Lipid Annotator: Towards Accurate Annotation in Non-Targeted Liquid Chromatography High-Resolution Tandem Mass Spectrometry (LC-HRMS/MS) Lipidomics Using a Rapid and User-Friendly Software

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Lipid Annotator Software
2.1.1. User-Workflow
2.1.2. Lipid Annotator Libraries
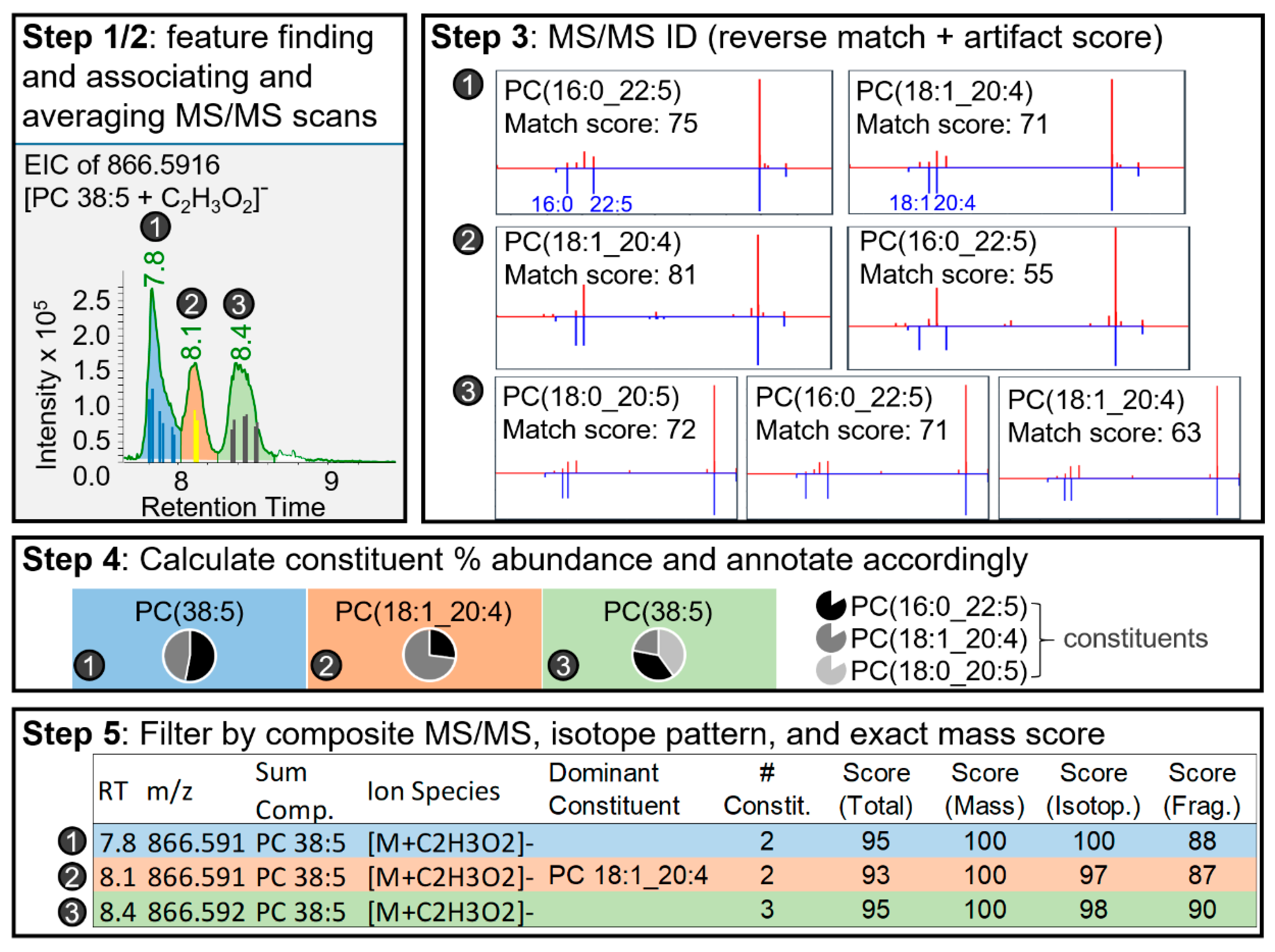
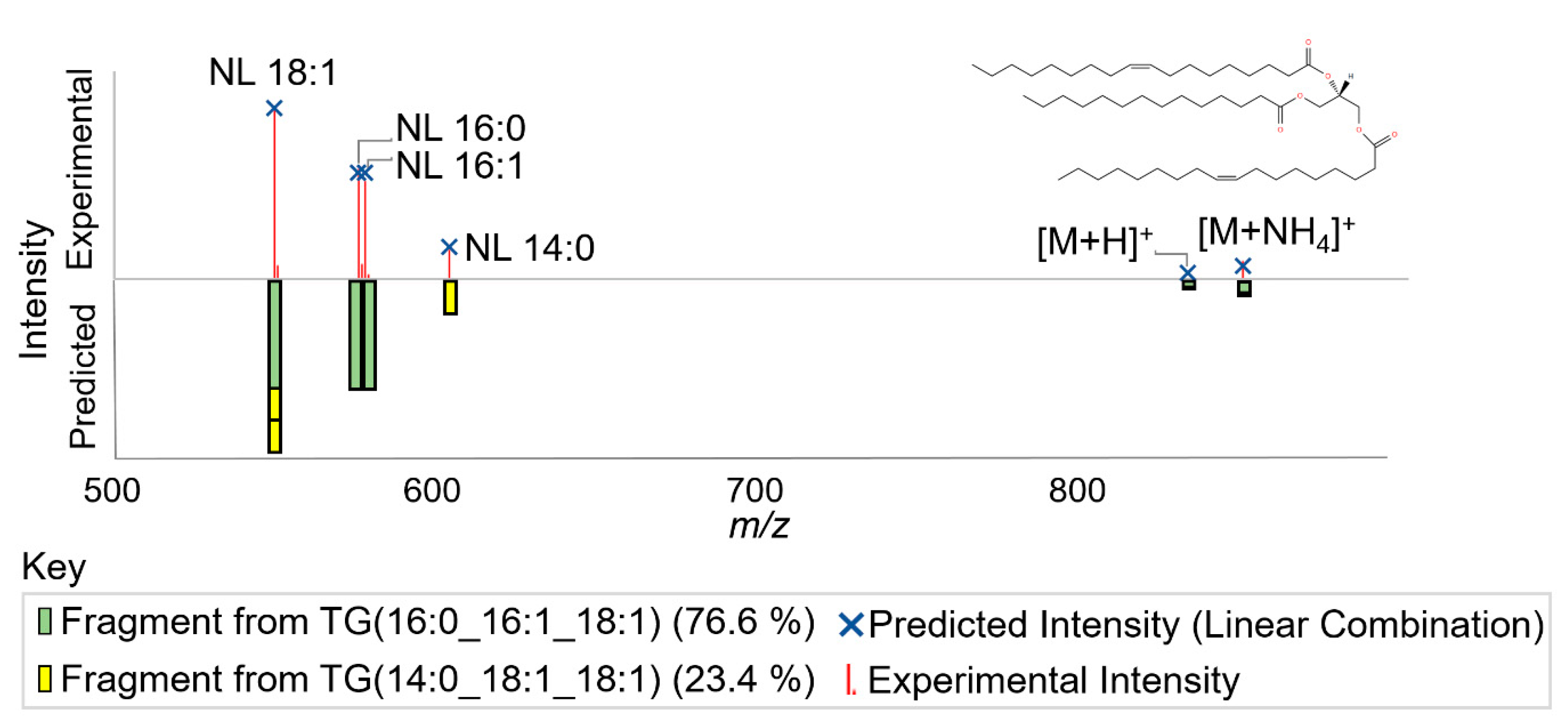
2.1.3. Lipid Annotator Annotation Algorithm
