
Structure of Benthic Microbial Communities in the Northeastern Part of the Barents Sea

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
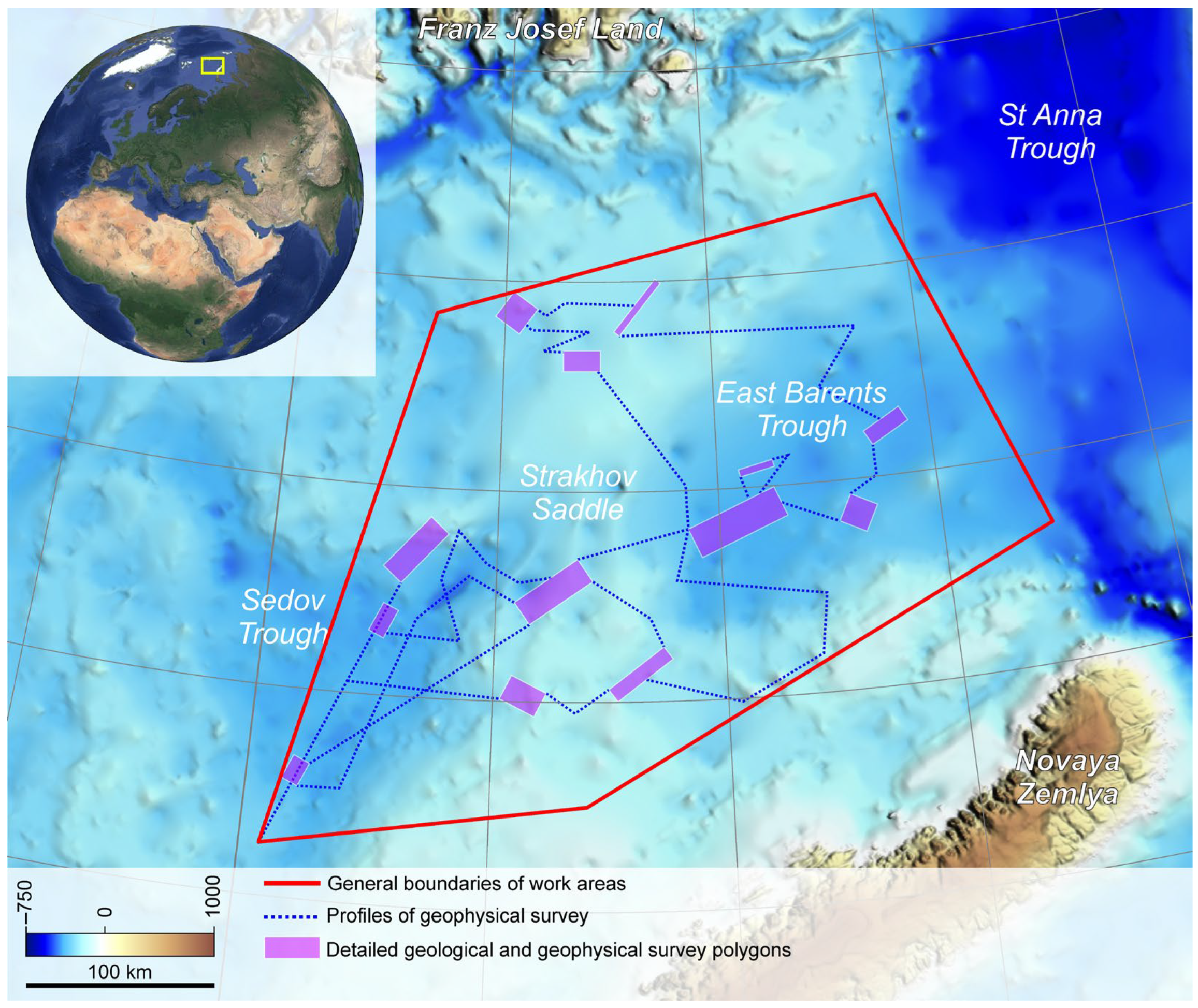
2.1. Study Area
2.2. Sample Collection
2.3. DNA Extraction, 16S rRNA Gene-Based Profiling, and qPCR
2.4. Bioinformatics and Statistical Analysis
3. Results
3.1. Characteristics of the Sampling Sites, the Bottom Water, and Upper Part of the Sediments
3.2. Microbial Community Composition
3.2.1. Total Numbers of Prokaryotes in the Bottom Water and Sediments of Barents Sea
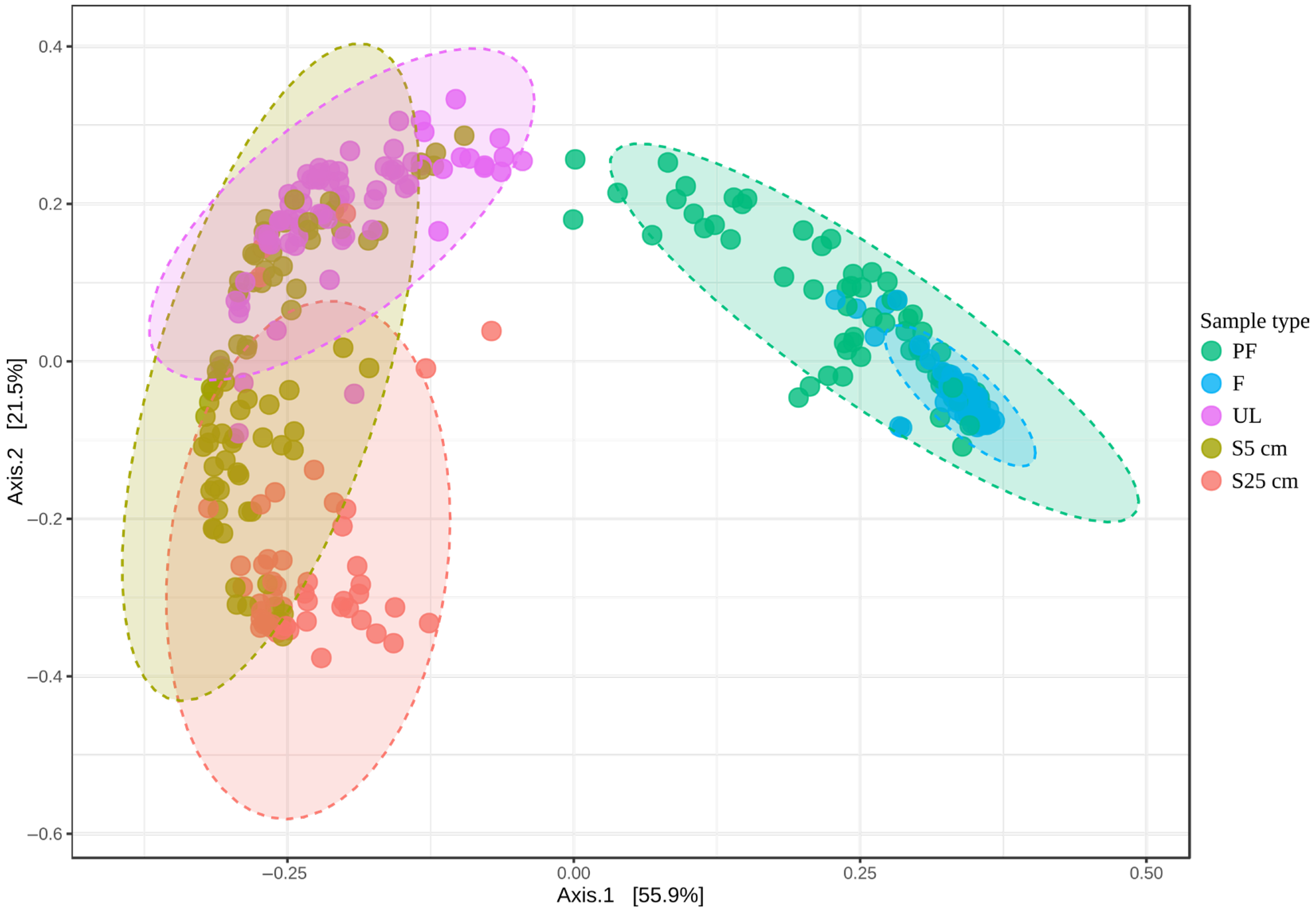
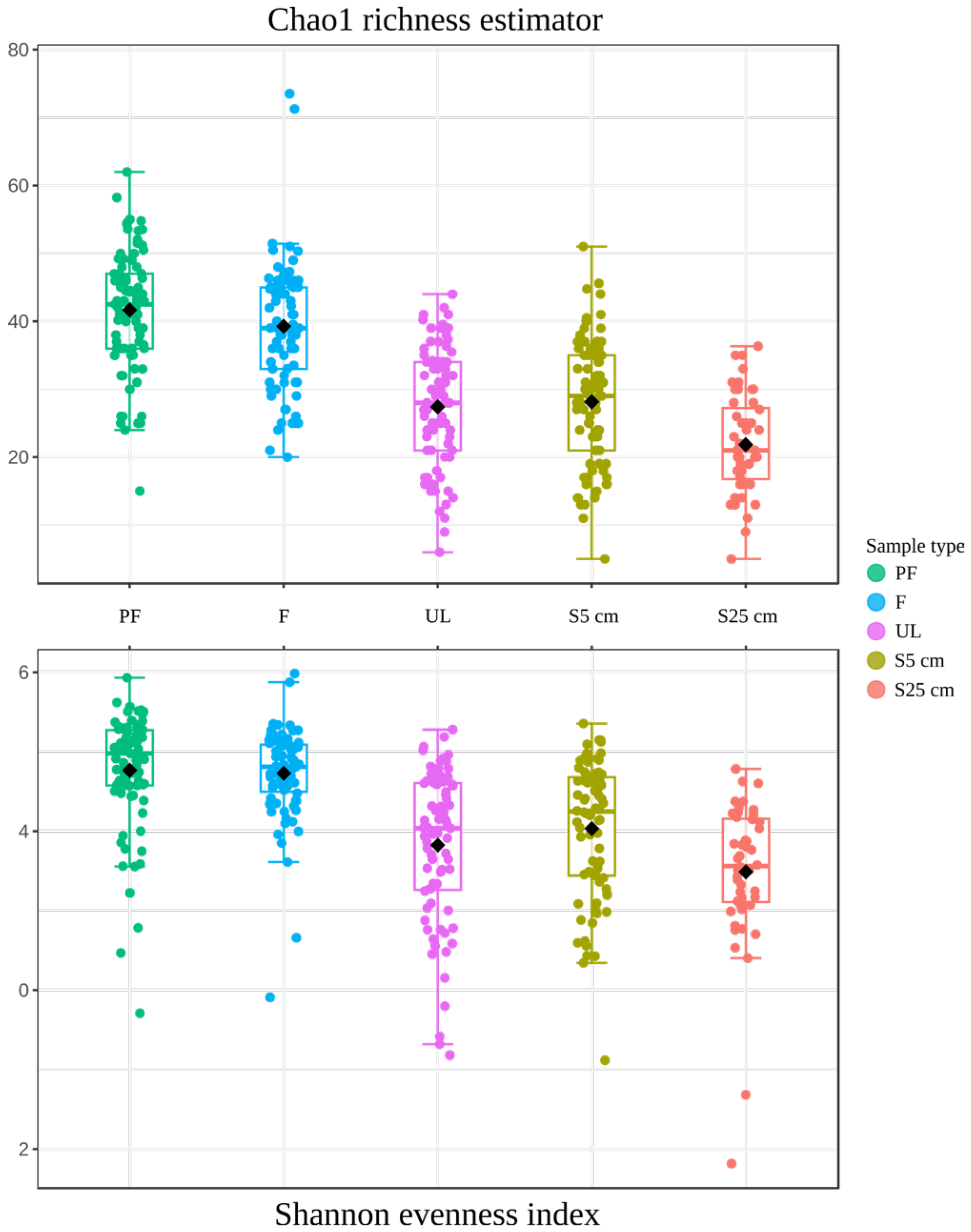
3.2.2. Comparison of Microbial Communities of the Different Types of Samples
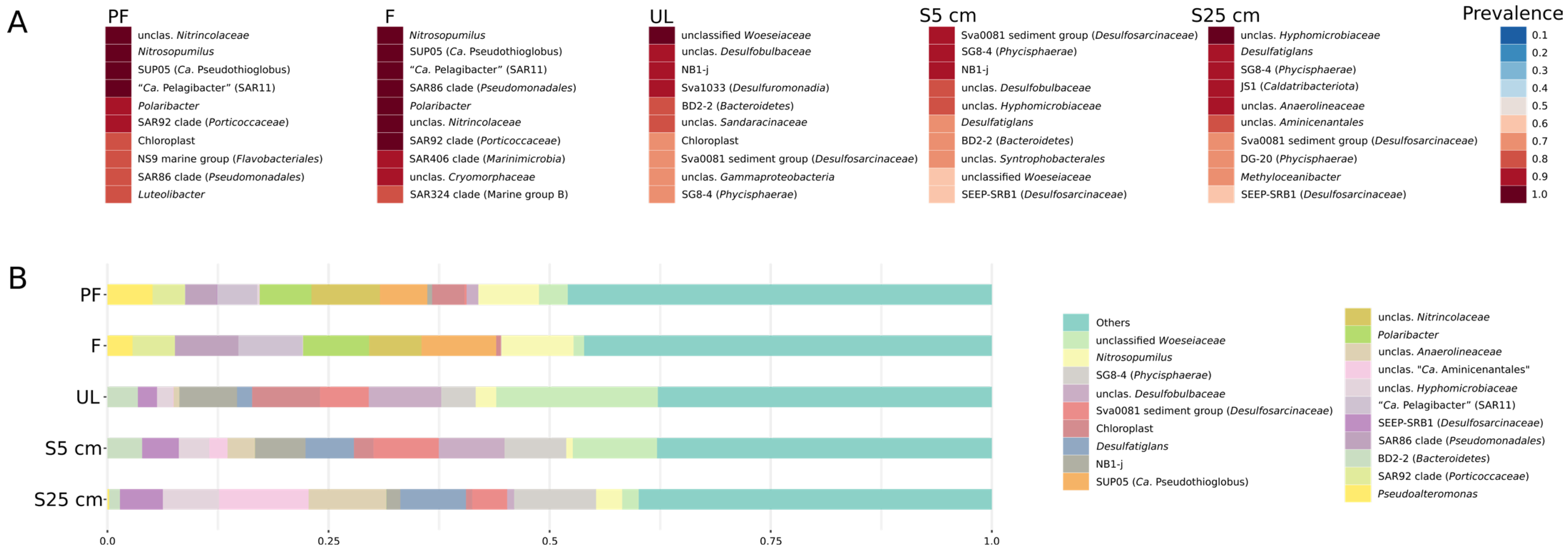
3.2.3. Microorganisms Prevailing in the Bottom Water Samples
3.2.4. Microorganisms Prevailing in the Sediment Samples
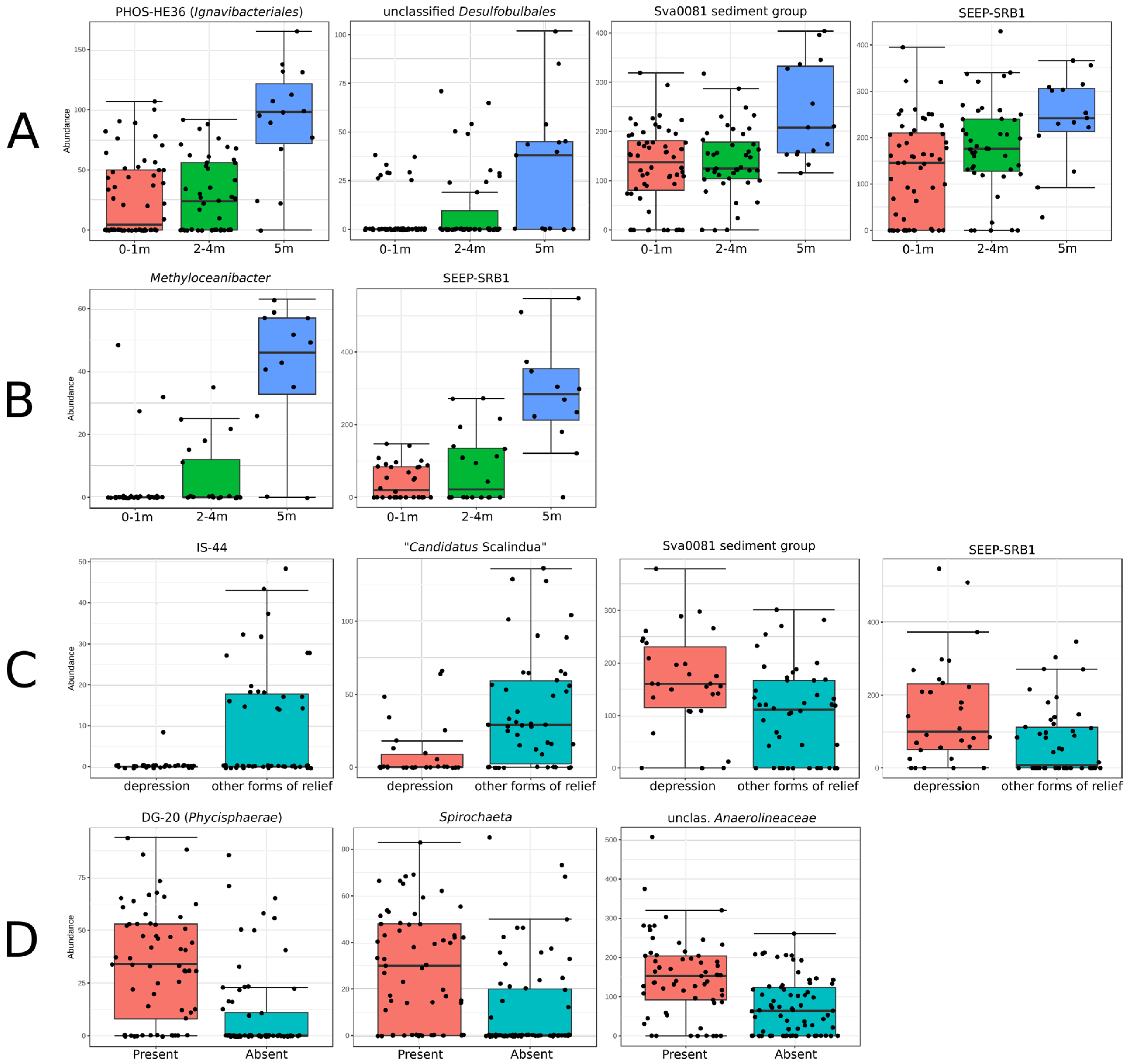
3.3. Correlation between the Structure of Benthic Microbial Communities and Environmental Parameters
3.4. Correlations between Microbial Community Composition and Total Microbial Abundance
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gray, J.; Elliott, M. Ecology of Marine Sediments: From Science to Management; Oxford University Press: Oxford, UK, 2009; ISBN 978-0-19-856902-2. [Google Scholar]
- Timmermans, M.-L.; Marshall, J. Understanding Arctic Ocean Circulation: A Review of Ocean Dynamics in a Changing Climate. J. Geophys. Res. Ocean. 2020, 125, e2018JC014378. [Google Scholar] [CrossRef]
- Stoupakova, A.V.; Kiryukhina, T.A.; Suslova, A.; Norina, D.A.; Mayer, N.M.; Pronina, N.; Mordasova, A. Oil and Gas Prospects in the Mesozoic Section of the Barents Sea Basin. Georesursy 2015, 61, 13–27. [Google Scholar] [CrossRef]
- Doré, A.G. Barents Sea Geology, Petroleum Resources and Commercial Potential. Arctic 1995, 48, 207–221. [Google Scholar] [CrossRef]
- Yakovlev, I.; Stein, Y.; Barkov, A.; Filippova, K.; Fedotov, S. 3D Geological Model for a Gas-Saturated Reservoir Based on Simultaneous Deterministic Partial Stack Inversion. First Break 2010, 28. [Google Scholar] [CrossRef]
