | 25 | |||||
| VapC-5 | 100 | 23 | 18 | 21 | 24 | 20 | 18 | 21 | 21 | 19 | 21 | ||||||
| VapC-6 | 100 | 16 | 18 | 32 | 19 | 16 | 18 | 21 | 21 | 18 | |||||||
| VapC-7 | 100 | 32 | 18 | 19 | 18 | 32 | 18 | 22 | 32 | ||||||||
| VapC-8 | 100 | 23 | 19 | 20 | 100 | 15 | 23 | 100 | |||||||||
| VapC-9 | 100 | 17 | 20 | 23 | 19 | 25 | 23 | ||||||||||
| VapC-10 | 100 | 20 | 19 | 12 | 21 | 19 | |||||||||||
| VapC-11 | 100 | 20 | 20 | 17 | 20 | ||||||||||||
| VapC-12 | 100 | 15 | 23 | 100 | |||||||||||||
| VapC-13 | 100 | 20 | 15 | ||||||||||||||
| VapC-14 | 100 | 23 | |||||||||||||||
| VapC-15 | 100 | ||||||||||||||||
| VapC of L. interrogans Serovar Copenhageni str. Fiocruz L1-130 | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Leptospira spp. | LIC10866 VapC-1 | LIC12116 VapC-2 | LIC12660 VapC-3 | LIC12713 VapC-4 | |||||||||||||
| Identity (%) | Positives (%) | Cover (%) | C-value | Identity (%) | Positives (%) | Cover (%) | C-value | Identity (%) | Positives (%) | Cover (%) | C-value | Identity (%) | Positives (%) | Cover (%) | C-value | ||
| Pathogenic | L. interrogans L. interrogans | 100 | 100 | 100 | 1.00 | 100 | 100 | 100 | 1.00 | 100 | 100 | 100 | 1.00 | 100 | 100 | 100 | 1.00 |
| L. kirschneri | 99 | 99 | 100 | 0.99 | 97 | 98 | 100 | 0.98 | 50 | 66 | 97 | 0.56 | 97 | 97 | 100 | 0.97 | |
| L. noguchii | - | - | - | - | 97 | 99 | 100 | 0.98 | 44 | 64 | 97 | 0.52 | - | - | - | - | |
| L. borgpetersenii | - | - | - | - | - | - | - | - | 48 | 68 | 97 | 0.56 | - | - | - | - | |
| L. weilii | - | - | - | - | 87 | 95 | 97 | 0.88 | 90 | 95 | 100 | 0.93 | 48 | 66 | 100 | 0.57 | |
| L. santarosai | - | - | - | - | 83 | 90 | 100 | 0.87 | 66 | 83 | 100 | 0.75 | 64 | 75 | 100 | 0.70 | |
| L. alexanderi | 93 | 95 | 100 | 0.94 | 85 | 94 | 100 | 0.90 | 89 | 94 | 100 | 0.92 | - | - | - | - | |
| L. alstoni | - | - | - | - | 86 | 95 | 100 | 0.91 | 90 | 96 | 100 | 0.93 | 61 | 73 | 97 | 0.65 | |
| L. kmetyi | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | |
| Intermediary | L. wolffii | - | - | - | - | - | - | - | - | 44 | 68 | 97 | 0.54 | 45 | 62 | 100 | 0.54 |
| L. licerasiae | - | - | - | - | - | - | - | - | 70 | 87 | 99 | 0.78 | 44 | 60 | 100 | 0.52 | |
| L. inadai | - | - | - | - | - | - | - | - | 63 | 84 | 99 | 0.73 | 65 | 78 | 100 | 0.72 | |
| L. fainei | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | |
| L. broomii | - | - | - | - | - | - | - | - | - | - | - | - | 63 | 75 | 100 | 0.69 | |
| Saprophytic | L. wolbachii | - | - | - | - | - | - | - | - | 66 | 83 | 99 | 0.74 | 64 | 75 | 100 | 0.70 |
| L. meyeri | 44 | 70 | 95 | 0.54 | - | - | - | - | 66 | 84 | 99 | 0.74 | 62 | 74 | 100 | 0.68 | |
| L. biflexa | - | - | - | - | - | - | - | - | 62 | 82 | 99 | 0.71 | 28 | 55 | 93 | 0.39 | |
| L. vanthielii | 46 | 69 | 95 | 0.55 | - | - | - | - | 51 | 69 | 98 | 0.59 | 61 | 75 | 100 | 0.68 | |
| L. terpstrae | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | |
| L. yanagawae | - | - | - | - | - | - | - | - | 48 | 70 | 98 | 0.58 | - | - | - | - | |
 very highly conserved (0.85 ≤ C-value ≤ 1.0);
very highly conserved (0.85 ≤ C-value ≤ 1.0);  highly conserved (0.7 ≤ C-value ≤ 0.84);
highly conserved (0.7 ≤ C-value ≤ 0.84);  moderately conserved (0.4 ≤ C-value ≤ 0.69);
moderately conserved (0.4 ≤ C-value ≤ 0.69);  poorly conserved (C-value ≤ 0.39);
poorly conserved (C-value ≤ 0.39);  no hits. Positive and cover values below 50% were not included.