2.1.4. User Interface and Downstream Workflow
2.2. Application and Validation: Analysis of NIST SRM 1950 using Iterative Exclusion
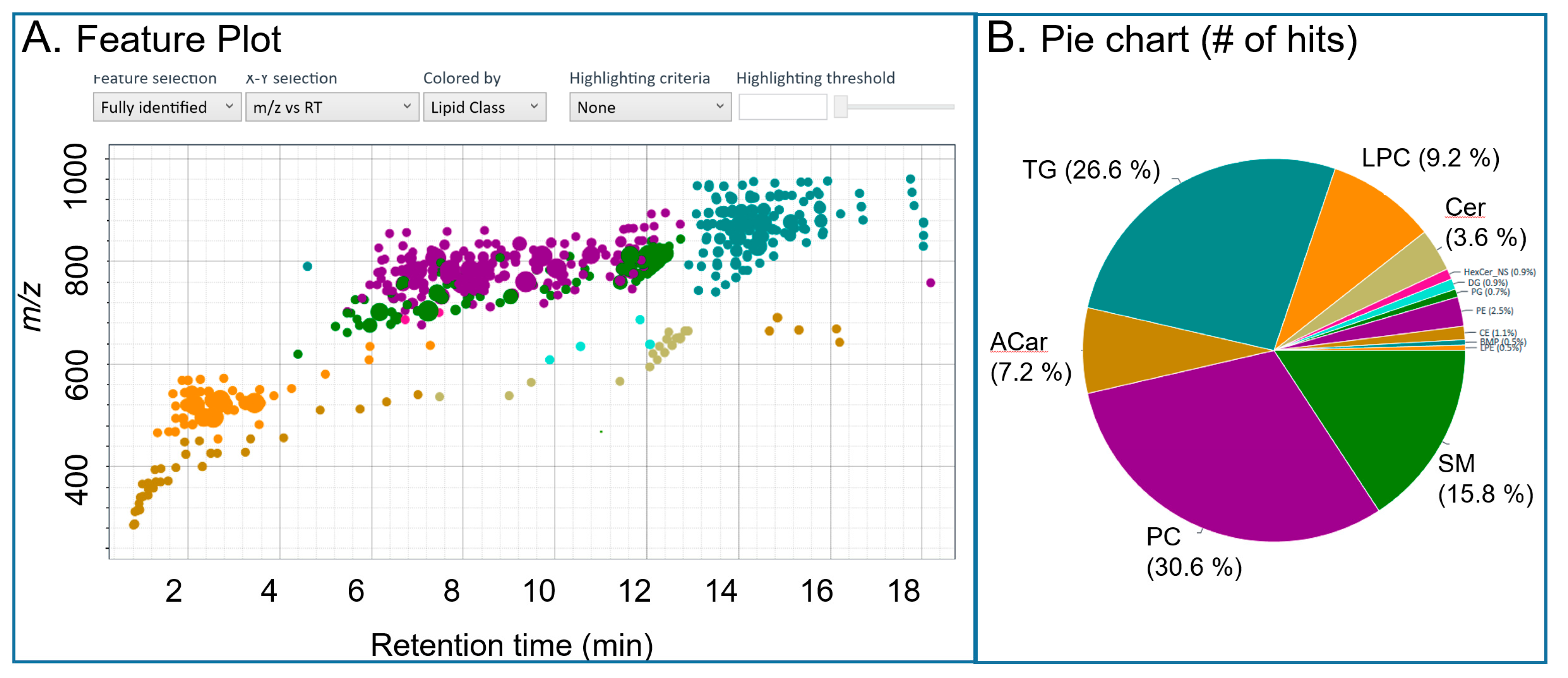
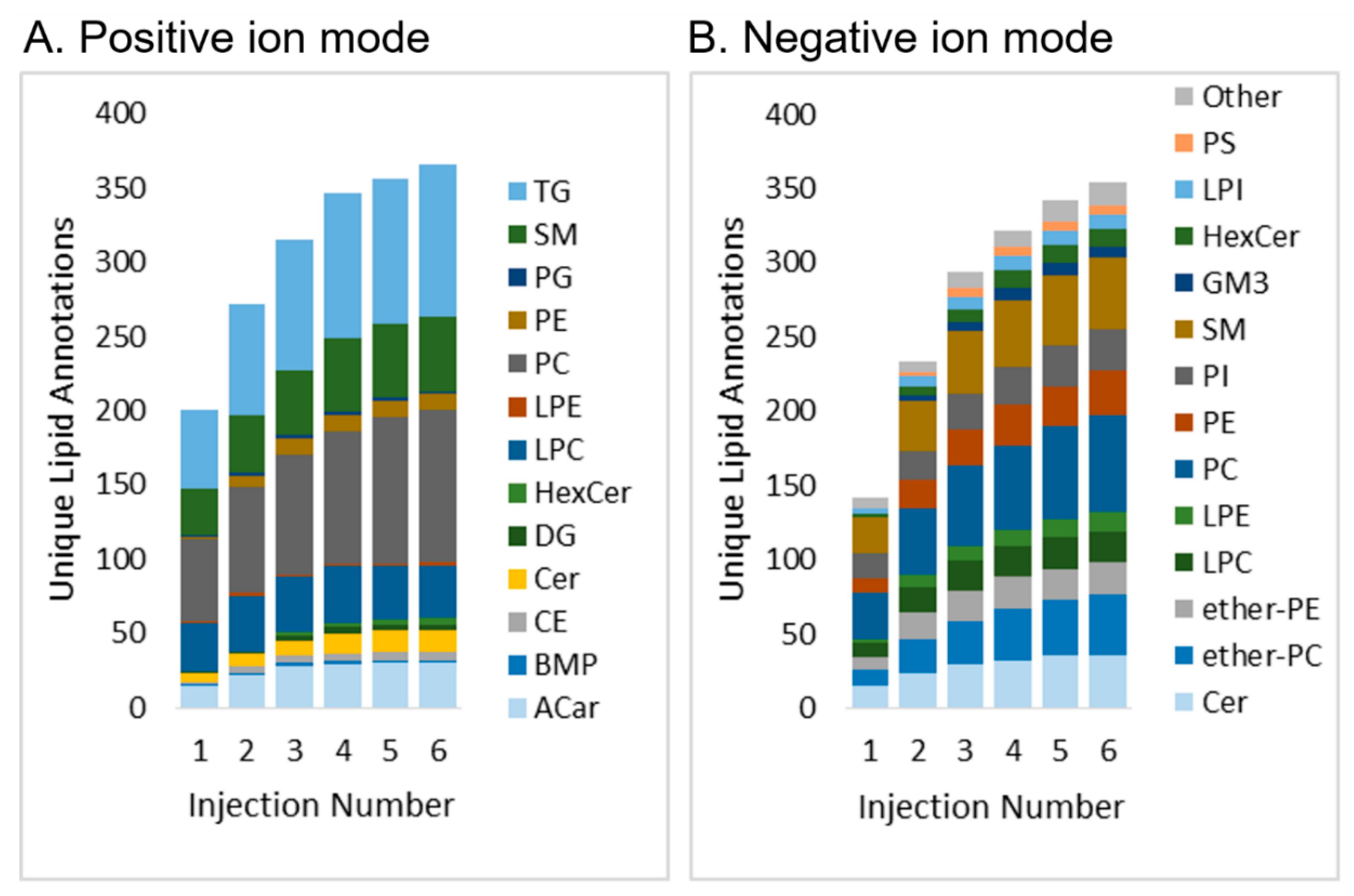