- Svendsen, J.I.; Alexanderson, H.; Astakhov, V.I.; Demidov, I.; Dowdeswell, J.A.; Funder, S.; Gataullin, V.; Henriksen, M.; Hjort, C.; Houmark-Nielsen, M.; et al. Late Quaternary Ice Sheet History of Northern Eurasia. Quat. Sci. Rev. 2004, 23, 1229–1271. [Google Scholar] [CrossRef]
- Hughes, A.L.C.; Gyllencreutz, R.; Lohne, Ø.S.; Mangerud, J.; Svendsen, J.I. The Last Eurasian Ice Sheets—A Chronological Database and Time-Slice Reconstruction, DATED-1. Boreas 2016, 45, 1–45. [Google Scholar] [CrossRef]
- Epshtein, O.G.; Dlugach, A.G.; Starovoytov, A.V. Cover of the Last Glaciation Deposits in the Eastern Barents Sea: Specificity of Composition, Thickness Distribution, Immensity, and Peculiarity of Structural Forms. Dokl. Earth Sci. 2019, 487, 898–901. [Google Scholar] [CrossRef]
- Dowdeswell, J.A.; Montelli, A.; Akhmanov, G.; Solovyeva, M.; Terekhina, Y.; Mironyuk, S.; Tokarev, M. Late Weichselian Ice-Sheet Flow Directions in the Russian Northern Barents Sea from High-Resolution Imagery of Submarine Glacial Landforms. Geology 2021, 49, 1484–1488. [Google Scholar] [CrossRef]
- Montelli, A.; Solovyeva, M.; Akhmanov, G.; Mazzini, A.; Piatilova, A.; Bakay, E.; Dowdeswell, J.A. The Geomorphic Record of Marine-Based Ice Dome Decay: Final Collapse of the Barents Sea Ice Sheet. Quat. Sci. Rev. 2023, 303, 107973. [Google Scholar] [CrossRef]
- Stevenson, M.A.; Faust, J.C.; Andrade, L.L.; Freitas, F.S.; Gray, N.D.; Tait, K.; Hendry, K.R.; Hilton, R.G.; Henley, S.F.; Tessin, A.; et al. Transformation of Organic Matter in a Barents Sea Sediment Profile: Coupled Geochemical and Microbiological Processes. Philos. Trans. A Math. Phys. Eng. Sci. 2020, 378, 20200223. [Google Scholar] [CrossRef]
- Ravenschlag, K.; Sahm, K.; Amann, R. Quantitative Molecular Analysis of the Microbial Community in Marine Arctic Sediments (Svalbard). Appl. Environ. Microbiol. 2001, 67, 387–395. [Google Scholar] [CrossRef]
