
Plasma Prostaglandin E2 Metabolite Levels Predict Type 2 Diabetes Status and One-Year Therapeutic Response Independent of Clinical Markers of Inflammation
 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design, Intake Appointment, and Plasma Sample Collection
2.2. Prostaglandin E Metabolite (PGEM) Assay
2.3. Statistical Analysis
3. Results
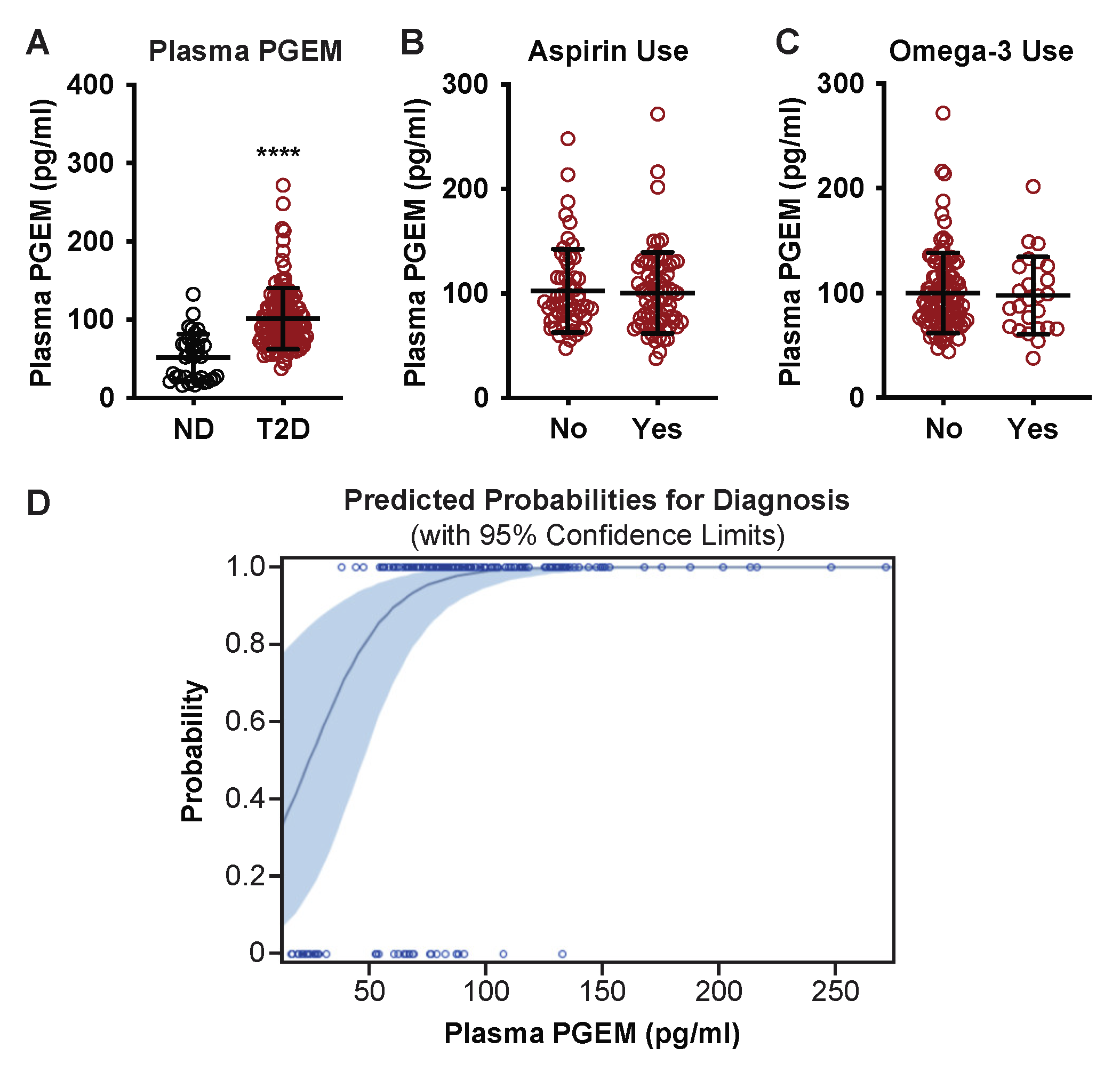
3.1. Plasma PGEM Is Increased Specifically in Subjects with T2D
3.2. In T2D Subjects, Plasma PGEM Is Only Weakly Correlated with Systemic Inflammation
3.3. Plasma PGEM Is a Strong Predictor of T2D Disease Status
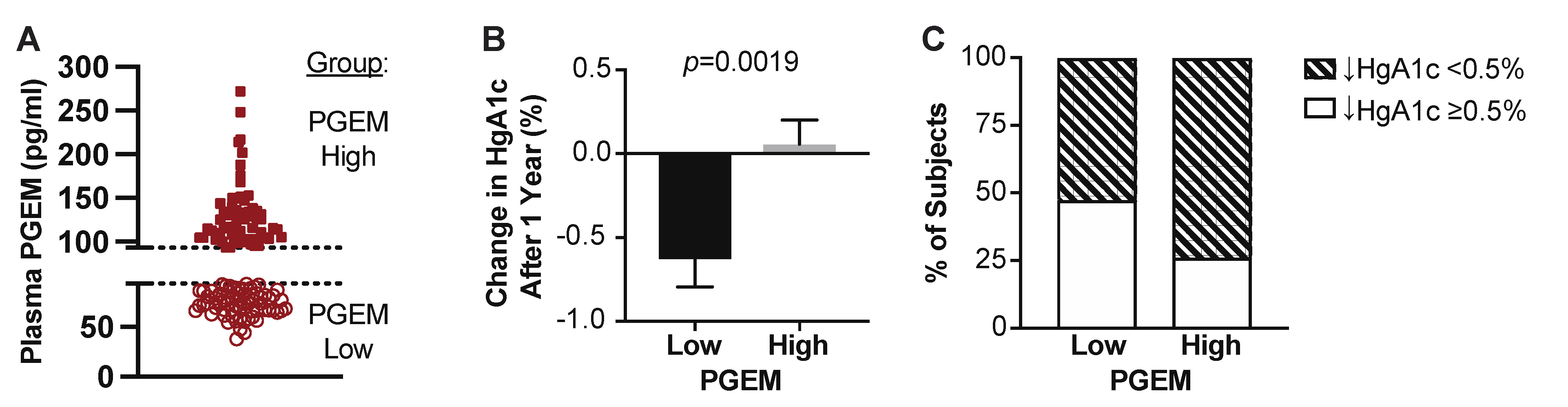
3.4. T2D Patients with High Plasma PGEM Levels Have Significantly Worse Blood Glucose Control One-Year Post-Enrollment
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Center for Diseease Control and Prevention, National Diabetes Statistics Report Website. Available online: https://www.cdc.gov/diabetes/data/statistics-report/index.html (accessed on 1 October 2022).
- Juarez, D.T.; Ma, C.; Kumasaka, A.; Shimada, R.; Davis, J. Failure to reach target glycated a1c levels among patients with diabetes who are adherent to their antidiabetic medication. Popul. Health Manag. 2014, 17, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Bacos, K.; Gillberg, L.; Volkov, P.; Olsson, A.H.; Hansen, T.; Pedersen, O.; Gjesing, A.P.; Eiberg, H.; Tuomi, T.; Almgren, P.; et al. Blood-based biomarkers of age-associated epigenetic changes in human islets associate with insulin secretion and diabetes. Nat. Commun. 2016, 7, 11089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirmira, R.G.; Sims, E.K.; Syed, F.; Evans-Molina, C. Biomarkers of beta-Cell Stress and Death in Type 1 Diabetes. Curr. Diabetes Rep. 2016, 16, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheen, A.J. Precision medicine: The future in diabetes care? Diabetes Res. Clin. Pract. 2016, 117, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Sims, E.K.; Evans-Molina, C.; Tersey, S.A.; Eizirik, D.L.; Mirmira, R.G. Biomarkers of islet beta cell stress and death in type 1 diabetes. Diabetologia 2018, 61, 2259–2265. [Google Scholar] [CrossRef] [Green Version]
- Thorens, B.; Rodriguez, A.; Cruciani-Guglielmacci, C.; Wigger, L.; Ibberson, M.; Magnan, C. Use of preclinical models to identify markers of type 2 diabetes susceptibility and novel regulators of insulin secretion—A step towards precision medicine. Mol. Metab. 2019, 27, S147–S154. [Google Scholar] [CrossRef]
- Wang, C.; Pan, Y.J.; Song, J.; Sun, Y.; Li, H.K.; Chen, L.; Hou, X.G. Serum Metrnl Level is Correlated with Insulin Resistance, But Not with beta-Cell Function in Type 2 Diabetics. Med. Sci. Monit. 2019, 25, 8968–8974. [Google Scholar] [CrossRef]