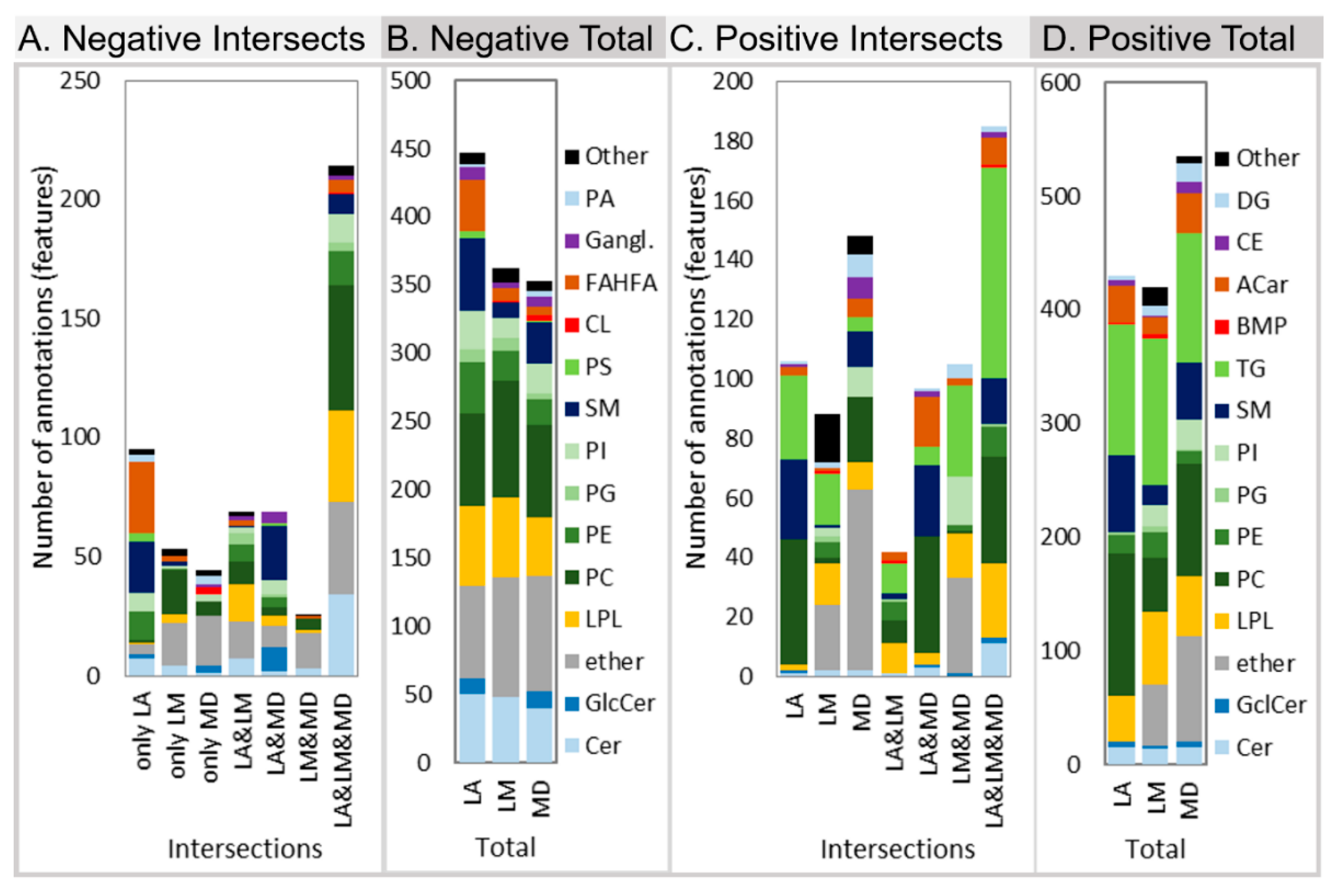
2.2.1. Lipid Coverage
2.2.2. Annotation Accuracy
- (1)
- internal and external standard solutions,
- (2)
- comparing annotations against other lipidomics software.