no hits. Positive and cover values below 50% were not included.| VapC of L. borgpetersenii Serovar Hardjo-bovis str. JB197—Conservation value (C-value) | ||||
|---|---|---|---|---|
| Leptospira spp. | LBJ_0624 VapC-1 | LBJ_0764 VapC-2 | LBJ_2077 VapC-3 | |
| Pathogenic | L. interrogans L. interrogans | 0.95 | 0.90 | 0.39 |
| L. kirschneri | 0.49 | 0.89 | 0.38 | |
| L. noguchii | 0.93 | - | 0.39 | |
| L. borgpetersenii | 1.00 | 1.00 | 1.00 | |
| L. weilii | 0.94 | 0.89 | 0.96 | |
| L. santarosai | 0.49 | 0.87 | 0.40 | |
| L. alexanderi | 0.52 | 0.48 | - | |
| L. alstoni | 0.95 | 0.88 | 0.44 | |
| L. kmetyi | - | - | - | |
| Intermediary | L. wolffii | 0.49 | - | - |
| L. licerasiae | 0.49 | - | 0.43 | |
| L. inadai | - | - | 0.38 | |
| L. fainei | - | - | - | |
| L. broomii | - | - | - | |
| Saprophytic | L. wolbachii | 0.51 | - | 0.38 |
| L. meyeri | 0.83 | - | - | |
| L. biflexa | 0.53 | - | 0.38 | |
| L. vanthielii | 0.82 | 0.47 | 0.38 | |
| L. terpstrae | - | - | 0.35 | |
| L. yanagawae | 0.51 | - | - | |
 very highly conserved (0.85 ≤ C-value ≤ 1.0);
very highly conserved (0.85 ≤ C-value ≤ 1.0);  highly conserved (0.7 ≤ C-value ≤ 0.84);
highly conserved (0.7 ≤ C-value ≤ 0.84);  moderately conserved (0.4 ≤ C-value ≤ 0.69);
moderately conserved (0.4 ≤ C-value ≤ 0.69);  poorly conserved (C-value ≤ 0.39);
poorly conserved (C-value ≤ 0.39);  no hits. Positive and cover values below 50% were not included.
