Lipidomic Analysis of Cells and Extracellular Vesicles from High- and Low-Metastatic Triple-Negative Breast Cancer

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
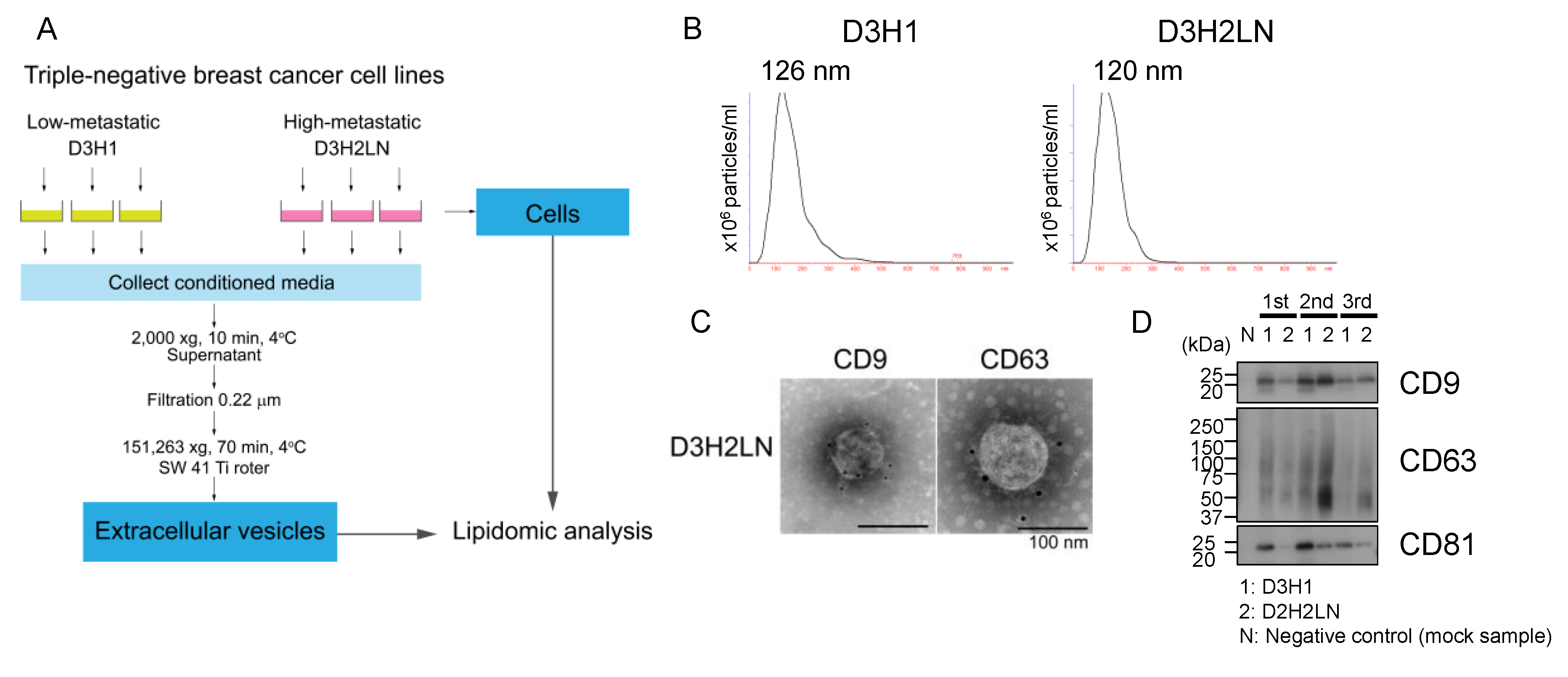
2.1. Characterization of EVs Isolated from High- And Low-Metastatic Breast Cancer Cells
