Establishment of Glycosaminoglycan Assays for Mucopolysaccharidoses

 ,
,  and
and
Abstract
:1. Introduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disorder | Deficient Enzyme | Trait | Chromosome | Primary storage GAG(s) | KS elevation in blood |
|---|---|---|---|---|---|
| MPS I (Hurler) | α-L-Iduronidase (IDUA) | AR | 4p16.3 | DS, HS | ↑↑↑ |
| MPS II (Hunter) | Iduronate-2-sulfatase (IDS) | XR | Xq28 | DS, HS | ↑↑↑ |
| MPS IIIA (Sanfillipo A) | Heparan-N-sulfatase (SGSH) | AR | 17q25.3 | HS | ↑ |
| MPS IIIB (Sanfillipo B) | α-N-Acetylglucoaminidase (NAGLU) | AR | 17q21 | HS | ↑ |
| MPS IIIC (Sanfillipo C) | α-Glucosaminidase acetyltransferase (HGSNAT) | AR | 8p11-q13 | HS | ↑ |
| MPS IIID (Sanfillipo D) | N-Acetylglucosamine 6-sulfatase (GNS) | AR | 12q14 | HS | NA |
| MPS IVA (Morquio A) | Galactose 6-sulfatase, N-acetylgalactosamine-6-sulfate sufatase (GALNS) | AR | 16q24.3 | C6S, KS | ↑↑↑ |
| MPS IVB (Morquio B) | β-Galactosidase (GLB1) | AR | 3p21.33 | KS | ↑ |
| MPS VI (Maroteaux-Lamy) | N-Acetylgalactosamine-4-sulfatase (G4S) | AR | 5q13.3 | C4S, DS | ↑↑ |
| MPS VII (Sly) | β-D-Glucuronidase (GUSB) | AR | 7q21-q22 | C4, 6S, DS, HS | ↑↑ |
2. ELISA
2.1. Background
2.2. Development of Sandwich ELISA Assay
2.2.1. Keratan Sulfate
2.2.2. Heparan Sulfate

3. History of GAG Assay by Tandem Mass Spectrometry (MS/MS)
4. MS/MS Method for Disaccharides
4.1. Standards and Enzymes
4.2. Sample Preparation
4.3. LC-MS/MS
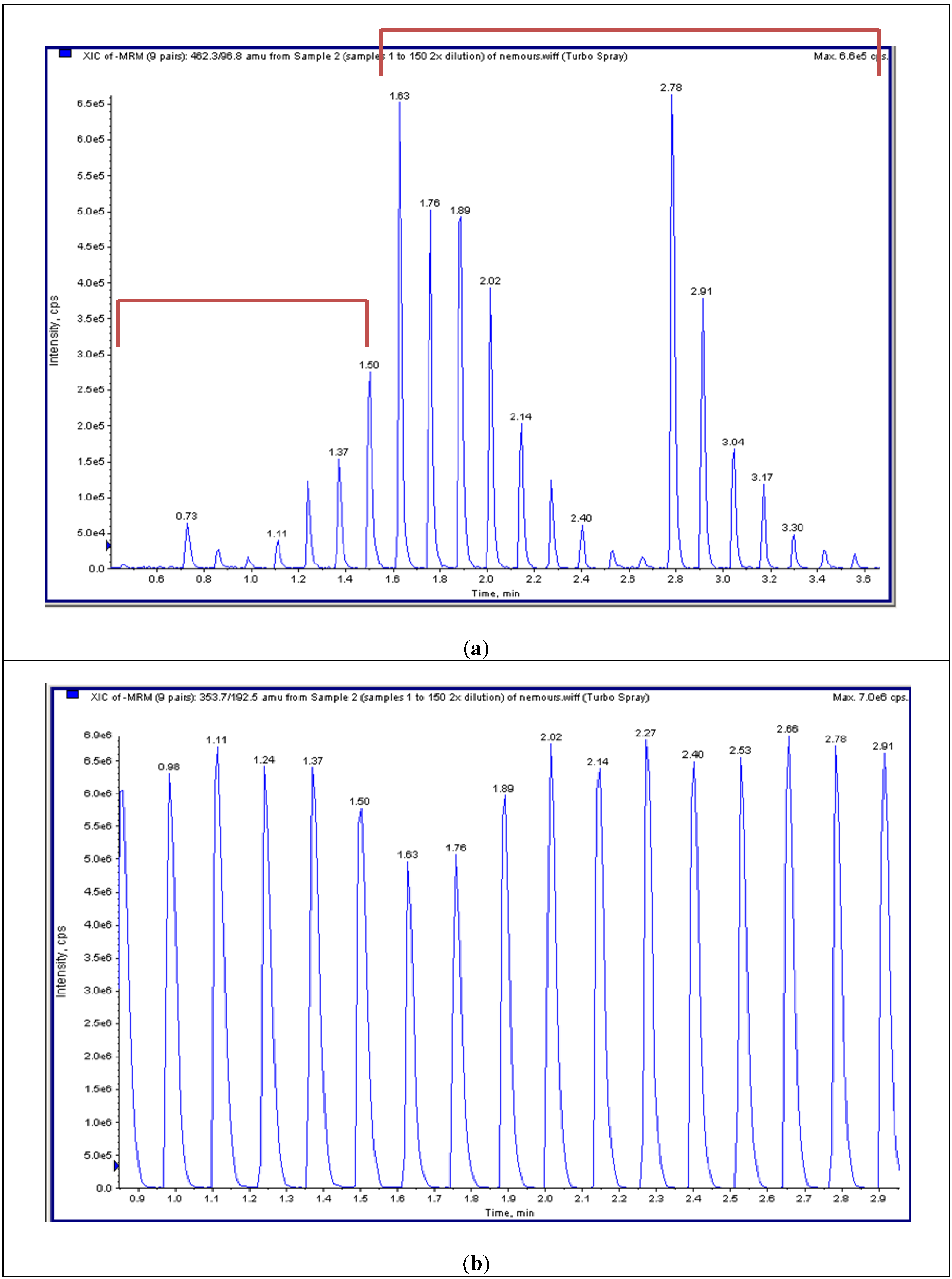
4.4. High-Throughput Tandem Mass Spectrometry (HT-MS/MS)