no hits. Positive and cover values below 50% were not included.| VapC of L. licerasiae Serovar Varillal str. VAR 010—Conservation value (C-value) | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Leptospira spp. | LEP1GSC185_0307—VapC-1 | LEP1GSC185_0418—VapC-2 | LEP1GSC185_0630—VapC-3 | LEP1GSC185_1922—VapC-4 | LEP1GSC185_2251—VapC-5 | LEP1GSC185_2580—VapC-6 | LEP1GSC185_3193—VapC-7 | LEP1GSC185_3530—VapC-8 * | LEP1GSC185_3550—VapC-9 | LEP1GSC185_3553—VapC-10 | LEP1GSC185_3557—VapC-11 | LEP1GSC185_3561—VapC-13 | LEP1GSC185_3566—VapC-14 | ||
| Pathogenic | L. interrogans L. interrogans | 0.48 | 0.36 | 0.66 | - | 0.53 | 0.80 | 0.54 | 0.64 | 0.78 | - | 0.87 | 0.82 | 0.92 | |
| L. kirschneri | - | - | 0.66 | - | 0.53 | 0.41 | 0.41 | 0.64 | 0.56 | - | 0.82 | - | 0.90 | ||
| L. noguchii | - | - | 0.65 | - | 0.20 | 0.46 | 0.42 | 0.63 | - | - | 0.54 | 0.85 | 0.92 | ||
| L. borgpetersenii | - | - | 0.48 | - | 0.20 | 0.79 | 0.46 | 0.67 | 0.55 | - | - | - | 0.92 | ||
| L. weilii | - | - | 0.48 | - | 0.67 | 0.80 | 0.45 | 0.66 | 0.93 | 0.91 | 0.86 | - | 0.77 | ||
| L. santarosai | - | 0.37 | 0.46 | - | 0.54 | - | 0.42 | 0.63 | 0.89 | - | 0.54 | 0.94 | 0.72 | ||
| L. alexanderi | - | - | 0.65 | - | 0.20 | 0.80 | 0.41 | 0.64 | 0.78 | - | 0.86 | - | - | ||
| L. alstoni | 0.65 | 0.40 | 0.48 | - | 0.50 | 0.79 | 0.75 | 0.66 | 0.78 | - | 0.87 | - | 0.77 | ||
| L. kmetyi | - | - | - | - | - | - | - | - | - | - | - | - | 0.92 | ||
| Intermediary | L. wolffii | 0.38 | 0.78 | - | 0.81 | 0.80 | 0.42 | 0.41 | - | 0.53 | - | 0.96 | - | 0.97 | |
| L. licerasiae | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | ||
| L. inadai | 0.77 | 0.38 | - | - | 0.55 | - | 0.39 | 0.87 | 0.88 | - | 0.94 | - | 0.91 | ||
| L. fainei | - | - | - | - | - | - | - | - | - | - | 0.93 | - | - | ||
| L. broomii | - | 0.41 | - | - | 0.56 | - | - | 0.40 | - | - | - | - | - | ||
| Saprophytic | L. wolbachii | - | 0.39 | - | - | 0.52 | 0.42 | 0.42 | 0.64 | 0.85 | - | 0.81 | - | 0.37 | |
| L. meyeri | - | 0.38 | 0.65 | - | 0.54 | 0.78 | 0.41 | 0.64 | 0.85 | - | 0.55 | - | 0.73 | ||
| L. biflexa | - | 0.42 | - | - | 0.37 | 0.77 | 0.40 | 0.66 | 0.80 | - | - | - | 0.73 | ||
| L. vanthielii | - | 0.39 | 0.61 | - | 0.53 | 0.78 | 0.41 | 0.65 | 0.56 | - | 0.54 | 0.75 | 0.37 | ||
| L. terpstrae | - | - | - | - | - | - | 0.41 | 0.66 | - | - | - | - | - | ||
| L. yanagawae | - | - | - | - | - | 0.42 | - | - | 0.56 | - | 0.55 | 0.74 | - | ||
 very highly conserved (0.85 ≤ C-value ≤ 1.0);
very highly conserved (0.85 ≤ C-value ≤ 1.0);  highly conserved (0.7 ≤ C-value ≤ 0.84);
highly conserved (0.7 ≤ C-value ≤ 0.84);  moderately conserved (0.4 ≤ C-value ≤ 0.69);
moderately conserved (0.4 ≤ C-value ≤ 0.69);  poorly conserved (C-value ≤ 0.39);
poorly conserved (C-value ≤ 0.39);  no hits. Positive and cover values below 50% were not included. * VapCs 8, 12, and 15 (LEP1GSC185:2580, LEP1GSC185:3559, and LEP1GSC185:3880) share the same amino-acid sequence. VapC-12 and VapC-15 were not shown here.
