

Comparative Genomic Analysis of Shrimp-Pathogenic Vibrio parahaemolyticus LC and Intraspecific Strains with Emphasis on Virulent Factors of Mobile Genetic Elements

Abstract
:1. Introduction
2. Materials and Methods
2.1. Strain Isolation and Cultivation
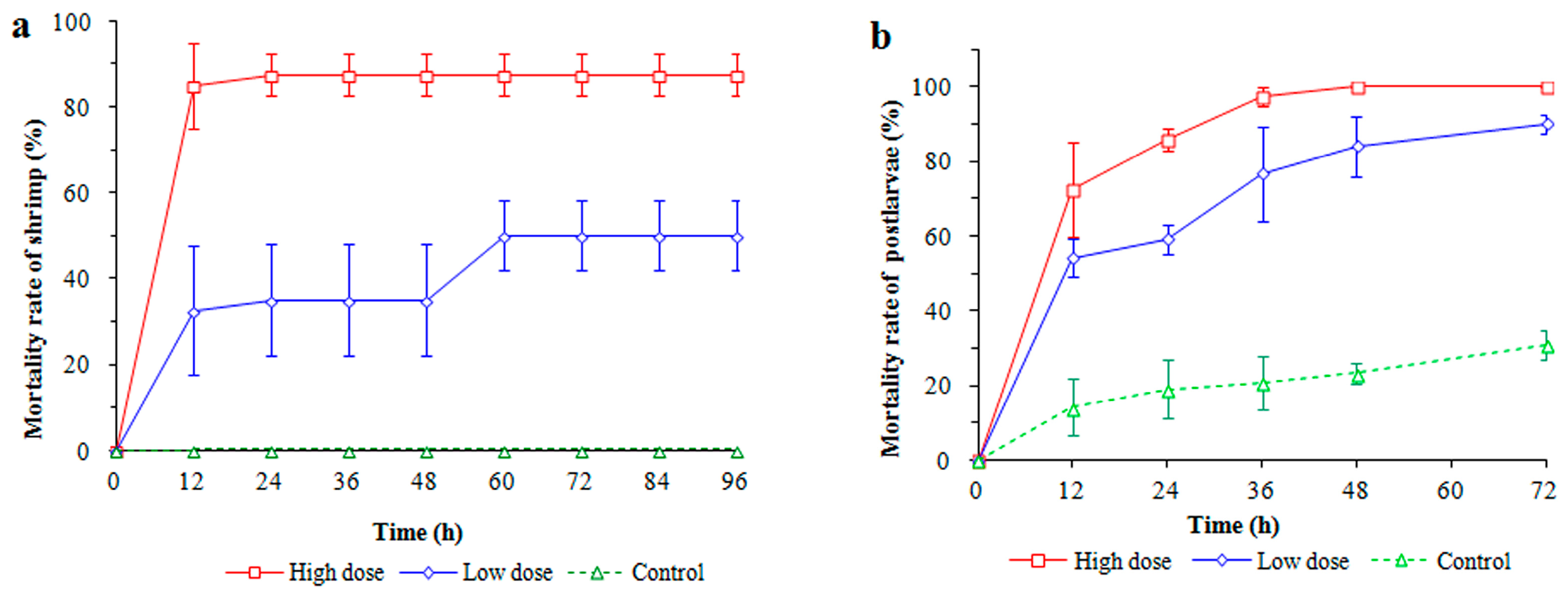
2.2. Infection of Shrimp with Strain LC
2.3. Genome Sequencing and Functional Annotation
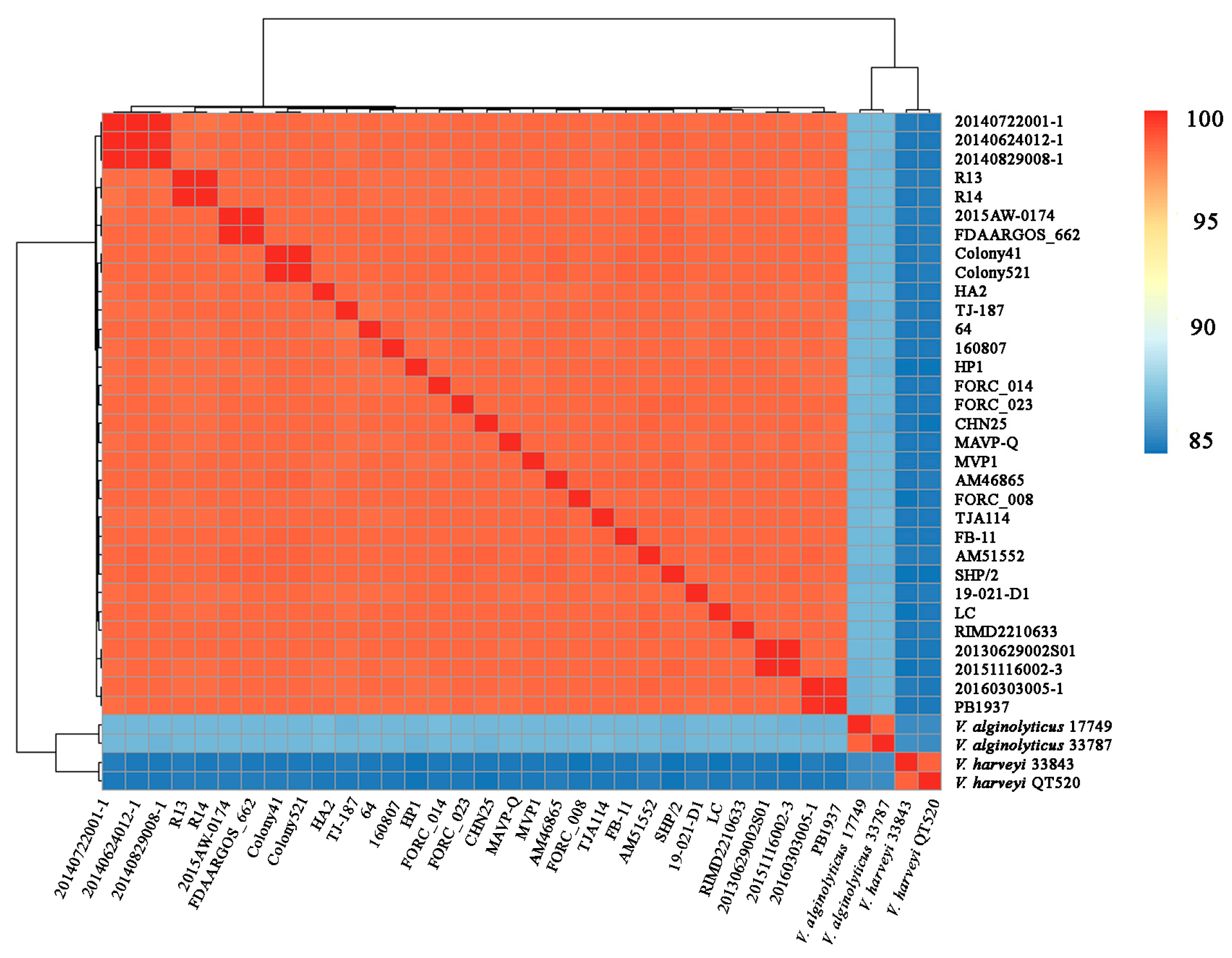
2.4. Calculation of Average Nucleotide Identity
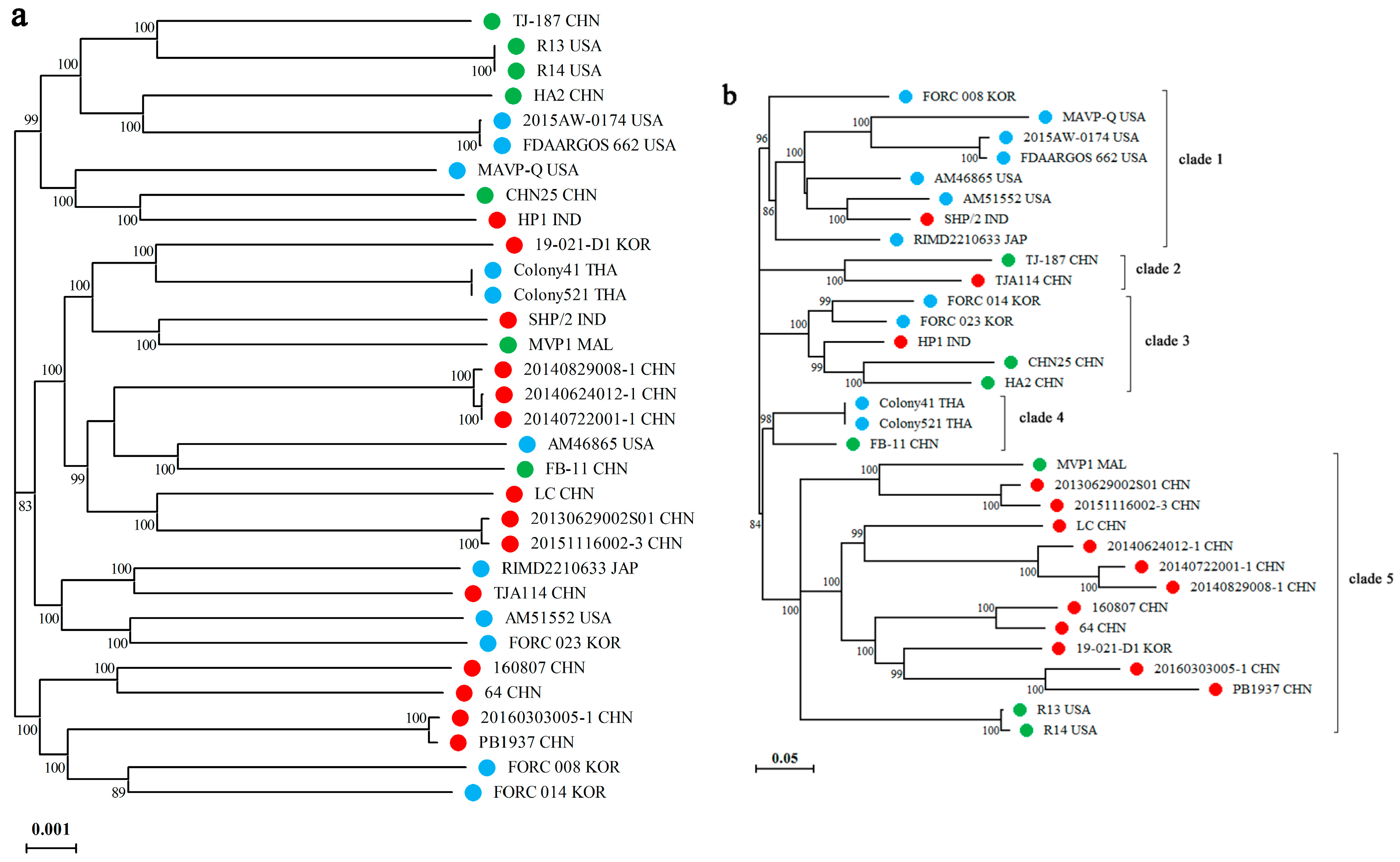
2.5. Analysis of Pangenome and Phylogeny
2.6. Identification of Virulence Factors
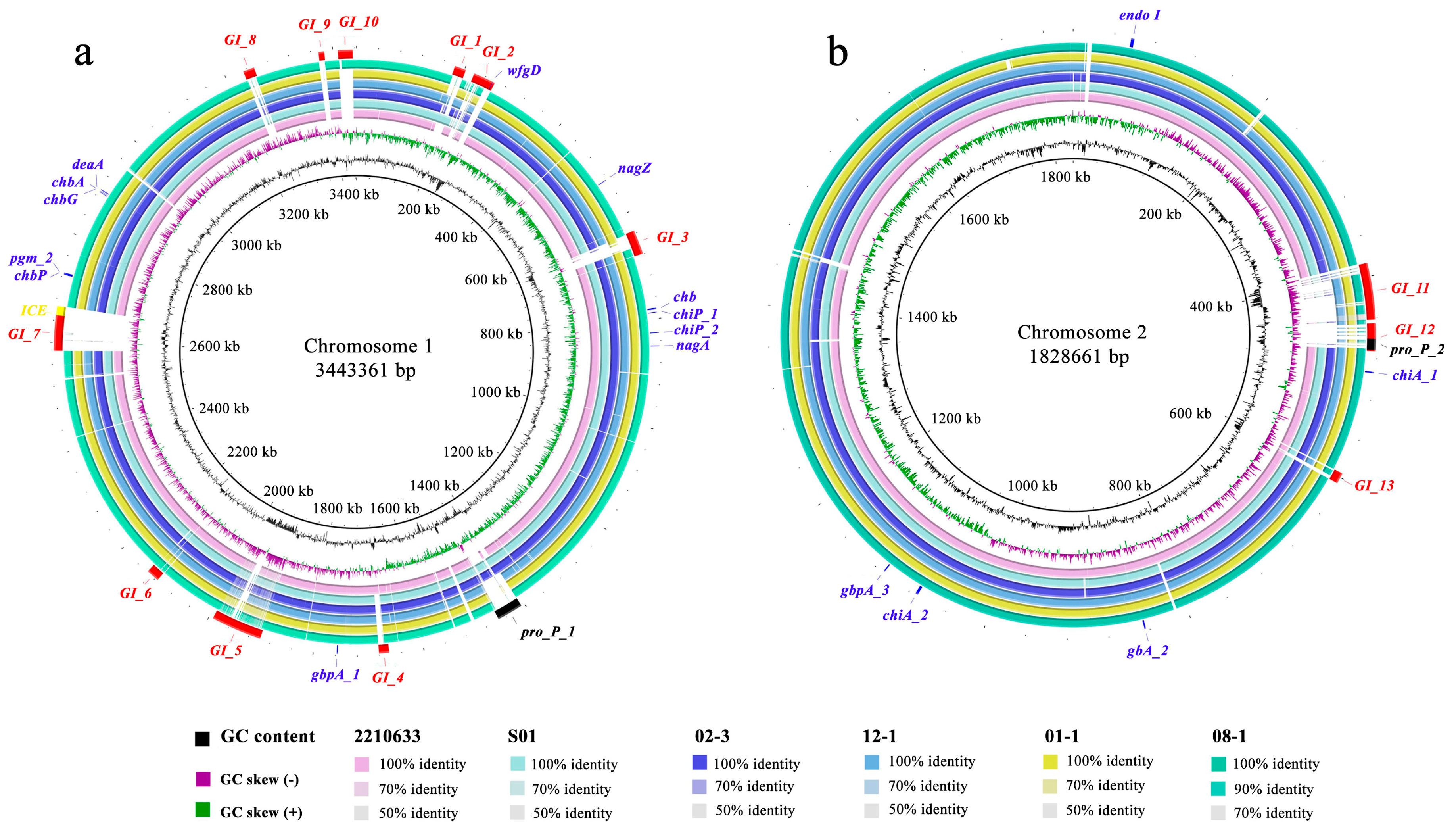
2.7. Comparison of Gene Contents of GIs and Plasmids
2.8. Statistical Analysis
3. Results
3.1. Shrimp Performance after Infection
3.2. Genome Feature and ANI Analysis
3.3. Phylogenome Analysis
3.4. Genome Comparison of LC with Six Closest Strains
3.5. General Virulence Factors
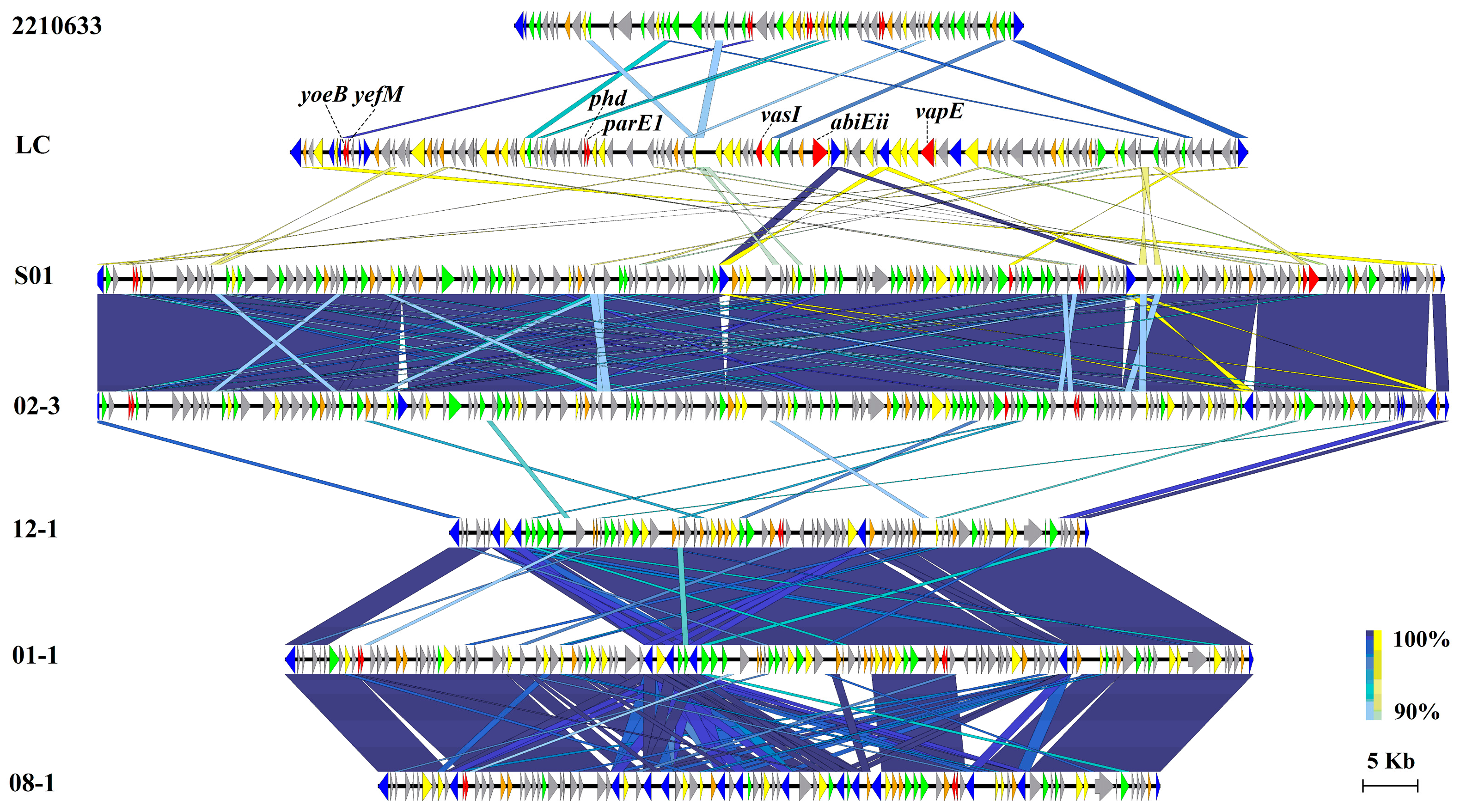
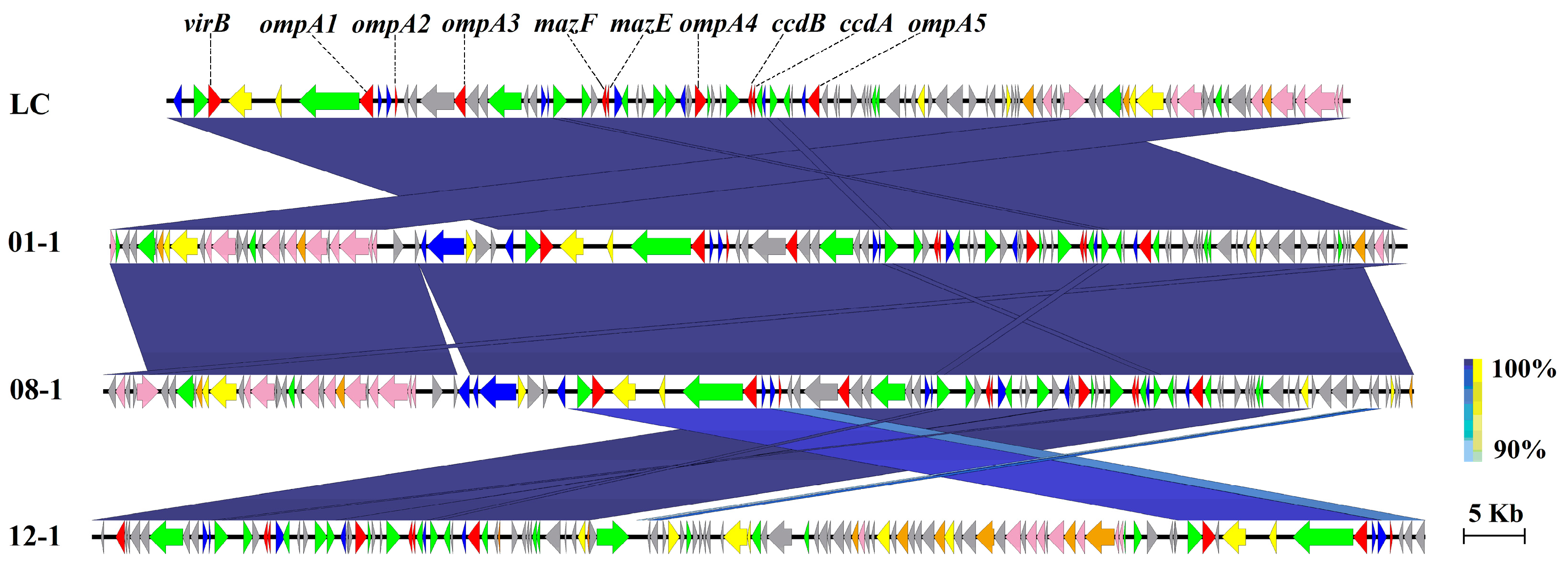
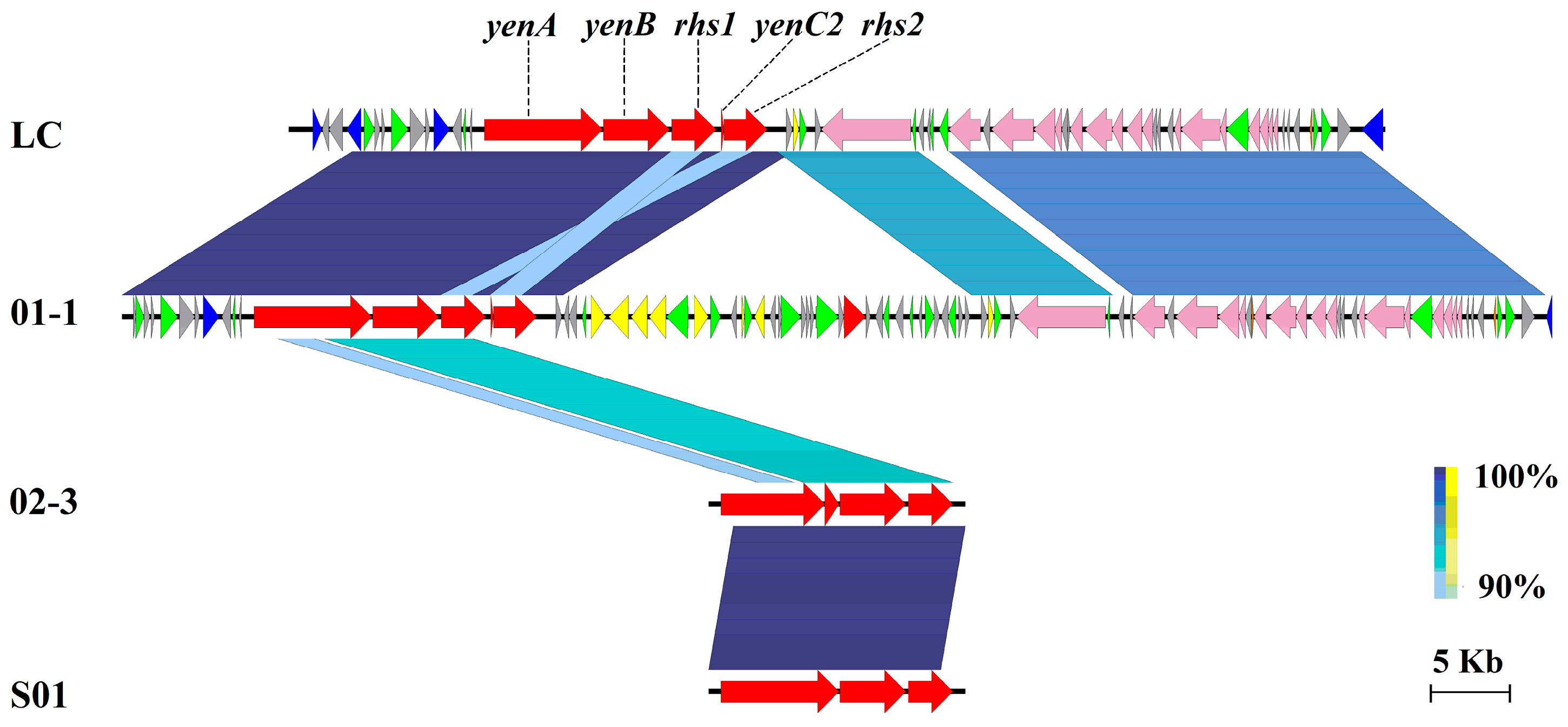
3.6. Virulent Factors Associated with MGEs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- de Souza Valente, C.; Wan, A.H.L. Vibrio and major commercially important vibriosis diseases in decapod crustaceans. J. Invertebr. Pathol. 2021, 181, 107527. [Google Scholar] [CrossRef]
- Li, L.; Wong, H.C.; Nong, W.; Cheung, M.K.; Law, P.T.; Kam, K.M.; Kwan, H.S. Comparative genomic analysis of clinical and environmental strains provides insight into the pathogenicity and evolution of Vibrio parahaemolyticus. BMC Genom. 2014, 15, 1135. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Urtaza, J.; van Aerle, R.; Abanto, M.; Haendiges, J.; Myers, R.A.; Trinanes, J.; Baker-Austin, C.; Gonzalez-Escalona, N. Genomic variation and evolution of Vibrio parahaemolyticus ST36 over the course of a transcontinental epidemic expansion. mBio 2017, 8, e01425-17. [Google Scholar] [CrossRef] [PubMed]