- Algora, C.; Gründger, F.; Adrian, L.; Damm, V.; Richnow, H.-H.; Krüger, M. Geochemistry and Microbial Populations in Sediments of the Northern Baffin Bay, Arctic. Geomicrobiol. J. 2013, 30, 690–705. [Google Scholar] [CrossRef]
- Sahm, K.; Berninger, U.-G. Abundance, Vertical Distribution, and Community Structure of Benthic Prokaryotes from Permanently Cold Marine Sediments (Svalbard, Arctic Ocean). Mar. Ecol. Prog. Ser. 1998, 165, 71–80. [Google Scholar] [CrossRef]
- Ravenschlag, K.; Sahm, K.; Pernthaler, J.; Amann, R. High Bacterial Diversity in Permanently Cold Marine Sediments. Appl. Environ. Microbiol. 1999, 65, 3982–3989. [Google Scholar] [CrossRef] [PubMed]
- Rapp, J. Bacterial Diversity in Sea Ice, Melt Ponds, Water Column, Ice Algal Aggregates and Deep-Sea Sediments of the Central Arctic Ocean. Master’ Thesis, University of Bremen, Bremen, Germany, 2014. [Google Scholar]
- Savvichev, A.S.; Rusanov, I.I.; Kadnikov, V.V.; Beletskii, A.V.; Ravin, N.V.; Pimenov, N.V. Microbial Community Composition and Rates of the Methane Cycle Microbial Processes in the Upper Sediments of the Yamal Sector of the Southwestern Kara Sea. Microbiology 2018, 87, 238–248. [Google Scholar] [CrossRef]
- Begmatov, S.; Savvichev, A.S.; Kadnikov, V.V.; Beletsky, A.V.; Rusanov, I.I.; Klyuvitkin, A.A.; Novichkova, E.A.; Mardanov, A.V.; Pimenov, N.V.; Ravin, N.V. Microbial Communities Involved in Methane, Sulfur, and Nitrogen Cycling in the Sediments of the Barents Sea. Microorganisms 2021, 9, 2362. [Google Scholar] [CrossRef]
- Tasianas, A.; Martens, I.; Bünz, S.; Mienert, J. Mechanisms Initiating Fluid Migration at Snøhvit and Albatross Fields, Barents Sea. Arktos 2016, 2, 26. [Google Scholar] [CrossRef]
- Rise, L.; Bellec, V.; Chand, S.; Bøe, R. Pockmarks in the Southwestern Barents Sea and Finnmark Fjords. Nor. J. Geol. 2015, 94, 263–282. [Google Scholar] [CrossRef]
- Ostanin, I.; Anka, Z.; Di Primio, R. Role of Faults in Hydrocarbon Leakage in the Hammerfest Basin, SW Barents Sea: Insights from Seismic Data and Numerical Modelling. Geosciences 2017, 7, 28. [Google Scholar] [CrossRef]