4.5. Disaccharide Determination Derived from GAGs
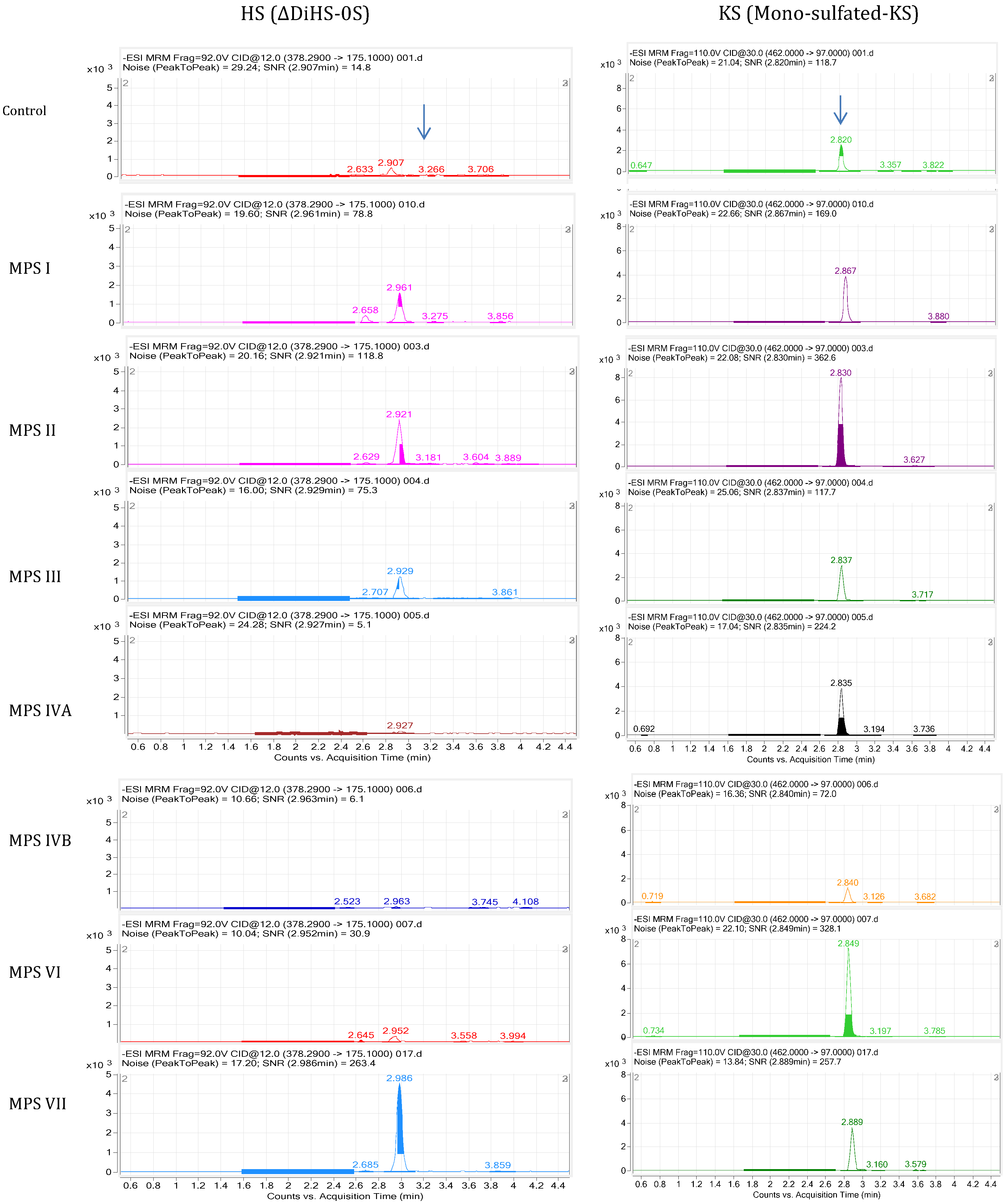
4.5.1. LC-MS/MS
4.5.2. HT-MS/MS


4.6. Analysis of Plasma and Serum Samples in MPS Patients by LC-MS/MS
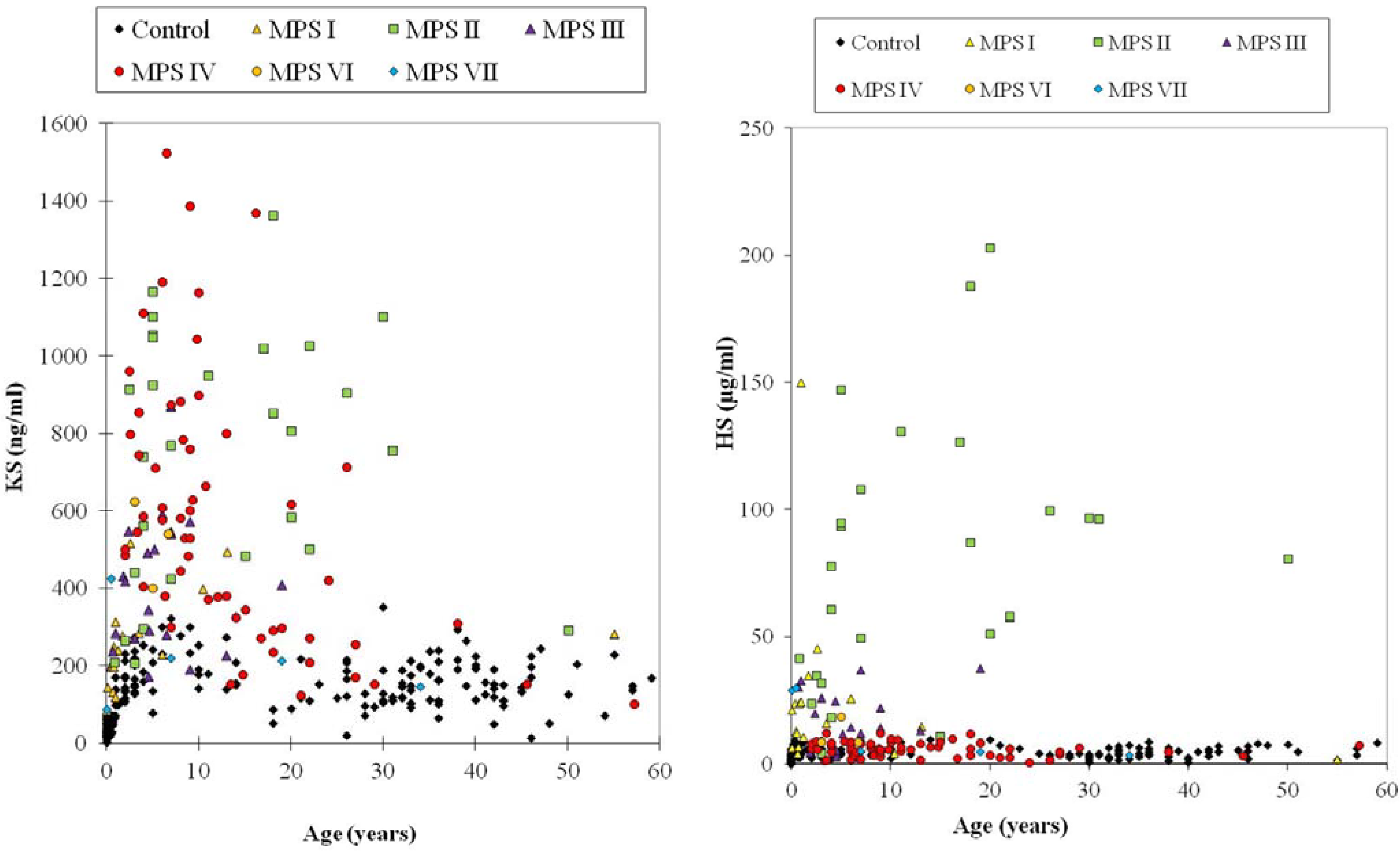
4.6.1. Keratan Sulfate
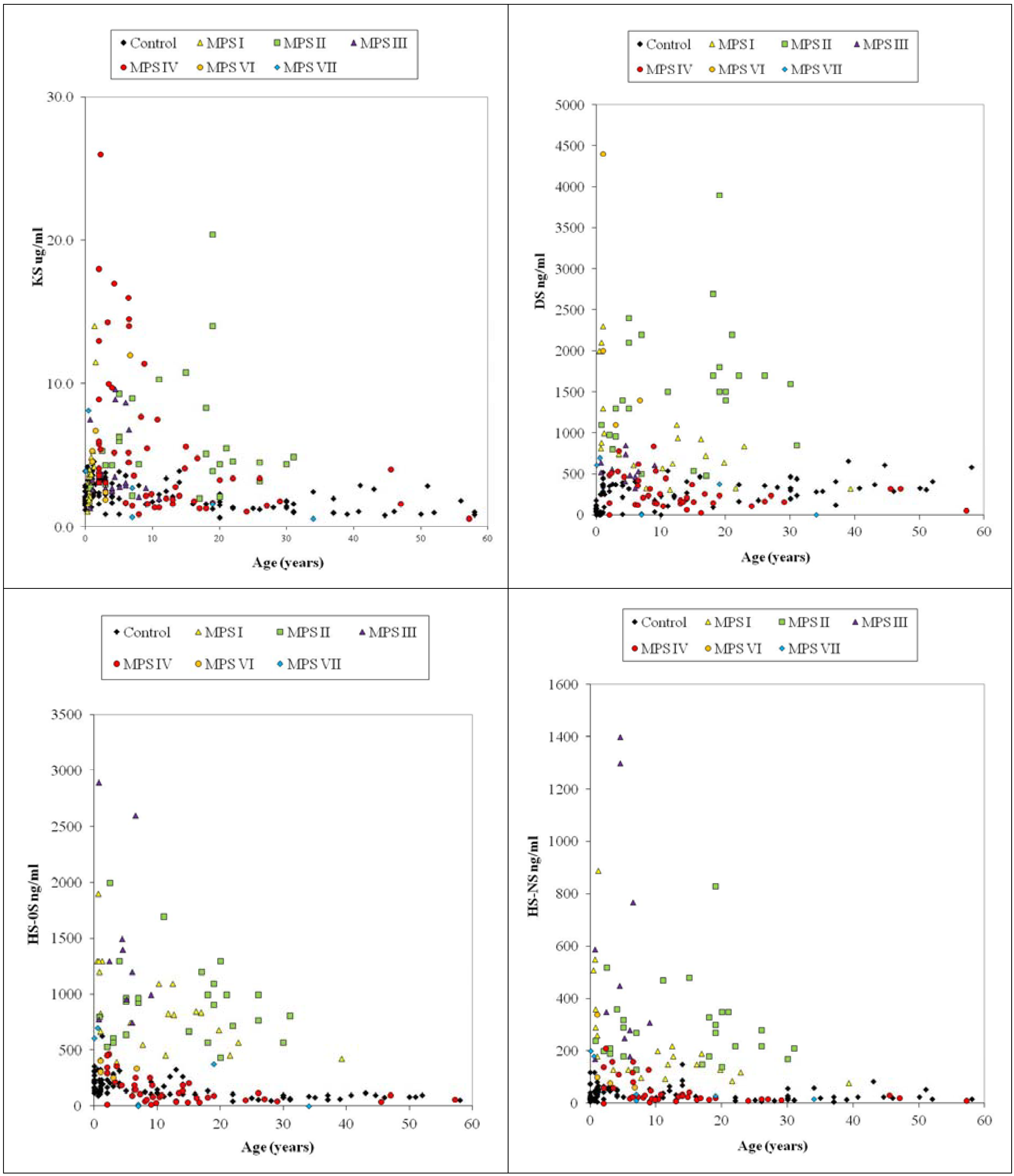
4.6.2. Dermatan Sulfate and Heparan Sulfate
4.6.3. Composition of DS and HS in Blood
4.7. Analysis of Plasma and Serum Samples in MPS Patients by LC-MS/MS

4.8. Newborn MPS
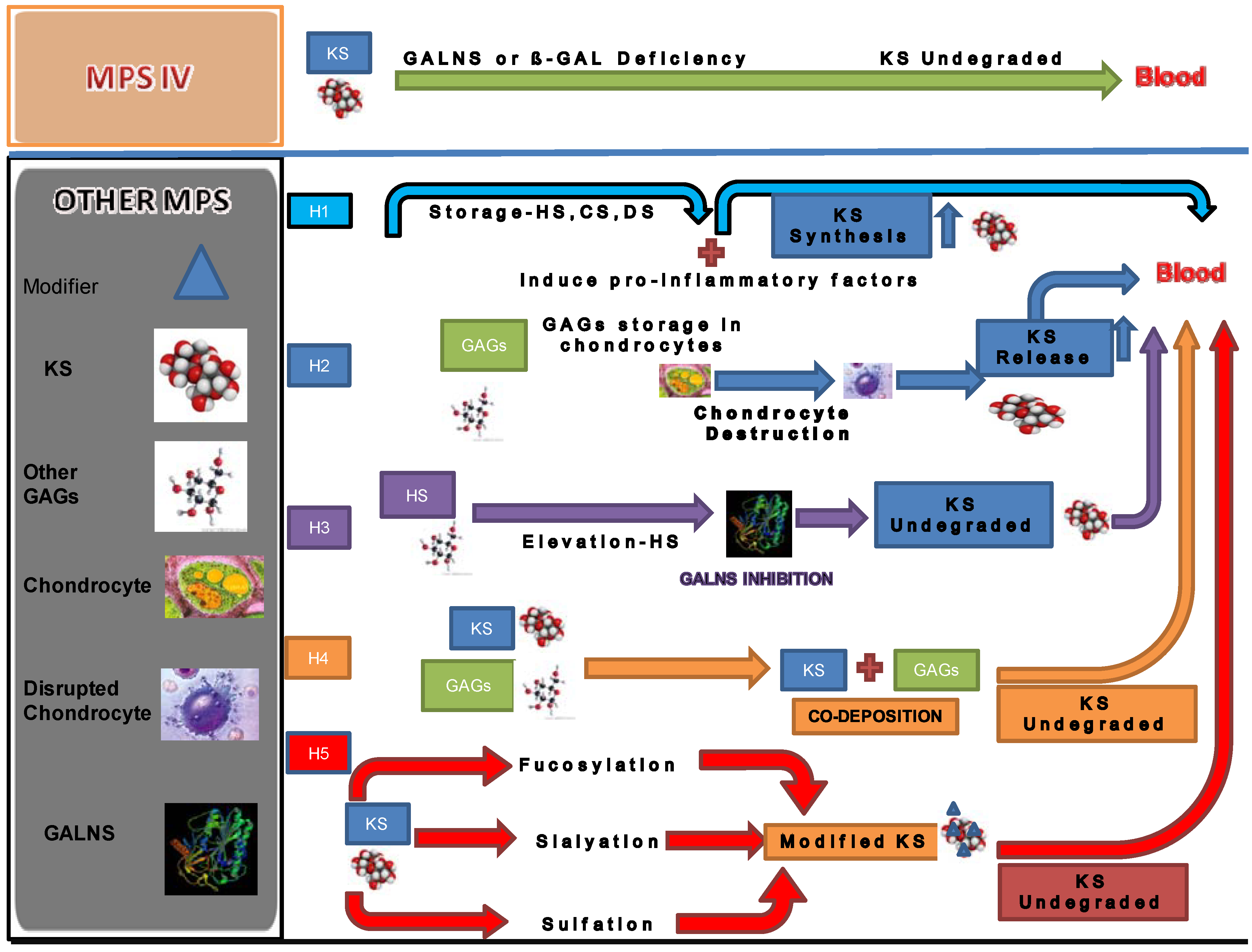
5. Secondary Elevation of KS
- (1)
- The synthesis of KS is stimulated by storage of other GAGs. Accumulation of GAGs can induce pro-inflammatory factors such as IL-1β, 6, and 10 and TNF-α [14]. Accumulated GAGs and/or pro-inflammatory factors promote the synthesis of KS secondarily.