- Bachand, P.T.; Tallman, J.J.; Powers, N.C.; Woods, M.; Azadani, D.N.; Zimba, P.V.; Turner, J.W. Genomic identification and characterization of co-occurring harveyi clade species following a vibriosis outbreak in Pacific white shrimp, Penaeus (litopenaeus) vannamei. Aquaculture 2020, 518, 734628. [Google Scholar] [CrossRef]
- Prithvisagar, K.S.; Krishna Kumar, B.; Kodama, T.; Rai, P.; Iida, T.; Karunasagar, I.; Karunasagar, I. Whole genome analysis unveils genetic diversity and potential virulence determinants in Vibrio parahaemolyticus associated with disease outbreak among cultured Litopenaeus vannamei (Pacific white shrimp) in India. Virulence 2021, 12, 1936–1949. [Google Scholar] [CrossRef]
- Ceccarelli, D.; Hasan, N.A.; Huq, A.; Colwell, R.R. Distribution and dynamics of epidemic and pandemic Vibrio parahaemolyticus virulence factors. Front. Cell. Infect. Microbiol. 2013, 3, 97. [Google Scholar] [CrossRef]
- Li, L.; Meng, H.; Gu, D.; Li, Y.; Jia, M. Molecular mechanisms of Vibrio parahaemolyticus pathogenesis. Microbiol. Res. 2019, 222, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Izutsu, K.; Kurokawa, K.; Tashiro, K.; Kuhara, S.; Hayashi, T.; Honda, T.; Iida, T. Comparative genomic analysis using microarray demonstrates a strong correlation between the presence of the 80-kilobase pathogenicity island and pathogenicity in Kanagawa phenomenon-positive Vibrio parahaemolyticus strains. Infect. Immun. 2008, 76, 1016–1023. [Google Scholar] [CrossRef]
- Kumar, V.; Roy, S.; Behera, B.K.; Bossier, P.; Das, B.K. Acute hepatopancreatic necrosis disease (AHPND): Virulence, pathogenesis and mitigation strategies in shrimp aquaculture. Toxins 2021, 13, 524. [Google Scholar] [CrossRef]
- Sanguanrut, P.; Munkongwongsiri, N.; Kongkumnerd, J.; Thawonsuwan, J.; Thitamadee, S.; Boonyawiwat, V.; Tanasomwang, V.; Flegel, T.W.; Sritunyalucks, K. A cohort study of 196 Thai shrimp ponds reveals a complex etiology for early mortality syndrome (EMS). Aquaculture 2018, 493, 26–36. [Google Scholar] [CrossRef]