- Pimenov, N.; Savvichev, A.; Rusanov, I.; Lein, A.; Egorov, A.; Gebruk, A.; Moskalev, L.; Vogt, P. Microbial Processes of Carbon Cycle as the Base of Food Chain of Håkon Mosby Mud Volcano Benthic Community. Geo-Mar. Lett. 1999, 19, 89–96. [Google Scholar] [CrossRef]
- Knittel, K.; Lösekann, T.; Boetius, A.; Kort, R.; Amann, R. Diversity and Distribution of Methanotrophic Archaea at Cold Seeps. Appl. Environ. Microbiol. 2005, 71, 467–479. [Google Scholar] [CrossRef]
- Astrom, E.; Carroll, M.; Ambrose, W.; Sen, A.; Silyakova, A.; Carroll, J. Methane Cold Seeps as Biological Oases in the High-Arctic Deep Sea: Cold Seeps in the High-Arctic Deep Sea. Limnol. Oceanogr. 2017, 63, S209–S231. [Google Scholar] [CrossRef]
- Khomyakova, M.A.; Zavarzina, D.G.; Merkel, A.Y.; Klyukina, A.A.; Pikhtereva, V.A.; Gavrilov, S.N.; Slobodkin, A.I. The First Cultivated Representatives of the Actinobacterial Lineage OPB41 Isolated from Subsurface Environments Constitute a Novel Order Anaerosomatales. Front. Microbiol. 2022, 13, 1047580. [Google Scholar] [CrossRef]
- Hugerth, L.W.; Wefer, H.A.; Lundin, S.; Jakobsson, H.E.; Lindberg, M.; Rodin, S.; Engstrand, L.; Andersson, A.F. DegePrime, a Program for Degenerate Primer Design for Broad-Taxonomic-Range PCR in Microbial Ecology Studies. Appl. Environ. Microbiol. 2014, 80, 5116–5123. [Google Scholar] [CrossRef]
- Merkel, A.Y.; Tarnovetskii, I.Y.; Podosokorskaya, O.A.; Toshchakov, S.V. Analysis of 16S rRNA Primer Systems for Profiling of Thermophilic Microbial Communities. Microbiology 2019, 88, 671–680. [Google Scholar] [CrossRef]
- Ansorge, R.; Birolo, G.; James, S.A.; Telatin, A. Dadaist2: A Toolkit to Automate and Simplify Statistical Analysis and Plotting of Metabarcoding Experiments. Int. J. Mol. Sci. 2021, 22, 5309. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Zhou, G.; Ewald, J.; Pang, Z.; Shiri, T.; Xia, J. MicrobiomeAnalyst 2.0: Comprehensive Statistical, Functional and Integrative Analysis of Microbiome Data. Nucleic Acids Res. 2023, 51, W310–W318. [Google Scholar] [CrossRef] [PubMed]