- Kimple, M.E.; Keller, M.P.; Rabaglia, M.R.; Pasker, R.L.; Neuman, J.C.; Truchan, N.A.; Brar, H.K.; Attie, A.D. Prostaglandin E2 receptor, EP3, is induced in diabetic islets and negatively regulates glucose- and hormone-stimulated insulin secretion. Diabetes 2013, 62, 1904–1912. [Google Scholar] [CrossRef] [Green Version]
- Neuman, J.C.; Schaid, M.D.; Brill, A.L.; Fenske, R.J.; Kibbe, C.R.; Fontaine, D.A.; Sdao, S.M.; Brar, H.K.; Connors, K.M.; Wienkes, H.N.; et al. Enriching Islet Phospholipids With Eicosapentaenoic Acid Reduces Prostaglandin E2 Signaling and Enhances Diabetic beta-Cell Function. Diabetes 2017, 66, 1572–1585. [Google Scholar] [CrossRef] [Green Version]
- Parazzoli, S.; Harmon, J.S.; Vallerie, S.N.; Zhang, T.; Zhou, H.; Robertson, R.P. Cyclooxygenase-2, not microsomal prostaglandin E synthase-1, is the mechanism for interleukin-1beta-induced prostaglandin E2 production and inhibition of insulin secretion in pancreatic islets. J. Biol. Chem. 2012, 287, 32246–32253. [Google Scholar] [CrossRef]
- Robertson, R.P. Dominance of cyclooxygenase-2 in the regulation of pancreatic islet prostaglandin synthesis. Diabetes 1998, 47, 1379–1383. [Google Scholar] [CrossRef]
- Sandhu, H.K.; Neuman, J.C.; Schaid, M.D.; Davis, S.E.; Connors, K.M.; Challa, R.; Guthery, E.; Fenske, R.J.; Patibandla, C.; Breyer, R.M.; et al. Rat prostaglandin EP3 receptor is highly promiscuous and is the sole prostanoid receptor family member that regulates INS-1 (832/3) cell glucose-stimulated insulin secretion. Pharm. Res. Perspect. 2021, 9, e00736. [Google Scholar] [CrossRef]
- Schaid, M.D.; Zhu, Y.; Richardson, N.E.; Patibandla, C.; Ong, I.M.; Fenske, R.J.; Neuman, J.C.; Guthery, E.; Reuter, A.; Sandhu, H.K.; et al. Systemic Metabolic Alterations Correlate with Islet-Level Prostaglandin E2 Production and Signaling Mechanisms That Predict beta-Cell Dysfunction in a Mouse Model of Type 2 Diabetes. Metabolites 2021, 11, 58. [Google Scholar] [CrossRef]
- Sjoholm, A. Prostaglandins inhibit pancreatic beta-cell replication and long-term insulin secretion by pertussis toxin-insensitive mechanisms but do not mediate the actions of interleukin-1 beta. Biochim. Biophys. Acta 1996, 1313, 106–110. [Google Scholar] [CrossRef] [Green Version]
- Tran, P.O.; Gleason, C.E.; Poitout, V.; Robertson, R.P. Prostaglandin E(2) mediates inhibition of insulin secretion by interleukin-1beta. J. Biol. Chem. 1999, 274, 31245–31248. [Google Scholar] [CrossRef] [Green Version]
- Tran, P.O.; Gleason, C.E.; Robertson, R.P. Inhibition of interleukin-1beta-induced COX-2 and EP3 gene expression by sodium salicylate enhances pancreatic islet beta-cell function. Diabetes 2002, 51, 1772–1778. [Google Scholar] [CrossRef] [Green Version]