- Powell, A.; Baker-Austin, C.; Wagley, S.; Bayley, A.; Hartnell, R. Isolation of pandemic Vibrio parahaemolyticus from UK water and shellfish produce. Microb. Ecol. 2013, 65, 924–927. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Xu, H.; Su, Y.; Liu, S.; Xu, L.; Guo, Z.; Wu, J.; Cheng, C.; Feng, J. Horizontal gene transfer contributes to virulence and antibiotic resistance of Vibrio harveyi 345 based on complete genome sequence analysis. BMC Genom. 2019, 20, 761. [Google Scholar] [CrossRef]
- Jones, J.L.; Lüdeke, C.H.; Bowers, J.C.; Garrett, N.; Fischer, M.; Parsons, M.B.; Bopp, C.A.; DePaola, A. Biochemical, serological, and virulence characterization of clinical and oyster Vibrio parahaemolyticus isolates. J. Clin. Microbiol. 2012, 50, 2343–2352. [Google Scholar] [CrossRef]
- Ruwandeepika, H.A.D.; Jayaweera, T.S.P.; Bhowmick, P.P.; Karunasagar, I.; Bossier, P.; Defoirdt, T. Pathogenesis, virulence factors and virulence regulation of vibrios belonging to the Harveyi clade. Rev. Aquacult. 2012, 4, 59–74. [Google Scholar] [CrossRef]
- Lee, C.T.; Chen, I.T.; Yang, Y.T.; Ko, T.P.; Huang, Y.T.; Huang, J.Y.; Huang, M.F.; Lin, S.J.; Chen, C.Y.; Lin, S.S.; et al. The opportunistic marine pathogen Vibrio parahaemolyticus becomes virulent by acquiring a plasmid expressing a deadly toxin. Proc. Natl. Acad. Sci. USA 2015, 112, 10798–10803. [Google Scholar] [CrossRef] [PubMed]
- Le Roux, F.; Blokesch, M. Eco-evolutionary dynamics linked to horizontal gene transfer in vibrios. Annu. Rev. Microbiol. 2018, 72, 89–110. [Google Scholar] [CrossRef]
- Xue, M.; Huang, X.; Xue, J.; He, R.; Liang, G.; Liang, H.; Liu, J.; Wen, C. Comparative genomic analysis of seven Vibrio alginolyticus strains isolated from shrimp larviculture water with emphasis on chitin utilization. Front. Microbiol. 2022, 13, 925747. [Google Scholar] [CrossRef]
- Dhillon, B.K.; Chiu, T.A.; Laird, M.R.; Langille, M.G.; Brinkman, F.S. IslandViewer update: Improved genomic island discovery and visualization. Nucleic Acids Res. 2013, 41, W129–W132. [Google Scholar] [CrossRef]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef]