- Lagkouvardos, I.; Fischer, S.; Kumar, N.; Clavel, T. Rhea: A Transparent and Modular R Pipeline for Microbial Profiling Based on 16S rRNA Gene Amplicons. PeerJ 2017, 5, e2836. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA Ribosomal RNA Gene Database Project: Improved Data Processing and Web-Based Tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, L.; Holler, T.; Knittel, K.; Meyerdierks, A.; Amann, R. Identification of the Dominant Sulfate-Reducing Bacterial Partner of Anaerobic Methanotrophs of the ANME-2 Clade. Environ. Microbiol. 2010, 12, 2327–2340. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Ma, Y.; Feng, C.; Cai, L.; Li, W. Diversity of Pelagic and Benthic Bacterial Assemblages in the Western Pacific Ocean. Front. Microbiol. 2020, 11, 1730. [Google Scholar] [CrossRef]
- Xue, C.-X.; Liu, J.; Lea-Smith, D.J.; Rowley, G.; Lin, H.; Zheng, Y.; Zhu, X.-Y.; Liang, J.; Ahmad, W.; Todd, J.D.; et al. Insights into the Vertical Stratification of Microbial Ecological Roles across the Deepest Seawater Column on Earth. Microorganisms 2020, 8, 1309. [Google Scholar] [CrossRef]
- Hoshino, T.; Doi, H.; Uramoto, G.-I.; Wörmer, L.; Adhikari, R.R.; Xiao, N.; Morono, Y.; D’Hondt, S.; Hinrichs, K.-U.; Inagaki, F. Global Diversity of Microbial Communities in Marine Sediment. Proc. Natl. Acad. Sci. USA 2020, 117, 27587–27597. [Google Scholar] [CrossRef]
- Kerrigan, Z.; D’Hondt, S. Patterns of Relative Bacterial Richness and Community Composition in Seawater and Marine Sediment Are Robust for Both Operational Taxonomic Units and Amplicon Sequence Variants. Front. Microbiol. 2022, 13, 796758. [Google Scholar] [CrossRef]
- Santiago, B.C.F.; de Souza, I.D.; Cavalcante, J.V.F.; Morais, D.A.A.; da Silva, M.B.; Pasquali, M.A.d.B.; Dalmolin, R.J.S. Metagenomic Analyses Reveal the Influence of Depth Layers on Marine Biodiversity on Tropical and Subtropical Regions. Microorganisms 2023, 11, 1668. [Google Scholar] [CrossRef] [PubMed]