- (2)
- The elevation of KS is a secondary consequence caused by skeletal dysplasia. Accumulation of other GAGs could cause inflammation and thereby damage cartilage and its ECM leading to increased secretion of KS into the circulation. Degradation of PGs occurs early in joint damage. The fragments of PGs are released into the synovial fluid and subsequently the blood [72,73]. This hypothesis is supported by the fact that KS levels in patients with MPS I, II, and VI are more elevated, compared with that in MPS III that results in less marked skeletal dysplasia. KS elevation is more prominent in patients with a severe form of MPS II than in an attenuated phenotype [28,31]. An MPS VII mouse model has a severe skeletal abnormality and blood KS level is elevated more than other mouse models of MPS that have less marked skeletal abnormalities [74]. Elevated levels of KS were also seen in an MPS I mouse model but was limited in MPS IIIA and MPS IVA mice. These findings suggest that blood KS elevation is caused by release of KS from chondrocytes damaged due accumulation of other GAGs and subsequent inflammation. Paradoxically, in the MPS IVA mouse model a severe bone dysplasia is not seen and KS elevation in blood is limited despite the absence of functional GALNS enzyme. Thus, although KS would be expected to accumulate in cartilage of MPS IV mice, it is primarily the severity of bone dysplasia that correlates with levels of KS measured in blood.
- (3)
- GALNS activity is inhibited by HS that accumulates in patients with MPS. HS is known to directly inhibit GALNS enzyme activity in vitro. MPS IIIA, I and VII mice have increasing levels of HS and also increasing levels of serum KS, indicating a correlation between HS inhibition of GALNS and increased KS [74]. Patients with a severe form of MPS II have a high level of HS and a more prominent increase of KS at a young age. These findings support the hypothesis that inhibition of the GALNS enzyme by elevated HS causes a secondary elevation of KS levels in MPS I, II, III, and VII patients.
- (4)
- Polymer KS interacts and co-deposits with other accumulated GAGs. Co-deposition with other GAGs hinders the interaction between KS and enzymes that catabolize KS. Elevation of KS was less striking in urine than in blood for MPS other than MPS IV. The lower elevation of urine KS in patients with other forms of MPS could be explained by aggregation of KS with other GAGs or unknown factors in the bloodstream. The aggregates may be too large to be cleared into the urine. In this scenario, undegraded KS not filtered out by the kidney remain retained in the blood.
- (5)
- Changes in the pattern of fucosylation, sialylation, and sulfation on KS secondary to other GAG accumulation makes KS resistant to degradation. KS derived from articular cartilage contains sialic acid and fucose. This hypothesis is supported by the fact that sialic acid residues, present as chain caps, and fucose residues inhibit degradation of KS molecules [75].

6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wraith, J.E. The mucopolysaccharidoses: A clinical review and guide to management. Arch. Dis. Child. 1995, 72, 263–267. [Google Scholar] [CrossRef]
- Neufeld, E.; Muenzer, J. The Mucopolysaccharidoses. In The Metabolic and Molecular Bases of Inherited Disease, 8th ed.; Scriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D., Eds.; McGraw-Hill: New York, 2001; pp. 3421–3452. [Google Scholar]
- Muenzer, J. Early initiation of enzyme replacement therapy for the mucopolysaccharidoses. Mol. Genet. Metab. 2014, 111, 63–72. [Google Scholar] [CrossRef]
- Tomatsu, S.; Montano, A.M.; Oikawa, H.; Dung, V.C.; Hashimoto, A.; Oguma, T.; Gutiérrez, M.L.; Takahashi, T.; Shimada, T.; Orii, T.; et al. Enzyme replacement therapy in newborn mucopolysaccharidosis IVA mice: Early treatment rescues bone lesions? Mol. Genet. MeTab. 2014. [Google Scholar] [CrossRef]
- Hendriksz, C.J.; Burton, B.; Fleming, T.R.; Harmatz, P.; Hughes, D.; Jones, S.A.; Lin, S.P.; Mengel, E.; Scarpa, M.; Valayannopoulos, V.; et al. Efficacy and safety of enzyme replacement therapy with BMN 110 (elosulfase alfa) for Morquio A syndrome (mucopolysaccharidosis IVA): A phase 3 randomized placebo-controlled study. J. Inherit. Metab. Dis. 2014. [Google Scholar] [CrossRef]
- Prasad, V.K.; Kurtzberg, J. Transplant outcomes in mucopolysaccharidoses. Semin. Hematol. 2010, 47, 59–69. [Google Scholar] [CrossRef]
- Patel, P.; Suzuki, Y.; Tanaka, A.; Yabe, H.; Kato, S.; Shimada, T.; Mason, R.W.; Orii, K.E.; Fukao, T.; Orii, T.; et al. Impact of enzyme replacement therapy and hematopoietic stem cell therapy on growth in patients with Hunter syndrome. Mol. Genet. Metab. Rep. 2014, 1, 184–196. [Google Scholar]
- Tanaka, A.; Okuyama, T.; Suzuki, Y.; Sakai, N.; Takakura, H.; Sawada, T.; Tanaka, T.; Otomo, T.; Ohashi, T.; Ishige, M.W; et al. Long-term efficacy of hematopoietic stem cell transplantation on brain involvement in patients with mucopolysaccharidosis type II: A nationwide survey in Japan. Mol. Genet. Metab. 2012, 107, 513–520. [Google Scholar] [CrossRef]
- Chinen, Y.; Higa, T.; Tomatsu, S.; Suzuki, Y.; Orii, T.; Hyakuna, N. Long-term therapeutic efficacy of allogenic bone marrow transplantation in a patient with mucopolysaccharidosis IVA. Mol. Genet. Metab. Rep. 2014, 1, 31–41. [Google Scholar]