- Makino, K.; Oshima, K.; Kurokawa, K.; Yokoyama, K.; Uda, T.; Tagomori, K.; Iijima, Y.; Najima, M.; Nakano, M.; Yamashita, A.; et al. Genome sequence of Vibrio parahaemolyticus: A pathogenic mechanism distinct from that of V. cholerae. Lancet 2003, 361, 743–749. [Google Scholar] [CrossRef]
- Jain, C.; Rodriguez-R, L.M.; Phillippy, A.M.; Konstantinidis, K.T.; Aluru, S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 2018, 9, 5114. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Liu, B.; Zheng, D.; Zhou, S.; Chen, L.; Yang, J. VFDB 2022: A general classification scheme for bacterial virulence factors. Nucleic Acids Res. 2022, 50, D912–D917. [Google Scholar] [CrossRef]
- Alikhan, N.F.; Petty, N.K.; Ben Zakour, N.L.; Beatson, S.A. BLAST Ring Image Generator (BRIG): Simple prokaryote genome comparisons. BMC Genom. 2011, 12, 402. [Google Scholar] [CrossRef]
- Sullivan, M.J.; Petty, N.K.; Beatson, S.A. Easyfig: A genome comparison visualizer. Bioinformatics 2011, 27, 1009–1010. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Ni, P.; Yang, Q.; Hu, H.; Wang, Q.; Ye, S.; Liu, Y. Delineating the key virulence factors and intraspecies divergence of Vibrio harveyi via whole-genome sequencing. Can. J. Microbiol. 2021, 67, 231–248. [Google Scholar] [CrossRef]
- Kazakov, T.; Kuznedelov, K.; Semenova, E.; Mukhamedyarov, D.; Datsenko, K.A.; Metlitskaya, A.; Vondenhoff, G.H.; Tikhonov, A.; Agarwal, V.; Nair, S.; et al. The RimL transacetylase provides resistance to translation inhibitor microcin C. J. Bacteriol. 2014, 196, 3377–3385. [Google Scholar] [CrossRef] [PubMed]
- Lobato-Márquez, D.; Díaz-Orejas, R.; García-Del Portillo, F. Toxin-antitoxins and bacterial virulence. FEMS Microbiol. Rev. 2016, 40, 592–609. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.Y.; Lee, B.J. Structure, biology, and therapeutic application of toxin-antitoxin systems in pathogenic bacteria. Toxins 2016, 8, 305. [Google Scholar] [CrossRef]