- Kirchman, D.L.; Cottrell, M.T.; Lovejoy, C. The Structure of Bacterial Communities in the Western Arctic Ocean as Revealed by Pyrosequencing of 16S rRNA Genes. Environ. Microbiol. 2010, 12, 1132–1143. [Google Scholar] [CrossRef]
- Li, Y.; Wang, Z.; Lin, X. Microbial Community Structure of Arctic Seawater as Revealed by Pyrosequencing. Acta Oceanol. Sin. 2016, 35, 78–84. [Google Scholar] [CrossRef]
- Wilson, B.; Müller, O.; Nordmann, E.-L.; Seuthe, L.; Bratbak, G.; Øvreås, L. Changes in Marine Prokaryote Composition with Season and Depth over an Arctic Polar Year. Front. Mar. Sci. 2017, 4, 95. [Google Scholar] [CrossRef]
- Thiele, S.; Vader, A.; Thomson, S.; Saubrekka, K.; Petelenz, E.; Müller, O.; Bratbak, G.; Øvreås, L. Seasonality of the Bacterial and Archaeal Community Composition of the Northern Barents Sea. Front. Microbiol. 2023, 14, 1213718. [Google Scholar] [CrossRef] [PubMed]
- Thiele, S.; Vader, A.; Thomson, S.; Saubrekka, K.; Petelenz, E.; Armo, H.R.; Müller, O.; Olsen, L.; Bratbak, G.; Øvreås, L. The Summer Bacterial and Archaeal Community Composition of the Northern Barents Sea. Prog. Oceanogr. 2023, 215, 103054. [Google Scholar] [CrossRef]
- Du, Z.-J.; Wang, Z.-J.; Zhao, J.-X.; Chen, G.-J. Woeseia oceani Gen. Nov., Sp. Nov., a Chemoheterotrophic Member of the Order Chromatiales, and Proposal of Woeseiaceae Fam. Nov. Int. J. Syst. Evol. Microbiol. 2016, 66, 107–112. [Google Scholar] [CrossRef]
- Mußmann, M.; Pjevac, P.; Krüger, K.; Dyksma, S. Genomic Repertoire of the Woeseiaceae/JTB255, Cosmopolitan and Abundant Core Members of Microbial Communities in Marine Sediments. ISME J. 2017, 11, 1276–1281. [Google Scholar] [CrossRef]
- Wunder, L.C.; Aromokeye, D.A.; Yin, X.; Richter-Heitmann, T.; Willis-Poratti, G.; Schnakenberg, A.; Otersen, C.; Dohrmann, I.; Römer, M.; Bohrmann, G.; et al. Iron and Sulfate Reduction Structure Microbial Communities in (Sub-)Antarctic Sediments. ISME J. 2021, 15, 3587–3604. [Google Scholar] [CrossRef]