- Pawelzik, S.C.; Avignon, A.; Idborg, H.; Boegner, C.; Stanke-Labesque, F.; Jakobsson, P.J.; Sultan, A.; Back, M. Urinary prostaglandin D2 and E2 metabolites associate with abdominal obesity, glucose metabolism, and triglycerides in obese subjects. Prostaglandins Other Lipid Mediat. 2019, 145, 106361. [Google Scholar] [CrossRef]
- Poreba, M.; Rostoff, P.; Siniarski, A.; Mostowik, M.; Golebiowska-Wiatrak, R.; Nessler, J.; Undas, A.; Gajos, G. Relationship between polyunsaturated fatty acid composition in serum phospholipids, systemic low-grade inflammation, and glycemic control in patients with type 2 diabetes and atherosclerotic cardiovascular disease. Cardiovasc. Diabetol. 2018, 17, 29. [Google Scholar] [CrossRef] [Green Version]
- Tans, R.; Bande, R.; van Rooij, A.; Molloy, B.J.; Stienstra, R.; Tack, C.J.; Wevers, R.A.; Wessels, H.; Gloerich, J.; van Gool, A.J. Evaluation of cyclooxygenase oxylipins as potential biomarker for obesity-associated adipose tissue inflammation and type 2 diabetes using targeted multiple reaction monitoring mass spectrometry. Prostaglandins Leukot. Essent. Fat. Acids 2020, 160, 102157. [Google Scholar] [CrossRef]
- Truchan, N.A.; Fenske, R.J.; Sandhu, H.K.; Weeks, A.M.; Patibandla, C.; Wancewicz, B.; Pabich, S.; Reuter, A.; Harrington, J.M.; Brill, A.L.; et al. Human Islet Expression Levels of Prostaglandin E2 Synthetic Enzymes, But Not Prostaglandin EP3 Receptor, Are Positively Correlated with Markers of beta-Cell Function and Mass in Nondiabetic Obesity. ACS Pharmacol. Transl. Sci. 2021, 4, 1338–1348. [Google Scholar] [CrossRef]
- Xia, F.; He, C.; Ren, M.; Xu, F.G.; Wan, J.B. Quantitative profiling of eicosanoids derived from n-6 and n-3 polyunsaturated fatty acids by twin derivatization strategy combined with LC-MS/MS in patients with type 2 diabetes mellitus. Anal. Chim. Acta 2020, 1120, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Vozarova, B.; Weyer, C.; Lindsay, R.S.; Pratley, R.E.; Bogardus, C.; Tataranni, P.A. High white blood cell count is associated with a worsening of insulin sensitivity and predicts the development of type 2 diabetes. Diabetes 2002, 51, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marz, W.; Scharnagl, H.; Winkler, K.; Tiran, A.; Nauck, M.; Boehm, B.O.; Winkelmann, B.R. Low-density lipoprotein triglycerides associated with low-grade systemic inflammation, adhesion molecules, and angiographic coronary artery disease: The Ludwigshafen Risk and Cardiovascular Health study. Circulation 2004, 110, 3068–3074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, P.C.; Lee, K.F.; So, W.Y.; Ng, M.H.; Chan, W.B.; Lo, M.K.; Chan, N.N.; Chan, J.C. White blood cell count is associated with macro- and microvascular complications in chinese patients with type 2 diabetes. Diabetes Care 2004, 27, 216–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Syrenicz, A.; Garanty-Bogacka, B.; Syrenicz, M.; Gebala, A.; Walczak, M. Low-grade systemic inflammation and the risk of type 2 diabetes in obese children and adolescents. Neuro Endocrinol. Lett. 2006, 27, 453–458. [Google Scholar]