- Jakóbkiewicz, J.B.; Piotrowska, E.; Gabig, M.C.; Borysiewicz, E.; Słomińska, M.W.; Narajczyk, M.; Węgrzyn, A.; Węgrzyn, G. Substrate reduction therapies for mucopolysaccharidoses. Curr. Pharm. Biotechnol. 2011, 12, 1860–1865. [Google Scholar] [CrossRef]
- Banecka, Z.M.; Jakóbkiewicz, J.B.; Gabig, M.C.; Borysiewicz, E.A.; Węgrzyn, G. Putative biological mechanisms of efficiency of substrate reduction therapies for mucopolysaccharidoses. Arch. Immunol. Ther. Exp. 2012, 60, 461–468. [Google Scholar] [CrossRef]
- Baldo, G.; Giugliani, R.; Matte, U. Gene delivery strategies for the treatment of mucopolysaccharidoses. Expert. Opin. Drug Deliv. 2014, 11, 449–459. [Google Scholar] [CrossRef]
- Alméciga-Díaz, C.J.; Montaño, A.M.; Tomatsu, S.; Barrera, L.A. Adeno-associated virus gene transfer in Morquio A disease-effect of promoters and sulfatase-modifying factor. FEBS J. 2010, 277, 3608–3619. [Google Scholar] [CrossRef]
- Simonaro, C.M.; Ge, Y.; Eliyahu, E.; He, X.; Jepsen, K.J.; Schuchman, E.H. Involvement of the Toll-like receptor 4 pathway and use of TNF-alpha antagonists for treatment of the mucopolysaccharidoses. Proc. Natl. Acad. Sci. USA 2010, 107, 222–227. [Google Scholar]
- Schuchman, E.H.; Ge, Y.; Lai, A.; Borisov, Y.; Faillace, M.; Eliyahu, E.; He, X.; Iatridis, J.; Vlassara, H.; Striker, G.; et al. Pentosan polysulfate: A novel therapy for the mucopolysaccharidoses. PLoS One 2013. [Google Scholar] [CrossRef]
- Iozzo, R.V. Matrix proteoglycans: from molecular design to cellular function. Annu. Rev. Biochem. 1998, 67, 609–652. [Google Scholar] [CrossRef]
- De Jong, J.G.; Wevers, R.A.; Laarakkers, C.; Poorthuis, B.J. Dimethylmethylene blue based spectrophotometry of glycosaminoglycans in untreated urine: A rapid screening procedure for mucopolysaccharidoses. Clin. Chem. 1989, 35, 1472–1477. [Google Scholar]
- Whitley, C.B.; Ridnour, M.D.; Draper, K.A.; Dutton, C.M.; Neglia, J.P. Diagnostic test for mucopolysaccharidosis. I. Direct method for quantifying excessive urinary glycosaminoglycan excretion. Clin. Chem. 1989, 35, 374–379. [Google Scholar]
- Whitley, C.B.; Draper, K.A.; Dutton, C.M.; Brown, P.A.; Severson, S.L.; France, L.A. Diagnostic test for mucopolysaccharidosis. II. Rapid quantification of glycosaminoglycan in urine samples collected on a paper matrix. Clin. Chem. 1989, 35, 2074–2081. [Google Scholar]
- De Jong, J.G.; Hasselman, J.J.; van Landeghem, A.A.; Vader, H.L.; Wevers, R.A. The spot test is not a reliable screening procedure for mucopolysaccharidoses. Clin. Chem. 1991, 37, 572–575. [Google Scholar]
- Iwata, S.; Sukegawa, K.; Kokuryu, M.; Tomatsu, S.; Kondo, N.; Iwasa, S.; Orii, T. Glycosaminoglycans in neonatal urine. Arch. Dis. Child. Fetal. Ed. Neonatal. 2000. [Google Scholar] [CrossRef]
- Iwata, S.; Sukegawa, K.; Sasaki, T.; Kokuryu, M.; Yamasita, S.; Noma, A.; Iwasa, S.; Kondo, N.; Orii, T. Mass screening test for mucopolysaccharidoses using the 1,9-dimethylmethylene blue method: positive interference from paper diapers. Clin. Chim. Acta. 1997, 264, 245–250. [Google Scholar] [CrossRef]
- Karlsson, M.; Edfors-Lilja, I.; Bjornsson, S. Binding and detection of glycosaminoglycans immobilized on membranes treated with cationic detergents. Anal. Biochem. 2000, 286, 51–58. [Google Scholar] [CrossRef]
- Yoshida, K.; Miyauchi, S.; Kikuchi, H.; Tawada, A.; Tokuyasu, K. Analysis of unsaturated disaccharides from glycosaminoglycuronan by high-performance liquid chromatography. Anal. Biochem. 1989, 177, 327–332. [Google Scholar] [CrossRef]
- Kinoshita, A.; Sugahara, K. Microanalysis of glycosaminoglycan-derived oligosaccharides labeled with a fluorophore 2-aminobenzamide by high-performance liquid chromatography: Application to disaccharide composition analysis and exosequencing of oligosaccharides. Anal. Biochem. 1999, 269, 367–378. [Google Scholar] [CrossRef]
- Yamada, H.; Miyauchi, S.; Morita, M.; Yoshida, Y.; Yoshihara, Y.; Kikuchi, T.; Washimi, O.; Washimi, Y.; Terada, N.; Seki, T.; et al. Content and sulfation pattern of keratan sulfate in hip osteoarthritis using high performance liquid chromatography. J. Rheumatol. 2000, 27, 1721–1724. [Google Scholar]
- Tomatsu, S.; Okamura, K.; Taketani, T.; Orii, K.O.; Nishioka, T.; Gutierrez, M.A.; Velez-Castrillon, S.; Fachel, A.A.; Grubb, J.H.; Cooper, A; et al. Development and testing of new screening method for keratan sulfate in mucopolysaccharidosis IVA. Pediatr. Res. 2004, 55, 592–597. [Google Scholar] [CrossRef]
- Tomatsu, S.; Okamura, K.; Maeda, H.; Taketani, T.; Castrillon, S.V.; Gutierrez, M.A.; Nishioka, T.; Fachel, A.A.; Orii, K.O.; Grubb, J.H.; et al. Keratan sulphate levels in mucopolysaccharidoses and mucolipidoses. J. Inherit. Metab. Dis. 2005, 28, 187–202. [Google Scholar] [CrossRef]
- Tomatsu, S.; Gutierrez, M.A.; Ishimaru, T.; Peña, O.M.; Montaño, A.M.; Maeda, H.; Velez-Castrillon, S.; Nishioka, T.; Fachel, A.A.; Cooper, A.; et al. Heparan sulfate levels in mucopolysaccharidoses and mucolipidoses. J. Inherit. Metab. Dis. 2005, 28, 743–757. [Google Scholar] [CrossRef]
- Tomatsu, S.; Shimada, T.; Mason, R.W.; Kelly, J.; LaMarr, W.A.; Yasuda, E.; Shibata, Y.; Futatsumori, H.; Montaño, A.M.; Yamaguchi, S.; et al. Assay for Glycosaminoglycans by tandem mass spectrometry and its applications. Available online: http://dx.doi.org/10.4172/2155-9872.S2-006 (accessed on 18 June 2014).