3. Materials and Methods
3.1. Methods: Lipid Extraction and Data-Acquisition
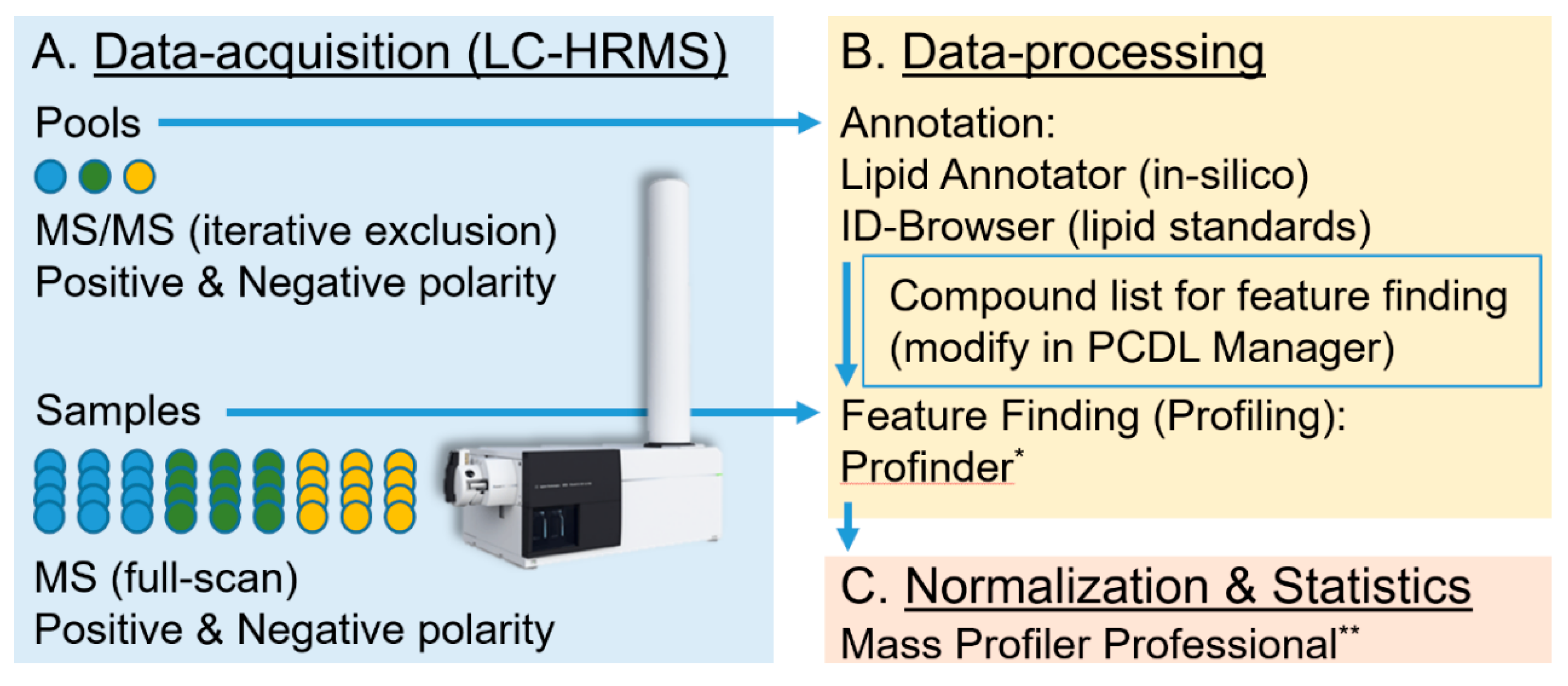
3.2. Methods: Data-Processing
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
Appendix A.1. Lipid Annotator Algorithms
Appendix A.1.1. Sum Composition Annotation Using Bayesian Theorem
Appendix A.1.2. Calculation of Lipid Relative Abundances to Fit Data using Non-Negative Least Squares Fit
Appendix A.1.3. Normalization of Probabilities
Appendix A.1.4. User Interface and Downstream Workflow
Appendix A.2. Software Settings
Appendix A.2.1. MS-DIAL Parameter Setting
Appendix A.2.2. LipidMatch Parameter Setting
Appendix B List of Acronyms
| Acronym | Definition |
| Acar | acylcarnitine |
| BMP | bis(monoacylglycero)phosphate |
| CCS | collision cross section |
| CE | cholesterol ester |
| Cer | ceramide |
| CL | cardiolipin |
| DG | diglyceride |
| EIC | reconstructed ion chromatogram |
| ether | plasmenyl/plasmanyl lipid |
| FAHFA | fatty acid ester of hydroxyl fatty acid |
| Gangl | ganglioside |
| GlcCer | glucosyl ceramide |
| GM3 | monosialodihexosylganglioside |
| HexCer_NS | hexosyl-ceramide |
| HRMS | high resolution mass spectrometry |
| ID | identification |
| LA | Lipid Annotator |
| LC | liquid chromatography |
| LM | LipidMatch |
| LPC | lysophosphatidylcholine |
| LPE | lysophosphatidylethanolamine |
| LPI | lysophosphatidylinisitol |
| LPL | lysophospholipid |
| M | molecular ion |
| MD | MS-DIAL |
| MG | monoglyceride |
| MPP | mass profiler professional |
| MS/MS | tandem mass spectrometry |
| NIST | National Institute of Standards and Technology |
| NL | neutral loss |
| PA | phosphatidic acid |
| PC | phosphatidylcholine |
| PCDL | Personal Compound Database and Library |
| PE | phosphatidylethanolamine |
| PG | phosphatidylglycerol |
| PI | phosphatidylinositol |
| PS | phosphatidylserine |
| Q-TOF | quadrupole time of flight |
| RAM | random access memory |
| SM | sphingomyelin |
| SRM | standard reference material |
| TG | triglyceride |
References
- Gross, R.W.; Han, X. Lipidomics in diabetes and the metabolic syndrome. Meth. Enzymol. 2007, 433, 73–90. [Google Scholar] [PubMed]