no hits. Positive and cover values below 50% were not included. * VapCs 8, 12, and 15 (LEP1GSC185:2580, LEP1GSC185:3559, and LEP1GSC185:3880) share the same amino-acid sequence. VapC-12 and VapC-15 were not shown here.| VapC of L. biflexa serovar Patoc strain Patoc 1 (Ames)—Conservation value (C-value) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Leptospira spp. | LBF_0418 VapC-1 | LBF_2142 VapC-2 | LBF_ 2175 VapC-3 | LBF_2183 VapC-4 | LBF_2185 VapC-5 | LBF_2276 VapC-6 | LBF_2813 VapC-7 | LBF_3285 VapC-8 | |
| Pathogenic | L. interrogans L. interrogans | 0.40 | 0.60 | - | 0.78 | 0.72 | 0.83 | 0.83 | 0.59 |
| L. kirschneri | 0.40 | - | - | 0.78 | 0.55 | - | 0.83 | 0.73 | |
| L. noguchii | - | 0.61 | - | 0.77 | 0.48 | 0.47 | 0.54 | 0.52 | |
| L. borgpetersenii | 0.35 | - | 0.27 | 0.78 | 0.55 | 0.89 | - | 0.73 | |
| L. weilii | 0.42 | 0.59 | 0.60 | 0.78 | 0.83 | 0.89 | 0.83 | 0.73 | |
| L. santarosai | 0.41 | 0.62 | - | 0.78 | 0.78 | 0.55 | 0.53 | 0.75 | |
| L. alexanderi | 0.35 | 0.59 | 0.60 | 0.77 | 0.72 | 0.88 | 0.84 | 0.72 | |
| L. alstoni | - | - | 0.59 | 0.79 | 0.72 | 0.85 | 0.83 | 0.73 | |
| L. kmetyi | - | - | - | - | - | - | - | - | |
| Intermediary | L. wolffii | - | - | - | - | 0.52 | 0.44 | 0.80 | 0.75 |
| L. licerasiae | 0.44 | - | - | 0.56 | 0.80 | 0.79 | 0.82 | 0.55 | |
| L. inadai | 0.41 | - | - | 0.63 | 0.79 | 0.33 | 0.80 | 0.50 | |
| L. fainei | - | - | - | - | - | - | 0.79 | - | |
| L. broomii | 0.42 | - | - | - | - | - | - | - | |
| Saprophytic | L. wolbachii | 0.40 | 0.94 | 0.99 | 0.88 | 0.92 | 0.45 | 0.87 | 0.92 |
| L. meyeri | 0.43 | 0.95 | 0.96 | 0.96 | 0.84 | 0.97 | 0.52 | 0.91 | |
| L. biflexa | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | |
| L. vanthielii | 0.43 | 0.95 | - | 0.91 | 0.56 | 0.97 | 0.53 | 0.93 | |
| L. terpstrae | - | - | - | 0.91 | - | - | - | - | |
| L. yanagawae | - | 0.95 | 0.97 | - | 0.57 | 0.44 | 0.53 | 0.95 | |
 very highly conserved (0.85 ≤ C-value ≤ 1.0);
very highly conserved (0.85 ≤ C-value ≤ 1.0);  highly conserved (0.7 ≤ C-value ≤ 0.84);
highly conserved (0.7 ≤ C-value ≤ 0.84);  moderately conserved (0.4 ≤ C-value ≤ 0.69);
moderately conserved (0.4 ≤ C-value ≤ 0.69);  poorly conserved (C-value ≤ 0.39);
poorly conserved (C-value ≤ 0.39);  no hits. Positive and cover values below 50% were not included.
no hits. Positive and cover values below 50% were not included.© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lopes, A.P.Y.; Azevedo, B.O.P.; Emídio, R.C.; Damiano, D.K.; Nascimento, A.L.T.O.; Barazzone, G.C. In Silico Analysis of Genetic VapC Profiles from the Toxin-Antitoxin Type II VapBC Modules among Pathogenic, Intermediate, and Non-Pathogenic Leptospira. Microorganisms 2019, 7, 56. https://doi.org/10.3390/microorganisms7020056
Lopes APY, Azevedo BOP, Emídio RC, Damiano DK, Nascimento ALTO, Barazzone GC. In Silico Analysis of Genetic VapC Profiles from the Toxin-Antitoxin Type II VapBC Modules among Pathogenic, Intermediate, and Non-Pathogenic Leptospira. Microorganisms. 2019; 7(2):56. https://doi.org/10.3390/microorganisms7020056
Chicago/Turabian StyleLopes, Alexandre P. Y., Bruna O. P. Azevedo, Rebeca C. Emídio, Deborah K. Damiano, Ana L. T. O. Nascimento, and Giovana C. Barazzone. 2019. "In Silico Analysis of Genetic VapC Profiles from the Toxin-Antitoxin Type II VapBC Modules among Pathogenic, Intermediate, and Non-Pathogenic Leptospira" Microorganisms 7, no. 2: 56. https://doi.org/10.3390/microorganisms7020056