- Równicki, M.; Lasek, R.; Trylska, J.; Bartosik, D. Targeting Type II toxin-antitoxin systems as antibacterial strategies. Toxins 2020, 12, 568. [Google Scholar] [CrossRef]
- Ma, D.; Gu, H.; Shi, Y.; Huang, H.; Sun, D.; Hu, Y. Edwardsiella piscicida YefM-YoeB: A Type II toxin-antitoxin system that is related to antibiotic resistance, biofilm formation, serum survival, and host infection. Front. Microbiol. 2021, 12, 646299. [Google Scholar] [CrossRef]
- Zhou, J.; Du, X.J.; Liu, Y.; Gao, Z.Q.; Geng, Z.; Dong, Y.H.; Zhang, H. Insights into the neutralization and DNA binding of toxin-antitoxin system ParESO-CopASO by structure-function studies. Microorganisms 2021, 9, 2506. [Google Scholar] [CrossRef]
- Dy, R.L.; Przybilski, R.; Semeijn, K.; Salmond, G.P.; Fineran, P.C. A widespread bacteriophage abortive infection system functions through a Type IV toxin-antitoxin mechanism. Nucleic Acids Res. 2014, 42, 4590–4605. [Google Scholar] [CrossRef]
- McConnell, M.J.; Actis, L.; Pachón, J. Acinetobacter baumannii: Human infections, factors contributing to pathogenesis and animal models. FEMS Microbiol. Rev. 2013, 37, 130–155. [Google Scholar] [CrossRef]
- Confer, A.W.; Ayalew, S. The OmpA family of proteins: Roles in bacterial pathogenesis and immunity. Vet. Microbiol. 2013, 163, 207–222. [Google Scholar] [CrossRef] [PubMed]
- Bunpa, S.; Chaichana, N.; Teng, J.L.L.; Lee, H.H.; Woo, P.C.Y.; Sermwittayawong, D.; Sawangjaroen, N.; Sermwittayawong, N. Outer membrane protein A (OmpA) is a potential virulence factor of Vibrio alginolyticus strains isolated from diseased fish. J. Fish Dis. 2020, 43, 275–284. [Google Scholar] [CrossRef]
- Bowen, D.; Rocheleau, T.A.; Blackburn, M.; Andreev, O.; Golubeva, E.; Bhartia, R.; ffrench-Constant, R.H. Insecticidal toxins from the bacterium Photorhabdus luminescens. Science 1998, 280, 2129–2132. [Google Scholar] [CrossRef] [PubMed]