- Lv, J.; Zhang, L.; Yan, F.; Wang, X. Clinical lipidomics: A new way to diagnose human diseases. Clin. Transl. Med. 2018, 7, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, F.; Zhao, H.; Zeng, Y. Lipidomics: A promising cancer biomarker. Clin. Transl. Med. 2018, 7, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bi, H.; Fu, D.; Wang, L.; Han, X. Lipid Nanotube Formation Using Space-Regulated Electric Field above Interdigitated Electrodes. ACS Nano. 2014, 8, 3961–3969. [Google Scholar] [CrossRef] [PubMed]
- Depalo, N.; Leo, V.D.; Corricelli, M.; Gristina, R.; Valente, G.; Casamassima, E.; Comparelli, R.; Laquintana, V.; Denora, N.; Fanizza, E.; et al. Lipid-based systems loaded with PbS nanocrystals: Near infrared emitting trackable nanovectors. J. Mater. Chem. B 2017, 5, 1471–1481. [Google Scholar] [CrossRef]
- Gopalakrishnan, G.; Danelon, C.; Izewska, P.; Prummer, M.; Bolinger, P.-Y.; Geissbühler, I.; Demurtas, D.; Dubochet, J.; Vogel, H. Multifunctional lipid/quantum dot hybrid nanocontainers for controlled targeting of live cells. Angew. Chem. Int. Ed. Engl. 2006, 45, 5478–5483. [Google Scholar] [CrossRef]
- Chang, Y.; Zhang, L.; Lu, X.; Zhao, C.; Zhu, Z.; Wang, F.; Zhang, J.; Chen, S.; Zhao, Y.; Xu, G. A simultaneous extraction method for metabolome and lipidome and its application in cry1Ac and sck-transgenic rice leaf treated with insecticide based on LC–MS analysis. Metabolomics 2014, 10, 1197–1209. [Google Scholar] [CrossRef]
- Welti, R.; Shah, J.; Li, W.; Li, M.; Chen, J.; Burke, J.J.; Fauconnier, M.-L.; Chapman, K.; Chye, M.-L.; Wang, X. Plant lipidomics: Discerning biological function by profiling plant complex lipids using mass spectrometry. Front. Biosci. 2007, 12, 2494–2506. [Google Scholar] [CrossRef] [Green Version]
- Christie, B. Lipid Matters-Archive of Older Blogs-2017. Available online: http://www.lipidhome.co.uk/info/blogarch-2017.htm (accessed on 26 March 2019).
- Kind, T.; Liu, K.-H.; Yup Lee, D.; DeFelice, B.; Meissen, J.K.; Fiehn, O. LipidBlast-in-silico tandem mass spectrometry database for lipid identification. Nat. Methods 2013, 10, 755–758. [Google Scholar] [CrossRef] [Green Version]
- Bowden, J.A.; Ulmer, C.Z.; Jones, C.M.; Koelmel, J.P.; Yost, R.A. NIST lipidomics workflow questionnaire: An assessment of community-wide methodologies and perspectives. Metabolomics 2018, 14, 53. [Google Scholar] [CrossRef]
- Lipid Maps-External Software. Available online: https://www.lipidmaps.org/resources/tools/index.php?tab=software (accessed on 26 March 2019).