- Gkrania-Klotsas, E.; Ye, Z.; Cooper, A.J.; Sharp, S.J.; Luben, R.; Biggs, M.L.; Chen, L.K.; Gokulakrishnan, K.; Hanefeld, M.; Ingelsson, E.; et al. Differential white blood cell count and type 2 diabetes: Systematic review and meta-analysis of cross-sectional and prospective studies. PLoS ONE 2010, 5, e13405. [Google Scholar] [CrossRef] [Green Version]
- Placzkowska, S.; Pawlik-Sobecka, L.; Kokot, I.; Sowinski, D.; Wrzosek, M.; Piwowar, A. Associations between basic indicators of inflammation and metabolic disturbances. Postep. Hig. Med. Dosw. 2014, 68, 1374–1382. [Google Scholar] [CrossRef]
- Shiny, A.; Bibin, Y.S.; Shanthirani, C.S.; Regin, B.S.; Anjana, R.M.; Balasubramanyam, M.; Jebarani, S.; Mohan, V. Association of neutrophil-lymphocyte ratio with glucose intolerance: An indicator of systemic inflammation in patients with type 2 diabetes. Diabetes Technol. 2014, 16, 524–530. [Google Scholar] [CrossRef]
- Fiorentino, T.V.; Hribal, M.L.; Perticone, M.; Andreozzi, F.; Sciacqua, A.; Perticone, F.; Sesti, G. Unfavorable inflammatory profile in adults at risk of type 2 diabetes identified by hemoglobin A1c levels according to the American Diabetes Association criteria. Acta Diabetol. 2015, 52, 349–356. [Google Scholar] [CrossRef]
- Sung, K.C.; Ryu, S.; Sung, J.W.; Kim, Y.B.; Won, Y.S.; Cho, D.S.; Kim, S.H.; Liu, A. Inflammation in the Prediction of Type 2 Diabetes and Hypertension in Healthy Adults. Arch. Med. Res. 2017, 48, 535–545. [Google Scholar] [CrossRef]
- Das, A.K.; Kalra, S.; Tiwaskar, M.; Bajaj, S.; Seshadri, K.; Chowdhury, S.; Sahay, R.; Indurkar, S.; Unnikrishnan, A.G.; Phadke, U.; et al. Expert Group Consensus Opinion: Role of Anti-inflammatory Agents in the Management of Type-2 Diabetes (T2D). J. Assoc. Physicians India 2019, 67, 65–74. [Google Scholar]
- Mahdiani, A.; Kheirandish, M.; Bonakdaran, S. Correlation Between White Blood Cell Count and Insulin Resistance in Type 2 Diabetes. Curr. Diabetes Rev. 2019, 15, 62–66. [Google Scholar] [CrossRef]
- Shin, G.; Jang, K.; Kim, M.; Lee, J.H.; Yoo, H.J. Inflammatory Markers and Plasma Fatty Acids in Predicting WBC Level Alterations in Association With Glucose-Related Markers: A Cross-Sectional Study. Front. Immunol. 2020, 11, 629. [Google Scholar] [CrossRef] [Green Version]
- Wan, Z.; Song, L.; Hu, L.; Lei, X.; Huang, Y.; Lv, Y.; Yu, S. The role of systemic inflammation in the association between serum 25-hydroxyvitamin D and type 2 diabetes mellitus. Clin. Nutr. 2021, 40, 3661–3667. [Google Scholar] [CrossRef]