- Tomatsu, S.; Montaño, AM.; Oguma, T.; Dung, V.C.; Oikawa, H.; Carvalho, T.G.; Gutiérrez, M.G.; Yamaguchi, S.; Suzuki, Y.; Fukushi, M.; et al. Validation of keratan sulfate level in Mucopolysaccharidosis IVA by liquid tandem mass spectrometry method. J. Inherit. Metab. Dis. 2010, 33, 35–42. [Google Scholar] [CrossRef]
- Shimada, T.; Tomatsu, S.; Mason, R.W.; Yasuda, Y.; Mackenzie, W.G.; Hossain, J.; Shibata, Y.; Montaño, A.M.; Kubaski, F.; Giugliani, R.; et al. Di-sulfated keratan sulfate as a novel biomarker for mucopolysaccharidosis IVA. J. Inherit. Metab. Dis. Rep. 2014, in press. [Google Scholar]
- Rattenbury, J.M.; Worthy, E.; Allen, J.C. Screening tests for glycosaminoglycans in urine: Experience from regional interlaboratory surveys. J. Clin. Pathol. 1988, 41, 936–939. [Google Scholar] [CrossRef]
- De Ruijter, J.; Valstar, M.J.; Narajczyk, M.; Wegrzyn, G.; Kulik, W.; Ijlst, L.; Wagemans, T.; van der Wal, W.M.; Wijburg, F.A. Genistein in Sanfilippo disease: A randomized controlled crossover trial. Ann. Neurol. 2012, 71, 110–120. [Google Scholar] [CrossRef]
- Bjornsson, S. Quantitation of proteoglycans as glycosaminoglycans in biological fluids using an alcian blue dot blot analysis. Anal. Biochem. 1998, 256, 229–237. [Google Scholar] [CrossRef]
- Thonar, E.J.; Lenz, M.E.; Klintworth, G.K.; Caterson, B.; Pachman, L.M.; Glickman, P.; Katz, R.; Huff, J.; Kuettner, K.E. Quantification of keratan sulfate in blood as a marker of cartilage catabolism. Arthritis. Rheum. 1985, 28, 1367–1376. [Google Scholar] [CrossRef]
- Tomatsu, S.; Montaño, A.M.; Oguma, T.; Dung, V.C.; Oikawa, H.; de Carvalho, T.G.; Gutiérrez, M.L.; Yamaguchi, S.; Suzuki, Y.; Fukushi, M.; et al. Dermatan sulfate and heparan sulfate as a biomarker for mucopolysaccharidosis I. J. Inherit. Metab. Dis. 2010, 33, 141–150. [Google Scholar] [CrossRef]
- Chai, W.; Luo, J.; Lim, C.K.; Lawson, A.M. Characterization of heparin oligosaccharide mixtures as ammonium salts using electrospray mass spectrometry. Anal. Chem. 1998, 70, 2060–2066. [Google Scholar] [CrossRef]
- Oguma, T.; Toyoda, H.; Toida, T.; Imanari, T. Analytical method of chondroitin/dermatan sulfates using high performance liquid chromatography/turbo ionspray ionization mass spectrometry: Application to analyses of the tumor tissue sections on glass slides. Biomed. Chromatogr. 2001, 5, 356–362. [Google Scholar]
- Oguma, T.; Toyoda, H.; Toida, T.; Imanari, T. Analytical method of heparan sulfates using high-performance liquid chromatography turbo-ionspray ionization tandem mass spectrometry. J. Chromatogr. B. Biomed. Sci. Appl. 2001, 754, 153–159. [Google Scholar] [CrossRef]
- Oguma, T.; Toyoda, H.; Toida, T.; Imanari, T. Analytical method for keratan sulfates by high-performance liquid chromatography/turbo-ionspray tandem mass spectrometry. Anal. Biochem. 2001, 290, 68–73. [Google Scholar] [CrossRef]
- Ramsay, S.L.; Meikle, P.J.; Hopwood, J.J. Determination of monosaccharides and disaccharides in mucopolysaccharidoses patients by electrospray ionisation mass spectrometry. Mol. Genet. Metab. 2003, 78, 193–204. [Google Scholar] [CrossRef]
- Fuller, M.; Rozaklis, T.; Ramsay, S.L.; Hopwood, J.J.; Meikle, P.J. Disease-specific markers for the mucopolysaccharidoses. Pediatr. Res. 2004, 56, 733–738. [Google Scholar] [CrossRef]
- Oguma, T.; Tomatsu, S.; Montano, A.M.; Okazaki, O. Analytical method for the determination of disaccharides derived from keratan, heparan, and dermatan sulfates in human serum and plasma by high-performance liquid chromatography/turbo ionspray ionization tandem mass spectrometry. Anal. Biochem. 2007, 368, 79–86. [Google Scholar] [CrossRef]
- Oguma, T.; Tomatsu, S.; Okazaki, O. Analytical method for determination of disaccharides derived from keratan sulfates in human serum and plasma by high-performance liquid chromatography/turbo-ionspray ionization tandem mass spectrometry. Biomed. Chromatogr. 2007, 21, 356–362. [Google Scholar] [CrossRef]
- Tomatsu, S.; Montano, A.M.; Oguma, T.; Dung, V.C.; Oikawa, H.; Gutierrez, M.L.; Yamaguchi, S.; Suzuki, Y.; Fukushi, M.; Barrera, L.A.; et al. Validation of disaccharide compositions derived from dermatan sulfate and heparan sulfate in mucopolysaccharidoses and mucolipidoses II and III by tandem mass spectrometry. Mol. Genet. Metab. 2010, 99, 124–131. [Google Scholar] [CrossRef]
- Martell, L.A.; Cunico, R.L.; Ohh, J.; Fulkerson, W.; Furneaux, R.; Foehr, E.D. Validation of an LC-MS/MS assay for detecting relevant disaccharides from keratan sulfate as a biomarker for Morquio A syndrome. Bioanalysis 2011, 3, 1855–1866. [Google Scholar] [CrossRef]
- De Ruijter, J.; de Ru, M.H.; Wagemans, T.; Ijlst, L.; Lund, A.M.; Orchard, P.J.; Schaefer, G.B.; Wijburg, F.A.; van Vlies, N. Heparan sulfate and dermatan sulfate derived disaccharides are sensitive markers for newborn screening for mucopolysaccharidoses types I, II and III. Mol. Genet. Metab. 2012, 107, 705–710. [Google Scholar] [CrossRef]