- Tsugawa, H.; Cajka, T.; Kind, T.; Ma, Y.; Higgins, B.; Ikeda, K.; Kanazawa, M.; VanderGheynst, J.; Fiehn, O.; Arita, M. MS-DIAL: Data-independent MS/MS deconvolution for comprehensive metabolome analysis. Nat. Methods 2015, 12, 523–526. [Google Scholar] [CrossRef] [PubMed]
- Hutchins, P.D.; Russell, J.D.; Coon, J.J. Mapping Lipid Fragmentation for Tailored Mass Spectral Libraries. J. Am. Soc. Mass Spectrom. 2019, 30, 659–668. [Google Scholar] [CrossRef] [PubMed]
- Kind, T.; Okazaki, Y.; Saito, K.; Fiehn, O. LipidBlast Templates As Flexible Tools for Creating New in-Silico Tandem Mass Spectral Libraries. Anal. Chem. 2014, 86, 11024–11027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartler, J.; Triebl, A.; Ziegl, A.; Trötzmüller, M.; Rechberger, G.N.; Zeleznik, O.A.; Zierler, K.A.; Torta, F.; Cazenave-Gassiot, A.; Wenk, M.R.; et al. Deciphering lipid structures based on platform-independent decision rules. Nat. Methods 2017, 14, 1171–1174. [Google Scholar] [CrossRef]
- Koelmel, J.P.; Kroeger, N.M.; Ulmer, C.Z.; Bowden, J.A.; Patterson, R.E.; Cochran, J.A.; Beecher, C.W.W.; Garrett, T.J.; Yost, R.A. LipidMatch: An automated workflow for rule-based lipid identification using untargeted high-resolution tandem mass spectrometry data. BMC Bioinform. 2017, 18, 331. [Google Scholar] [CrossRef]
- Kyle, J.E.; Crowell, K.L.; Casey, C.P.; Fujimoto, G.M.; Kim, S.; Dautel, S.E.; Smith, R.D.; Payne, S.H.; Metz, T.O. LIQUID: An-open source software for identifying lipids in LC-MS/MS-based lipidomics data. Bioinformatics 2017, 33, 1744–1746. [Google Scholar] [CrossRef] [Green Version]
- Liebisch, G.; Vizcaíno, J.A.; Köfeler, H.; Trötzmüller, M.; Griffiths, W.J.; Schmitz, G.; Spener, F.; Wakelam, M.J.O. Shorthand notation for lipid structures derived from mass spectrometry. J. Lipid Res. 2013, 54, 1523–1530. [Google Scholar] [CrossRef] [Green Version]
- Koelmel, J.P.; Ulmer, C.Z.; Jones, C.M.; Yost, R.A.; Bowden, J.A. Common cases of improper lipid annotation using high-resolution tandem mass spectrometry data and corresponding limitations in biological interpretation. Biochim. Biophys. Acta 2017, 1862, 766–770. [Google Scholar] [CrossRef]
- Hernández, B.; Pennington, S.R.; Parnell, A.C. Bayesian methods for proteomic biomarker development. EuPA Open Proteom. 2015, 9, 54–64. [Google Scholar] [CrossRef]
- Serang, O.; MacCoss, M.J.; Noble, W.S. Efficient Marginalization to Compute Protein Posterior Probabilities from Shotgun Mass Spectrometry Data. J. Proteome. Res. 2010, 9, 5346–5357. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.F.; Arnold, R.J.; Li, Y.; Radivojac, P.; Sheng, Q.; Tang, H. A Bayesian Approach to Protein Inference Problem in Shotgun Proteomics. J. Comput. Biol. 2009, 16, 1183–1193. [Google Scholar] [CrossRef] [PubMed]
- Witting, M.; Ruttkies, C.; Neumann, S.; Schmitt-Kopplin, P. LipidFrag: Improving reliability of in silico fragmentation of lipids and application to the Caenorhabditis elegans lipidome. PLoS ONE 2017, 12, e0172311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, J.; Shi, X.; Zhang, X.; Kim, S.; Shen, C. An empirical Bayes model using a competition score for metabolite identification in gas chromatography mass spectrometry. BMC Bioinform. 2011, 12, 392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koelmel, J.P.; Kroeger, N.M.; Gill, E.L.; Ulmer, C.Z.; Bowden, J.A.; Patterson, R.E.; Yost, R.A.; Garrett, T.J. Expanding Lipidome Coverage Using LC-MS/MS Data-Dependent Acquisition with Automated Exclusion List Generation. J. Am. Soc. Mass Spectrom. 2017, 28, 908–917. [Google Scholar] [CrossRef] [Green Version]
- Pluskal, T.; Castillo, S.; Villar-Briones, A.; Orešič, M. MZmine 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinform. 2010, 11, 395. [Google Scholar] [CrossRef] [Green Version]
- Olivon, F.; Grelier, G.; Roussi, F.; Litaudon, M.; Touboul, D. MZmine 2 Data-Preprocessing To Enhance Molecular Networking Reliability. Anal. Chem. 2017, 89, 7836–7840. [Google Scholar] [CrossRef]
- Chong, J.; Soufan, O.; Li, C.; Caraus, I.; Li, S.; Bourque, G.; Wishart, D.S.; Xia, J. MetaboAnalyst 4.0: Towards more transparent and integrative metabolomics analysis. Nucleic. Acids Res. 2018, 46, W486–W494. [Google Scholar] [CrossRef] [Green Version]
- Ulmer, C.Z.; Koelmel, J.P.; Ragland, J.M.; Garrett, T.J.; Bowden, J.A. LipidPioneer: A Comprehensive User-Generated Exact Mass Template for Lipidomics. J. Am. Soc. Mass Spectrom. 2017, 28, 562–565. [Google Scholar] [CrossRef] [Green Version]
- Joyce, J. Bayes’ Theorem. In The Stanford Encyclopedia of Philosophy; Zalta, E.N., Ed.; Metaphysics Research Lab, Stanford University: Stanford, CA, USA, 2003; Available online: https://plato.stanford.edu/entries/bayes-theorem/ (accessed on 7 March 2020).