- Gordillo-Moscoso, A.; Ruiz, E.; Carnero, M.; Reguillo, F.; Rodriguez, E.; Tejerina, T.; Redondo, S. Relationship between serum levels of triglycerides and vascular inflammation, measured as COX-2, in arteries from diabetic patients: A translational study. Lipids Health Dis. 2013, 12, 62. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.Y.; Zhang, X.J.; Liu, Y.M.; Geng, L.Y.; Guan, L.Y.; Li, X.H. Comparison of the triglyceride glucose index and blood leukocyte indices as predictors of metabolic syndrome in healthy Chinese population. Sci. Rep. 2021, 11, 10036. [Google Scholar] [CrossRef]
- Welty, F.K. How do elevated triglycerides and low HDL-cholesterol affect inflammation and atherothrombosis? Curr. Cardiol. Rep. 2013, 15, 400. [Google Scholar] [CrossRef] [Green Version]
- Vane, J.R.; Botting, R.M. The mechanism of action of aspirin. Thromb. Res. 2003, 110, 255–258. [Google Scholar] [CrossRef]
- Brill, A.L.; Wisinski, J.A.; Cadena, M.T.; Thompson, M.F.; Fenske, R.J.; Brar, H.K.; Schaid, M.D.; Pasker, R.L.; Kimple, M.E. Synergy Between Galphaz Deficiency and GLP-1 Analog Treatment in Preserving Functional beta-Cell Mass in Experimental Diabetes. Mol. Endocrinol. 2016, 30, 543–556. [Google Scholar] [CrossRef] [Green Version]
- Carboneau, B.A.; Allan, J.A.; Townsend, S.E.; Kimple, M.E.; Breyer, R.M.; Gannon, M. Opposing effects of prostaglandin E2 receptors EP3 and EP4 on mouse and human beta-cell survival and proliferation. Mol. Metab. 2017, 6, 548–559. [Google Scholar] [CrossRef]
- Fenske, R.J.; Cadena, M.T.; Harenda, Q.E.; Wienkes, H.N.; Carbajal, K.; Schaid, M.D.; Laundre, E.; Brill, A.L.; Truchan, N.A.; Brar, H.; et al. The Inhibitory G Protein alpha-Subunit, Galphaz, Promotes Type 1 Diabetes-Like Pathophysiology in NOD Mice. Endocrinology 2017, 158, 1645–1658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neuman, J.C.; Kimple, M.E. The EP3 Receptor: Exploring a New Target for Type 2 Diabetes Therapeutics. J. Endocrinol. Diabetes Obes. 2013, 1, 1002. [Google Scholar] [PubMed]
- Robertson, R.P. The COX-2/PGE2/EP3/Gi/o/cAMP/GSIS Pathway in the Islet: The Beat Goes On. Diabetes 2017, 66, 1464–1466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaid, M.D.; Wisinski, J.A.; Kimple, M.E. The EP3 Receptor/Gz Signaling Axis as a Therapeutic Target for Diabetes and Cardiovascular Disease. AAPS J. 2017, 19, 1276–1283. [Google Scholar] [CrossRef] [PubMed]
- Chakarov, S.; Bleriot, C.; Ginhoux, F. Role of adipose tissue macrophages in obesity-related disorders. J. Exp. Med. 2022, 219, e20211948. [Google Scholar] [CrossRef]