- Dyksma, S.; Lenk, S.; Sawicka, J.E.; Mußmann, M. Uncultured Gammaproteobacteria and Desulfobacteraceae Account for Major Acetate Assimilation in a Coastal Marine Sediment. Front. Microbiol. 2018, 9, 3124. [Google Scholar] [CrossRef]
- Dyksma, S.; Pjevac, P.; Ovanesov, K.; Mussmann, M. Evidence for H2 Consumption by Uncultured Desulfobacterales in Coastal Sediments. Environ. Microbiol. 2018, 20, 450–461. [Google Scholar] [CrossRef]
- Ruff, S.E.; Biddle, J.F.; Teske, A.P.; Knittel, K.; Boetius, A.; Ramette, A. Global Dispersion and Local Diversification of the Methane Seep Microbiome. Proc. Natl. Acad. Sci. USA 2015, 112, 4015–4020. [Google Scholar] [CrossRef]
- Dekas, A.E.; Connon, S.A.; Chadwick, G.L.; Trembath-Reichert, E.; Orphan, V.J. Activity and Interactions of Methane Seep Microorganisms Assessed by Parallel Transcription and FISH-NanoSIMS Analyses. ISME J. 2016, 10, 678–692. [Google Scholar] [CrossRef]
- Spring, S.; Bunk, B.; Spröer, C.; Rohde, M.; Klenk, H.P. Genome biology of a novel lineage of planctomycetes widespread in anoxic aquatic environments. Environ. Microbiol. 2018, 20, 2438–2455. [Google Scholar] [CrossRef]
- Pradel, N.; Fardeau, M.L.; Tindall, B.J.; Spring, S. Anaerohalosphaera lusitana gen. nov., sp. nov., and Limihaloglobus sulfuriphilus gen. nov., sp. nov., isolated from solar saltern sediments, and proposal of Anaerohalosphaeraceae fam. nov. within the order Sedimentisphaerales. Int. J. Syst. Evol. Microbiol. 2020, 70, 1321–1330. [Google Scholar] [CrossRef]
- Kraft, B.; Canfield, D.E. Microbe Profile: Nitrosopumilus maritimus. Microbiology 2022, 168, 001207. [Google Scholar] [CrossRef]
- Shah, V.; Chang, B.X.; Morris, R.M. Cultivation of a Chemoautotroph from the SUP05 Clade of Marine Bacteria That Produces Nitrite and Consumes Ammonium. ISME J. 2017, 11, 263–271. [Google Scholar] [CrossRef]
- Spietz, R.L.; Lundeen, R.A.; Zhao, X.; Nicastro, D.; Ingalls, A.E.; Morris, R.M. Heterotrophic Carbon Metabolism and Energy Acquisition in Candidatus Thioglobus Singularis Strain PS1, a Member of the SUP05 Clade of Marine Gammaproteobacteria. Environ. Microbiol. 2019, 21, 2391–2401. [Google Scholar] [CrossRef]