- Auray-Blais, C.; Lavoie, P.; Zhang, H.; Gagnon, R.; Clarke, J.T.; Maranda, B.; Young, S.P.; An, Y.; Millington, D.S. An improved method for glycosaminoglycan analysis by LC-MS/MS of urine samples collected on filter paper. Clin. Chim. Acta. 2012, 413, 771–778. [Google Scholar] [CrossRef]
- Auray-Blais, C.; Bhérer, P.; Gagnon, R.; Young, S.P.; Zhang, H.H.; An, Y.; Clarke, J.T.; Millington, D.S. Efficient analysis of urinary glycosaminoglycans by LC–MS/MS in mucopolysaccharidoses type I, II and VI. Mol. Genet. Metab. 2011, 102, 49–56. [Google Scholar] [CrossRef]
- Lawrence, R.; Brown, J.R.; Al-Mafraji, K.; Lamanna, W.C.; Beitel, J.R.; Boons, G.J.; Esko, J.D.; Crawford, B.E. Disease-specific non-reducing end carbohydrate biomarkers for mucopolysaccharidoses. Nat. Chem. Biol. 2012, 8, 197–204. [Google Scholar]
- Tomatsu, S.; Montano, A.M.; Ohashi, A.; Gutierrez, M.A.; Oikawa, H.; Oguma, T.; Dung, V.C.; Nishioka, T.; Orii, T.; Sly, W.S. Enzyme replacement therapy in a murine model of Morquio A syndrome. Hum. Mol. Genet. 2008, 17, 815–824. [Google Scholar]
- Montaño, A.M.; Oikawa, H.; Tomatsu, S.; Nishioka, T.; Vogler, C.; Gutierrez, M.A.; Oguma, T.; Tan, Y.; Grubb, J.H.; Dung, V.C.; et al. Acidic amino acid tag enhances response to enzyme replacement in mucopolysaccharidosis type VII mice. Mol. Genet. Metab. 2008, 94, 178–189. [Google Scholar] [CrossRef]
- Hintze, J.P.; Tomatsu, S.; Fujii, T.; Montano, A.M.; Yamaguchi, S.; Suzuki, Y.; Fukushi, M.; Ishimaru, T.; Orii, T. Comparison of liquid chromatography-tandem mass spectrometry and sandwich ELISA for determination of keratan sulfate in plasma and urine. Biomark Insights 2011, 6, 69–78. [Google Scholar]
- De Ruijter, J.; Ijlst, L.; Kulik, W.; van Lenthe, H.; Wagemans, T.; van Vlies, N.; Wijburg, F.A. Heparan sulfate derived disaccharides in plasma and total urinary excretion of glycosaminoglycans correlate with disease severity in Sanfilippo disease. J. Inherit. Metab. Dis. 2013, 36, 271–279. [Google Scholar] [CrossRef]
- Tomatsu, S.; Montaño, A.M.; Dung, V.C.; Ohashi, A.; Oikawa, H.; Oguma, T.; Orii, T.; Barrera, L.; Sly, W.S. Enhancement of drug delivery: enzyme replacement therapy for murine Morquio A syndrome. Mol. Ther. 2010, 18, 1094–1102. [Google Scholar] [CrossRef]
- De Ru, M.H.; van der Tol, L.; van Vlies, N.; Bigger, B.W.; Hollak, C.E.; Ijlst, L.; Kulik, W.; van Lenthe, H.; Saif, M.A.; Wagemans, T.; et al. Plasma and urinary levels of dermatan sulfate and heparan sulfate derived disaccharides after long-term enzyme replacement therapy (ERT) in MPS I: correlation with the timing of ERT and with total urinary excretion of glycosaminoglycans. J. Inherit. Metab. Dis. 2013, 36, 247–255. [Google Scholar] [CrossRef]
- Quercia, A.K.; LaMarr, W.A.; Myung, J.; Ozbal, C.C.; Landro, J.A.; Lumb, K.J. Highthroughput screening by mass spectrometry: Comparison with the scintillation proximity assay with a focused-file screen of AKT1/PKB alpha. J. Biomol. Screen. 2007, 12, 473–480. [Google Scholar] [CrossRef]
- Leveridge, M.V.; Bardera, A.I.; LaMarr, W.; Billinton, A.; Bellenie, B.; Edge, C.; Francis, P.; Christodoulou, E.; Shillings, A.; Hibbs, M.; et al. Lead discovery for microsomal prostaglandin E synthase using a combination of high-throughput fluorescent-based assays and RapidFire mass spectrometry. J. Biomol. Screen. 2012, 17, 641–650. [Google Scholar] [CrossRef]
- Hutchinson, S.E.; Leveridge, M.V.; Heathcote, M.L.; Francis, P.; Williams, L; Gee, M.; Munoz-Muriedas, J.; Leavens, B.; Shillings, A.; Jones, E.; et al. Enabling lead discovery for histone lysine demethylases by highthroughput RapidFire mass spectrometry. J. Biomol. Screen. 2012, 17, 39–48. [Google Scholar] [CrossRef]
- Plant, M.; Dineen, T.; Cheng, A.; Long, A.M.; Chen, H.; Morgensterna, K.A. Screening for lysine-specific demethylase-1 inhibitors using a label-free high-throughput mass spectrometry assay. Anal. Biochem. 2011, 419, 217–227. [Google Scholar] [CrossRef]
- Atkinson, K.A.; Beretta, E.E.; Brown, J.A.; Castrodad, M.; Chen, Y.; Cosqrove, J.M.; Du, P.; Lithfield, J.; Makowshi, M.; Martin, k.; et al. N-benzylimidazole carboxamides as potent, orally active stearoylCoA desaturase-1 inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 1621–1625. [Google Scholar] [CrossRef]
- Highkin, M.K.; Yates, M.P.; Nemirovskiy, O.V.; Lamarr, W.A.; Munie, G.E. High-throughput screening assay for sphingosine kinase inhibitors in whole blood using RapidFire® mass spectrometry. J. Biomol. Screen. 2011, 16, 272–277. [Google Scholar] [CrossRef]
- Langsdorf, E.F.; Malikzay, A.; Lamarr, W.A.; Daubaras, D.; Kravec, C.; Zhang, R.; Hart, R.; Monsma, F.; Black, T; Ozbal, C.C.; et al. Screening for antibacterial inhibitors of the UDP-3-O-(R-3-hydroxymyristoyl)-N-acetylglucosaminedeacetylase (LpxC) using a high-throughput mass spectrometry assay. J. Biomol. Screen. 2010, 15, 52–61. [Google Scholar] [CrossRef]