- Busby, J.N.; Landsberg, M.J.; Simpson, R.M.; Jones, S.A.; Hankamer, B.; Hurst, M.R.; Lott, J.S. Structural analysis of Chi1 chitinase from Yen-Tc: The multisubunit insecticidal ABC toxin complex of Yersinia entomophaga. J. Mol. Biol. 2012, 415, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Hurst, M.R.H.; Jones, S.; Young, S.; Muetzel, S.; Calder, J.; van Koten, C. Assessment of toxicity and persistence of Yersinia entomophaga and its Yen-Tc associated toxin. Pest Manag. Sci. 2020, 76, 4301–4310. [Google Scholar] [CrossRef]
- Roderer, D.; Raunser, S. Tc toxin complexes: Assembly, membrane permeation, and protein translocation. Annu. Rev. Microbiol. 2019, 73, 247–265. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; You, Y.L.; Lai, Q.L.; Xu, L.M.; Li, F. Vibrio parahaemolyticus becomes highly virulent by producing Tc toxins. Aquaculture 2023, 576, 739817. [Google Scholar] [CrossRef]
- Tran, L.; Nunan, L.; Redman, R.M.; Mohney, L.L.; Pantoja, C.R.; Fitzsimmons, K.; Lightner, D.V. Determination of the infectious nature of the agent of acute hepatopancreatic necrosis syndrome affecting penaeid shrimp. Dis. Aquat. Organ. 2013, 105, 45–55. [Google Scholar] [CrossRef]
- Kehlet-Delgado, H.; Häse, C.C.; Mueller, R.S. Comparative genomic analysis of Vibrios yields insights into genes associated with virulence towards C. gigas larvae. BMC Genom. 2020, 21, 599. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, S.; Kumar, M.; Kumari, P.; Mahapatro, G.K.; Banerjee, N.; Sarin, N.B. Novel insecticidal chitinase from the insect pathogen Xenorhabdus nematophila. Int. J. Biol. Macromol. 2020, 159, 394–401. [Google Scholar] [CrossRef] [PubMed]
- Meibom, K.L.; Blokesch, M.; Dolganov, N.A.; Wu, C.Y.; Schoolnik, G.K. Chitin induces natural competence in Vibrio cholerae. Science 2005, 310, 1824–1827. [Google Scholar] [CrossRef]
- Borgeaud, S.; Metzger, L.C.; Scrignari, T.; Blokesch, M. The type VI secretion system of Vibrio cholerae fosters horizontal gene transfer. Science 2015, 347, 63–67. [Google Scholar] [CrossRef]