- Eslick, S.; Williams, E.J.; Berthon, B.S.; Wright, T.; Karihaloo, C.; Gately, M.; Wood, L.G. Weight Loss and Short-Chain Fatty Acids Reduce Systemic Inflammation in Monocytes and Adipose Tissue Macrophages from Obese Subjects. Nutrients 2022, 14, 765. [Google Scholar] [CrossRef]
- Johnson, A.R.; Milner, J.J.; Makowski, L. The inflammation highway: Metabolism accelerates inflammatory traffic in obesity. Immunol. Rev. 2012, 249, 218–238. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Mechanistic Insight into Oxidative Stress-Triggered Signaling Pathways and Type 2 Diabetes. Molecules 2022, 27, 950. [Google Scholar] [CrossRef]
- Bosma, K.J.; Andrei, S.R.; Katz, L.S.; Smith, A.A.; Dunn, J.C.; Ricciardi, V.F.; Ramirez, M.A.; Baumel-Alterzon, S.; Pace, W.A.; Carroll, D.T.; et al. Pharmacological blockade of the EP3 prostaglandin E2 receptor in the setting of type 2 diabetes enhances beta-cell proliferation and identity and relieves oxidative damage. Mol. Metab. 2021, 54, 101347. [Google Scholar] [CrossRef]
- Bosma, K.J.; Kaiser, C.E.; Kimple, M.E.; Gannon, M. Effects of Arachidonic Acid and Its Metabolites on Functional Beta-Cell Mass. Metabolites 2022, 12, 342. [Google Scholar] [CrossRef]
- Harsch, I.A.; Konturek, P.C. The Role of Gut Microbiota in Obesity and Type 2 and Type 1 Diabetes Mellitus: New Insights into “Old” Diseases. Med. Sci. 2018, 6, 32. [Google Scholar] [CrossRef] [Green Version]
- Nieuwdorp, M.; Gilijamse, P.W.; Pai, N.; Kaplan, L.M. Role of the microbiome in energy regulation and metabolism. Gastroenterology 2014, 146, 1525–1533. [Google Scholar] [CrossRef]
- Sikalidis, A.K.; Maykish, A. The Gut Microbiome and Type 2 Diabetes Mellitus: Discussing a Complex Relationship. Biomedicines 2020, 8, 8. [Google Scholar] [CrossRef] [Green Version]
- Baggio, L.L.; Drucker, D.J. Biology of incretins: GLP-1 and GIP. Gastroenterology 2007, 132, 2131–2157. [Google Scholar] [CrossRef]
- Yamane, S.; Inagaki, N. Regulation of glucagon-like peptide-1 sensitivity by gut microbiota dysbiosis. J. Diabetes Investig. 2018, 9, 262–264. [Google Scholar] [CrossRef]
- Martin, A.M.; Sun, E.W.; Rogers, G.B.; Keating, D.J. The Influence of the Gut Microbiome on Host Metabolism Through the Regulation of Gut Hormone Release. Front. Physiol. 2019, 10, 428. [Google Scholar] [CrossRef] [Green Version]
- Amer Diabet, A. 2. Classification and Diagnosis of Diabetes. Diabetes Care 2016, 39, S13–S22. [Google Scholar] [CrossRef] [Green Version]
- Owora, A.H. Diagnostic Validity and Clinical Utility of HbA1c Tests for Type 2 Diabetes Mellitus. Curr. Diabetes Rev. 2018, 14, 196–199. [Google Scholar] [CrossRef] [Green Version]
- Pfuetzner, A.; Weber, M.M.; Forst, T. A biomarker concept for assessment of insulin resistance, beta-cell function and chronic systemic inflammation in type 2 diabetes mellitus. Clin. Lab. 2008, 54, 485–490. [Google Scholar]