- Holt, T.G.; Choi, B.K.; Geoghagen, N.S.; Jensen, K.K.; Luo, Q.; LaMarr, W.A.; Makara, G.M.; Malwowitz, L.; Ozbal, C.C.; Xiong, Y.; et al. Labelfree high-throughput screening via mass spectrometry: a single cystathionine quantitative method for multiple applications. Assay Drug. Dev. Technol. 2009, 7, 495–506. [Google Scholar] [CrossRef]
- Wu, X.; Wang, J.; Tan, L.; Bui, J.; Gjerstad, E.; McMillan, K.; Zhang, W. In vitro ADME profiling using high-throughput rapidfire mass spectrometry: Cytochrome p450 inhibition and metabolic stability assays. J. Biomol. Screen. 2012, 17, 761–772. [Google Scholar] [CrossRef]
- Tomatsu, S.; Fujii, T.; Fukushi, M.; Oguma, T.; Shimada, T.; Maeda, M.; Kida, K.; Shibata, Y.; Futatsumori, H.; Montaño, A.M.; et al. Newborn screening and diagnosis of mucopolysaccharidoses. Minireview. Mol. Genet. Metab. 2013, 110, 42–53. [Google Scholar] [CrossRef]
- Shimada, T.; Tomatsu, S.; Yasuda, E.; Mason, R.W.; Mackenzie, W.G.; Shibata, Y.; Kubaski, F.; Giugliani, R.; Yamaguchi, S.; Suzuki, Y.; et al. Chondroitin 6-sulfate as a novel biomarker for mucopolysaccharidosis IVA and VII. J. Inher. Metab. Dis. Rep. 2014, in press. [Google Scholar]
- Caterson, B.; Christner, J.E.; Baker, J.R. Identification of a monoclonal antibody that specifically recognizes corneal and skeletal keratan sulfate. Monoclonal antibodies to cartilage proteoglycan. J. Biol. Chem. 1983, 258, 8848–8854. [Google Scholar]
- Mehmet, H.; Scudder, P.; Tang, P.W.; Hounsell, E.F.; Caterson, B.; Feizi, T. The antigenic determinants recognized by three monoclonal antibodies to keratan sulphate involve sulphated hepta- or larger oligosaccharides of the poly (N-acetyllactosamine) series. Eur. J. Biochem. 1986, 157, 385–391. [Google Scholar] [CrossRef]
- Shimada, T.; Kelly, J.; LaMarr, W.A.; van Vlies, N.; Yasuda, E.; Mason, R.W.; Mackenzie, W.; Kubaski, F.; Giugliani, R.; Chinen, T.; et al. Novel heparan sulfate assay by using automated high-throughput mass spectrometry: Application to monitoring and screening for mucopolysaccharidoses. Mol. Genet. Metab. 2014, in press. [Google Scholar]
- Dingle, J.T.; Horsfield, P.; Fell, H.B.; Barratt, M.E. Breakdown of proteoglycan and collagen induced in pig articular cartilage in organ culture. Ann. Rheum. Dis. 1975, 34, 303–311. [Google Scholar] [CrossRef]
- Ratcliffe, A.; Doherty, M.; Maini, R.N.; Hardingham, T.E. Increased concentrations of proteoglycan components in the synovial fluids of patients with acute but not chronic joint disease. Ann. Rheum. Dis. 1988, 47, 826–832. [Google Scholar] [CrossRef]
- Rowan, D.J.; Tomatsu, S.; Grubb, J.H.; Montaño, A.M.; Sly, W.S. Assessment of bone dysplasia by micro-CT and glycosaminoglycan levels in mouse models for mucopolysaccharidosis type I, IIIA, IVA, and VII. J. Inherit. Metab. Dis. 2013, 36, 235–246. [Google Scholar] [CrossRef]
- Tai, G.H.; Huckerby, T.N.; Nieduszynski, I.A. 600 MHz 1H NMR study of a fucose-containing heptasaccharide derived from a keratanase digestion of bovine articular cartilage keratan sulphate. Carbohydr. Res. 1994, 255, 303–309. [Google Scholar] [CrossRef]
- Crow, J.; Gibbs, D.A.; Cozens, W.; Spellacy, E.; Watts, R.W. Biochemical and histopathological studies on patients with mucopolysaccharidoses, two of whom had been treated by fibroblast transplantation. J. Clin. Pathol. 1983, 36, 415–430. [Google Scholar] [CrossRef]
- Wiesmann, U.N.; Spycher, M.A.; Meier, C.; Liebaers, I.; Herschkowitz, N. Prenatal mucopolysaccharidosis II (Hunter): A pathogenetic study. Pediatr. Res. 1980, 14, 749–756. [Google Scholar] [CrossRef]
- Beck, M.; Braun, S.; Coerdt, W.; Coerdt, W.; Merz, E.; Young, E.; Sewell, A. Fetal presentation of Morquio disease type A. Prenat. Diagn. 1992, 12, 1019–1029. [Google Scholar] [CrossRef]
- Rowan, D.J.; Tomatsu, S.; Grubb, J.H.; Haupt, B.; Montaño, A.M.; Oikawa, H.; Sosa, A.C.; Chen, A.; Sly, W.S. Long circulating enzyme replacement therapy rescues bone pathology in mucopolysaccharidosis VII murine model. Mol. Genet. Metab. 2012, 107, 161–172. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Tomatsu, S.; Shimada, T.; Mason, R.W.; Montaño, A.M.; Kelly, J.; LaMarr, W.A.; Kubaski, F.; Giugliani, R.; Guha, A.; Yasuda, E.; et al. Establishment of Glycosaminoglycan Assays for Mucopolysaccharidoses. Metabolites 2014, 4, 655-679. https://doi.org/10.3390/metabo4030655
Tomatsu S, Shimada T, Mason RW, Montaño AM, Kelly J, LaMarr WA, Kubaski F, Giugliani R, Guha A, Yasuda E, et al. Establishment of Glycosaminoglycan Assays for Mucopolysaccharidoses. Metabolites. 2014; 4(3):655-679. https://doi.org/10.3390/metabo4030655
Chicago/Turabian StyleTomatsu, Shunji, Tsutomu Shimada, Robert W. Mason, Adriana M. Montaño, Joan Kelly, William A. LaMarr, Francyne Kubaski, Roberto Giugliani, Aratrik Guha, Eriko Yasuda, and et al. 2014. "Establishment of Glycosaminoglycan Assays for Mucopolysaccharidoses" Metabolites 4, no. 3: 655-679. https://doi.org/10.3390/metabo4030655