- Kostiuk, B.; Unterweger, D.; Provenzano, D.; Pukatzki, S. T6SS intraspecific competition orchestrates Vibrio cholerae genotypic diversity. Int. Microbiol. 2017, 20, 130–137. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain Type | Strain Name (abbr.) | Countries of Origin | Genome (Mb) | G+C (%) | Replicons | No. Genes | Accession Numbers |
|---|---|---|---|---|---|---|---|
| Shrimp-pathogenic | LC | China | 5.50 | 45.38 | 2 Chr., 3 Pla. | 5142 | CP119301-CP119305 |
| 19-021-D1 | Korea | 5.58 | 45.35 | 2 Chr., 2 Pla. | 5272 | CP046411-CP046414 | |
| 160807 | China | 5.40 | 45.26 | 2 Chr., 1 Pla. | 4990 | CP033141-CP033143 | |
| 20151116002-3 (02-3) | China | 5.46 | 45.24 | 2 Chr., 2 Pla. | 5087 | CP034305-CP034308 | |
| 20160303005-1 | China | 5.97 | 45.15 | 2 Chr., 5 Pla. | 5662 | CP034298-CP034304 | |
| 20140722001-1 (01-1) | China | 5.56 | 45.27 | 2 Chr., 3 Pla. | 5214 | CP034289-CP034293 | |
| 20130629002S01 (S01) | China | 5.40 | 45.36 | 2 Chr., 2 Pla. | 5027 | CP020034-CP020037 | |
| 20140829008-1 (08-1) | China | 5.42 | 45.35 | 2 Chr., 2 Pla. | 5061 | CP034294-CP034297 | |
| 20140624012-1 (12-1) | China | 5.35 | 45.32 | 2 Chr., 2 Pla. | 4997 | CP034285-CP034288 | |
| HP1 | India | 5.08 | 45.31 | 2 Chr. | 4644 | CP069236-CP069237 | |
| SHP/2 | India | 5.25 | 45.36 | 2 Chr., 2 Pla. | 4817 | CP066156-CP066159 | |
| TJA114 | China | 5.57 | 45.20 | 2 Chr., 3 Pla. | 5215 | CP060087-CP060091 | |
| PB1937 | China | 5.58 | 45.34 | 2 Chr., 2 Pla. | 5361 | CP022243-CP022246 | |
| 64 | China | 5.43 | 45.27 | 2 Chr., 2 Pla. | 5305 | CP074415-CP074418 | |
| Human-pathogenic | RIMD2210633 (2210633) | Japan | 5.17 | 45.40 | 2 Chr. | 4738 | NC_004603, NC_004605 |
| FORC_008 | Korea | 5.04 | 45.44 | 2 Chr. | 4661 | CP009982-CP009983 | |
| FORC_014 | Korea | 5.29 | 45.35 | 2 Chr., 1 Pla. | 4943 | CP011406-CP011408 | |
| FORC_023 | Korea | 5.02 | 45.44 | 2 Chr. | 4605 | CP012950-CP012951 | |
| Colony521 | Thailand | 5.39 | 46.17 | 2 Chr. | 4452 | CP078661-CP078661 | |