- Connon, S.A.; Giovannoni, S.J. High-Throughput Methods for Culturing Microorganisms in Very-Low-Nutrient Media Yield Diverse New Marine Isolates. Appl. Environ. Microbiol. 2002, 68, 3878–3885. [Google Scholar] [CrossRef]
- Moore, E.R.; Weaver, A.J.; Davis, E.W.; Giovannoni, S.J.; Halsey, K.H. Metabolism of Key Atmospheric Volatile Organic Compounds by the Marine Heterotrophic Bacterium Pelagibacter HTCC1062 (SAR11). Environ. Microbiol. 2022, 24, 212–222. [Google Scholar] [CrossRef]
- Ghai, R.; Mizuno, C.M.; Picazo, A.; Camacho, A.; Rodriguez-Valera, F. Metagenomics Uncovers a New Group of Low GC and Ultra-Small Marine Actinobacteria. Sci. Rep. 2013, 3, 2471. [Google Scholar] [CrossRef]
- Long, A.M.; Hou, S.; Ignacio-Espinoza, J.C.; Fuhrman, J.A. Benchmarking Microbial Growth Rate Predictions from Metagenomes. ISME J. 2021, 15, 183–195. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stroeva, A.R.; Klyukina, A.A.; Vidishcheva, O.N.; Poludetkina, E.N.; Solovyeva, M.A.; Pyrkin, V.O.; Gavirova, L.A.; Birkeland, N.-K.; Akhmanov, G.G.; Bonch-Osmolovskaya, E.A.; et al. Structure of Benthic Microbial Communities in the Northeastern Part of the Barents Sea. Microorganisms 2024, 12, 387. https://doi.org/10.3390/microorganisms12020387
Stroeva AR, Klyukina AA, Vidishcheva ON, Poludetkina EN, Solovyeva MA, Pyrkin VO, Gavirova LA, Birkeland N-K, Akhmanov GG, Bonch-Osmolovskaya EA, et al. Structure of Benthic Microbial Communities in the Northeastern Part of the Barents Sea. Microorganisms. 2024; 12(2):387. https://doi.org/10.3390/microorganisms12020387
Chicago/Turabian StyleStroeva, Aleksandra R., Alexandra A. Klyukina, Olesya N. Vidishcheva, Elena N. Poludetkina, Marina A. Solovyeva, Vladislav O. Pyrkin, Liliya A. Gavirova, Nils-Kåre Birkeland, Grigorii G. Akhmanov, Elizaveta A. Bonch-Osmolovskaya, and et al. 2024. "Structure of Benthic Microbial Communities in the Northeastern Part of the Barents Sea" Microorganisms 12, no. 2: 387. https://doi.org/10.3390/microorganisms12020387