- Hutchins, P.D.; Russell, J.D.; Coon, J.J. LipiDex: An Integrated Software Package for High-Confidence Lipid Identification. Cell Syst. 2018, 6, 621–625. [Google Scholar] [CrossRef] [Green Version]
- Xu, F.; Zou, L.; Lin, Q.; Ong, C.N. Use of liquid chromatography/tandem mass spectrometry and online databases for identification of phosphocholines and lysophosphatidylcholines in human red blood cells. Rapid Commun. Mass Spectrom. 2009, 23, 3243–3254. [Google Scholar] [CrossRef] [PubMed]
- Gathungu, R.M.; Larrea, P.; SniatynskI, M.J.; Marur, V.R.; Bowden, J.A.; Koelmel, J.P.; Starke-Reed, P.; Hubbard, V.S.; Kristal, B.S. Optimization of ESI-Source Parameters for Lipidomics Reduces Misannotation of In-Source Fragments as Precursor Ions. Anal. Chem. 2018, 90, 13523–13532. [Google Scholar] [CrossRef] [PubMed]
- Yore, M.M.; Syed, I.; Moraes-Vieira, P.M.; Zhang, T.; Herman, M.A.; Homan, E.A.; Patel, R.T.; Lee, J.; Chen, S.; Peroni, O.D.; et al. Discovery of a class of endogenous mammalian lipids with anti-diabetic and anti-inflammatory effects. Cell 2014, 159, 318–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folch, J.; Lees, M.; Sloane Stanley, G.H. A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [PubMed]
- Sartain, M.; Salcedo, J.; Murali, A.; Li, X.; Stow, S.; Koelmel, J. Improving Coverage of the Plasma Lipidome Using Iterative MS/MS Data Acquisition Combined with Lipid Annotator Software and 6546 LC/Q-TOF. Agilent Application Note 2019, 5994–0775en. Available online: https://www.agilent.com/cs/library/applications/application-6546-q-tof-lipidome-5994-0775en-agilent.pdf (accessed on 7 March 2020).






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koelmel, J.P.; Li, X.; Stow, S.M.; Sartain, M.J.; Murali, A.; Kemperman, R.; Tsugawa, H.; Takahashi, M.; Vasiliou, V.; Bowden, J.A.; et al. Lipid Annotator: Towards Accurate Annotation in Non-Targeted Liquid Chromatography High-Resolution Tandem Mass Spectrometry (LC-HRMS/MS) Lipidomics Using a Rapid and User-Friendly Software. Metabolites 2020, 10, 101. https://doi.org/10.3390/metabo10030101
Koelmel JP, Li X, Stow SM, Sartain MJ, Murali A, Kemperman R, Tsugawa H, Takahashi M, Vasiliou V, Bowden JA, et al. Lipid Annotator: Towards Accurate Annotation in Non-Targeted Liquid Chromatography High-Resolution Tandem Mass Spectrometry (LC-HRMS/MS) Lipidomics Using a Rapid and User-Friendly Software. Metabolites. 2020; 10(3):101. https://doi.org/10.3390/metabo10030101
Chicago/Turabian StyleKoelmel, Jeremy P., Xiangdong Li, Sarah M. Stow, Mark J. Sartain, Adithya Murali, Robin Kemperman, Hiroshi Tsugawa, Mikiko Takahashi, Vasilis Vasiliou, John A. Bowden, and et al. 2020. "Lipid Annotator: Towards Accurate Annotation in Non-Targeted Liquid Chromatography High-Resolution Tandem Mass Spectrometry (LC-HRMS/MS) Lipidomics Using a Rapid and User-Friendly Software" Metabolites 10, no. 3: 101. https://doi.org/10.3390/metabo10030101