| Colony41 | Thailand | 5.39 | 46.17 | 2 Chr. | 4452 | CP076383-CP076384 | |
| FDAARGOS_662 | USA | 5.15 | 45.36 | 2 Chr. | 4758 | CP044070-CP044071 | |
| 2015AW-0174 | USA | 5.16 | 45.30 | 2 Chr. | 4773 | CP046753-CP046754 | |
| MAVP-Q | USA | 5.26 | 45.30 | 2 Chr. | 4912 | CP022472-CP022473 | |
| AM46865 | USA | 5.09 | 45.47 | 2 Chr. | 4712 | CP046761-CP046762 | |
| AM51552 | USA | 5.14 | 45.36 | 2 Chr. | 4716 | CP046759-CP046760 | |
| Non-pathogenic | R14 | USA | 5.44 | 45.27 | 2 Chr., 3 Pla. | 5006 | CP028141-CP028145 |
| R13 | USA | 5.44 | 45.27 | 2 Chr., 3 Pla. | 4999 | CP028342-CP028346 | |
| MVP1 | Malaysia | 5.39 | 45.27 | 2 Chr., 1 Pla. | 4945 | CP043421-CP043423 | |
| CHN25 | China | 5.44 | 45.19 | 2 Chr. 3 Pla. | 5048 | CP010883-CP010887 | |
| HA2 | China | 5.28 | 44.93 | 2 Chr. | 4869 | CP023709-CP023710 | |
| TJ-187 | China | 5.47 | 45.28 | 2 Chr., 3 Pla. | 5149 | CP068647-CP068651 | |
| FB-11 | China | 5.13 | 45.40 | 2 Chr. | 4709 | CP073068-CP073069 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xue, M.; Gao, Q.; Yan, R.; Liu, L.; Wang, L.; Wen, B.; Wen, C. Comparative Genomic Analysis of Shrimp-Pathogenic Vibrio parahaemolyticus LC and Intraspecific Strains with Emphasis on Virulent Factors of Mobile Genetic Elements. Microorganisms 2023, 11, 2752. https://doi.org/10.3390/microorganisms11112752
Xue M, Gao Q, Yan R, Liu L, Wang L, Wen B, Wen C. Comparative Genomic Analysis of Shrimp-Pathogenic Vibrio parahaemolyticus LC and Intraspecific Strains with Emphasis on Virulent Factors of Mobile Genetic Elements. Microorganisms. 2023; 11(11):2752. https://doi.org/10.3390/microorganisms11112752
Chicago/Turabian StyleXue, Ming, Qi Gao, Rui Yan, Lingping Liu, Ling Wang, Binyu Wen, and Chongqing Wen. 2023. "Comparative Genomic Analysis of Shrimp-Pathogenic Vibrio parahaemolyticus LC and Intraspecific Strains with Emphasis on Virulent Factors of Mobile Genetic Elements" Microorganisms 11, no. 11: 2752. https://doi.org/10.3390/microorganisms11112752