- Ahlqvist, E.; Storm, P.; Käräjämäki, A.; Martinell, M.; Dorkhan, M.; Carlsson, A.; Vikman, P.; Prasad, R.B.; Aly, D.M.; Almgren, P.; et al. Novel subgroups of adult-onset diabetes and their association with outcomes: A data-driven cluster analysis of six variables. Lancet Diabetes Endocrinol. 2018, 6, 361–369. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Wancewicz, B.; Schaid, M.; Tiambeng, T.N.; Wenger, K.; Jin, Y.; Heyman, H.; Thompson, C.J.; Barsch, A.; Cox, E.D.; et al. Ultrahigh-Resolution Mass Spectrometry-Based Platform for Plasma Metabolomics Applied to Type 2 Diabetes Research. J. Proteome Res. 2021, 20, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Kimple, M.E.; Neuman, J.C.; Linnemann, A.K.; Casey, P.J. Inhibitory G proteins and their receptors: Emerging therapeutic targets for obesity and diabetes. Exp. Mol. Med. 2014, 46, e102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neuman, J.C.; Fenske, R.J.; Kimple, M.E. Dietary polyunsaturated fatty acids and their metabolites: Implications for diabetes pathophysiology, prevention, and treatment. Nutr. Healthy Aging 2017, 4, 127–140. [Google Scholar] [CrossRef] [PubMed]
- Kimple, M.E.; Nixon, A.B.; Kelly, P.; Bailey, C.L.; Young, K.H.; Fields, T.A.; Casey, P.J. A role for G(z) in pancreatic islet beta-cell biology. J. Biol. Chem. 2005, 280, 31708–31713. [Google Scholar] [CrossRef] [Green Version]
- Kimple, M.E.; Moss, J.B.; Brar, H.K.; Rosa, T.C.; Truchan, N.A.; Pasker, R.L.; Newgard, C.B.; Casey, P.J. Deletion of GalphaZ protein protects against diet-induced glucose intolerance via expansion of beta-cell mass. J. Biol. Chem. 2012, 287, 20344–20355. [Google Scholar] [CrossRef] [Green Version]
- Schaid, M.D.; Green, C.L.; Peter, D.C.; Gallagher, S.J.; Guthery, E.; Carbajal, K.A.; Harrington, J.M.; Kelly, G.M.; Reuter, A.; Wehner, M.L.; et al. Agonist-independent Galphaz activity negatively regulates beta-cell compensation in a diet-induced obesity model of type 2 diabetes. J. Biol. Chem. 2021, 296, 100056. [Google Scholar] [CrossRef]
- Kaul, N.; Ali, S. Genes, Genetics, and Environment in Type 2 Diabetes: Implication in Personalized Medicine. DNA Cell Biol. 2016, 35, 1–12. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups | ND vs. T2D | Linear Regression vs. PGEM (T2D Only) | ||||
|---|---|---|---|---|---|---|
| Baseline Demographics | ||||||
| All | Non-Diabetic | T2D | p-Value | p-Value | R2 | |
| Subjects (n) | 167 | 35 | 132 | - | - | - |
| Male (n; %) | 85; 51% | 13; 37% | 72; 55% | - | - | - |
| Female (n; %) | 82; 49% | 22; 63% | 60; 45% | - | - | - |
| Race/Ethnicity = White/Non-Hispanic (n; %) | 153; 92% | 32; 91% | 121; 92% | - | - | - |
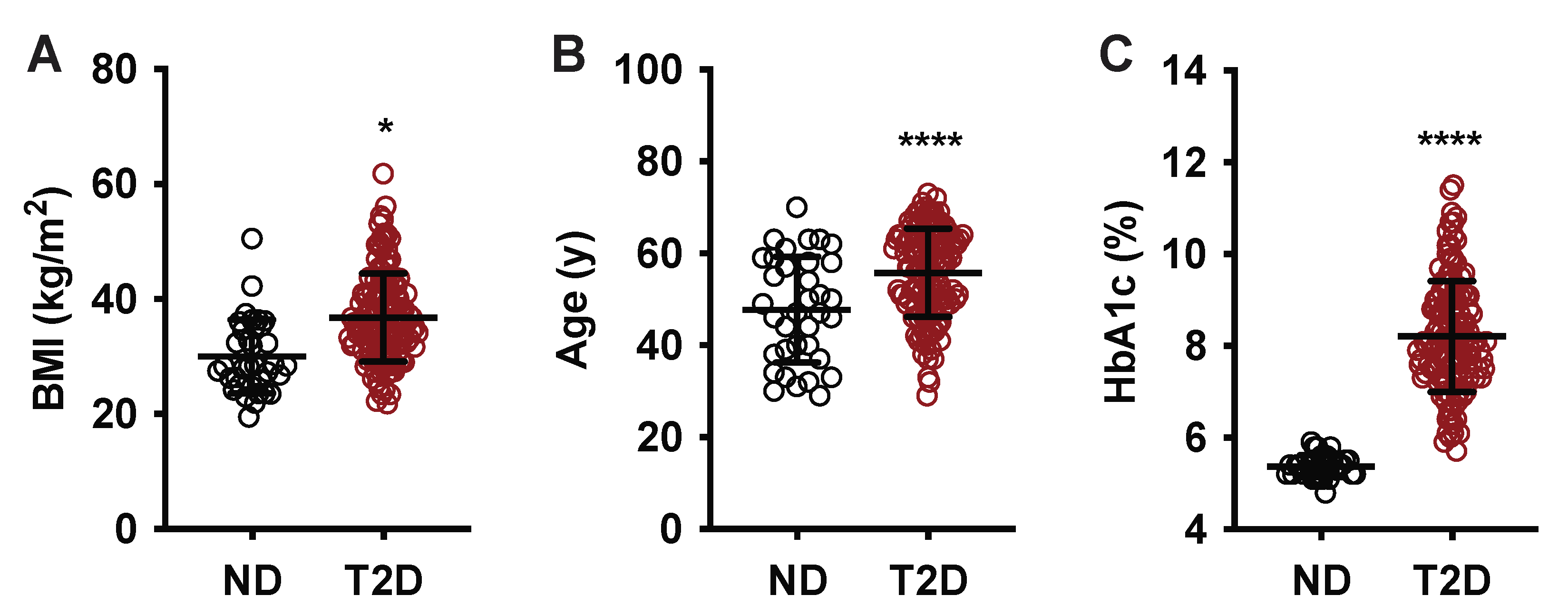
| Age (years ± SD; range) | 54.1 ± 10.5; 29–73 | 47.8 ± 11.5; 29–70 | 55.8 ± 9.6; 29–73 | <0.0001 | 0.84 | 0.0003 |
| Baseline Biometric and Laboratory Parameters | ||||||
| BMI (kg/m2 ± SD; range) | 35.2 ± 8.3; 19.5–61.9 | 29.9 ± 6.5; 19.5–50.5 | 36.8 ± 7.7; 21.8–61.9 | <0.0001 | 0.79 | 0.0005 |
| HbA1c (% ± SD; range) | 7.6 ± 1.6 | 5.4 ± 0.2 | 8.2 ± 1.2 | <0.0001 | 0.79 | 0.0006 |
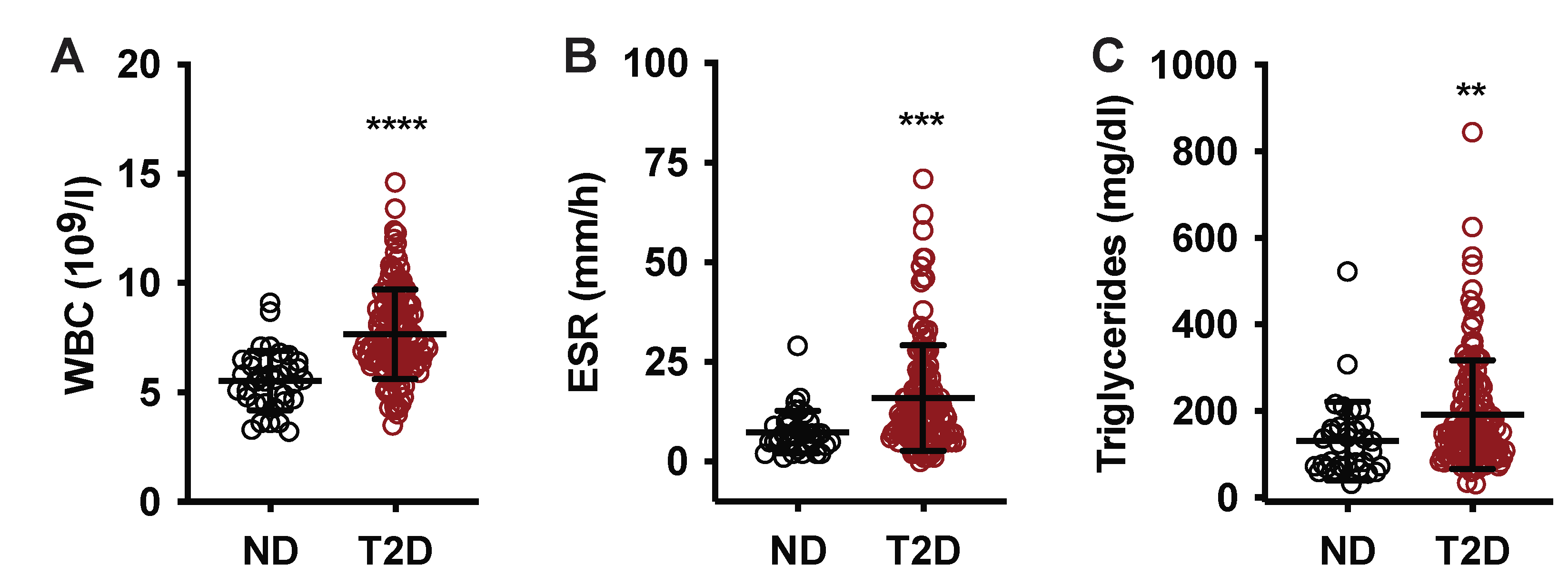
| WBC (109/L ± SD) | 7.2 ± 2.1 | 5.5 ± 1.4 | 7.7 ± 2.0 | <0.0001 | 0.25 | 0.01 |
| ESR, mm/h (mm/h ± SD) | 14.2 ± 12.5 | 7.5 ± 5.4 | 16.0 ± 13.2 | 0.0003 | 0.035 | 0.034 |
| Triglycerides (mg/dL ± SD) | 179.6 ± 121.3 | 131.2 ± 90.8 | 191.1 ± 125.5 | 0.009 | 0.79 | 0.007 |
| Plasma PGEM, (pg/mL ± SD) | 91.0 ± 42.5 | 51.6 ± 30.4 | 101.5 ± 39.1 | <0.0001 | - | - |
| Analysis of Maximum Likelihood Parameter Estimates | ||||||
|---|---|---|---|---|---|---|
| Parameter | DF | Estimate | Std. Error | 95% CI | ChiSq | Pr > ChiSq |
| Intercept | 1 | −15.8799 | 3.3533 | −22.4522 to −9.3076 | 22.43 | <0.0001 |
| PGEM | 1 | 0.0593 | 0.0141 | −0.0317 to 0.0869 | 17.70 | <0.0001 |
| Age | 1 | 0.1001 | 0.0375 | 0.0265 to 0.1737 | 7.71 | 0.0076 |
| BMI | 1 | 0.0765 | 0.0449 | −0.0166 to 0.1746 | 2.90 | 0.0886 |
| WBC | 1 | 0.7225 | 0.2341 | 0.2635 to 1.1814 | 9.52 | 0.0020 |
| Triglycerides | 1 | 0.0064 | 0.0042 | −0.0019 to 0.0147 | 2.25 | 0.1332 |
| Sex | 1 | −0.9199 | 0.6979 | −2.2878 to 0.4480 | 1.74 | 0.1875 |
| Aspirin use | 1 | −23.689 | 180, 132.4 | −353,077 to 353,023 | 0.00 | 0.9999 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fenske, R.J.; Weeks, A.M.; Daniels, M.; Nall, R.; Pabich, S.; Brill, A.L.; Peter, D.C.; Punt, M.; Cox, E.D.; Davis, D.B.; et al. Plasma Prostaglandin E2 Metabolite Levels Predict Type 2 Diabetes Status and One-Year Therapeutic Response Independent of Clinical Markers of Inflammation. Metabolites 2022, 12, 1234. https://doi.org/10.3390/metabo12121234
Fenske RJ, Weeks AM, Daniels M, Nall R, Pabich S, Brill AL, Peter DC, Punt M, Cox ED, Davis DB, et al. Plasma Prostaglandin E2 Metabolite Levels Predict Type 2 Diabetes Status and One-Year Therapeutic Response Independent of Clinical Markers of Inflammation. Metabolites. 2022; 12(12):1234. https://doi.org/10.3390/metabo12121234
Chicago/Turabian StyleFenske, Rachel J., Alicia M. Weeks, Michael Daniels, Randall Nall, Samantha Pabich, Allison L. Brill, Darby C. Peter, Margaret Punt, Elizabeth D. Cox, Dawn Belt Davis, and et al. 2022. "Plasma Prostaglandin E2 Metabolite Levels Predict Type 2 Diabetes Status and One-Year Therapeutic Response Independent of Clinical Markers of Inflammation" Metabolites 12, no. 12: 1234. https://doi.org/10.3390/metabo12121234