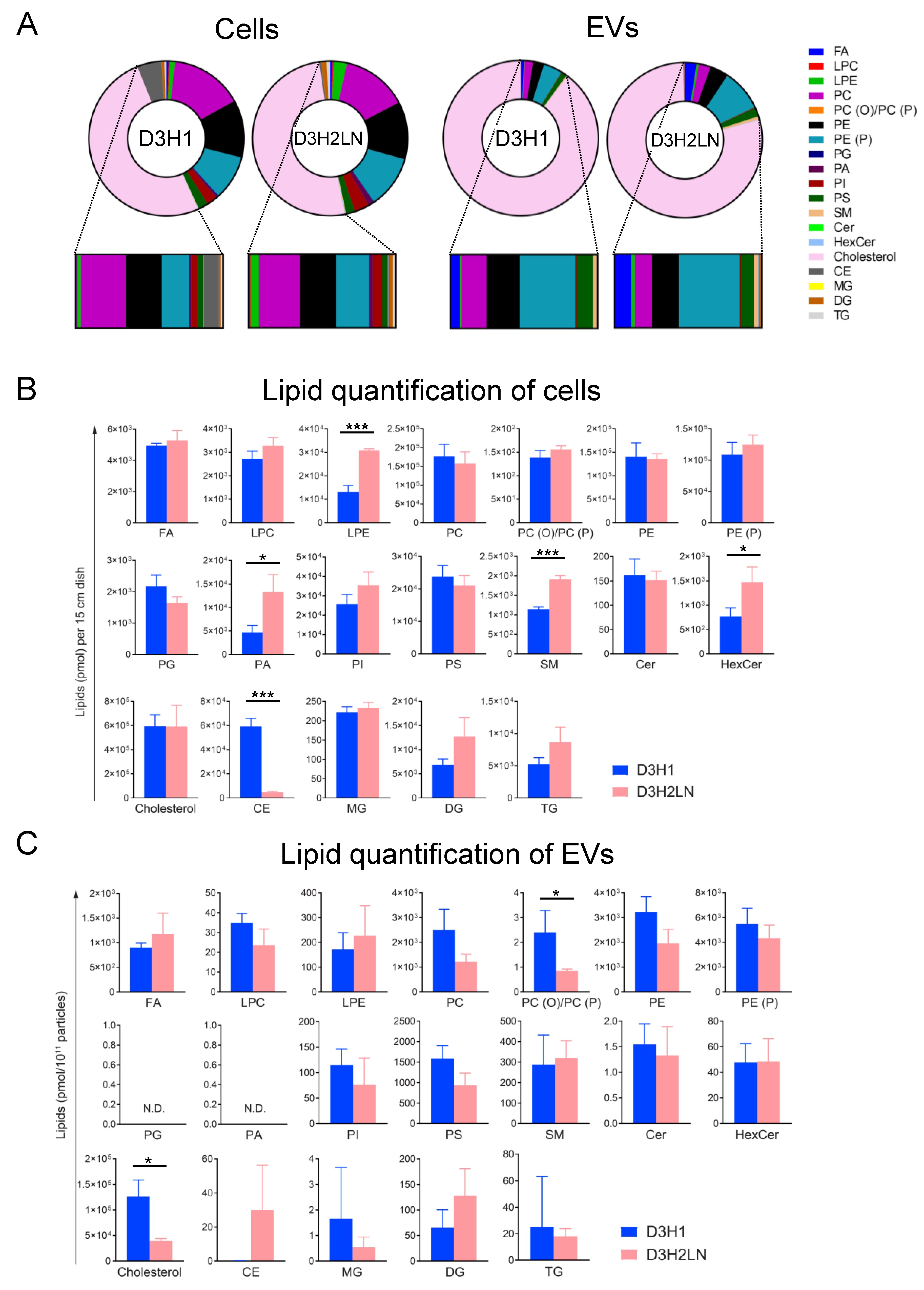
2.2. SFC/QqQMS-Based Lipidomic Analysis in Cells and EVs Derived from High- And Low-Metastatic TNBC Cell Lines
2.3. Comparison of Lipid Components between EVs and Their Secreting Cells
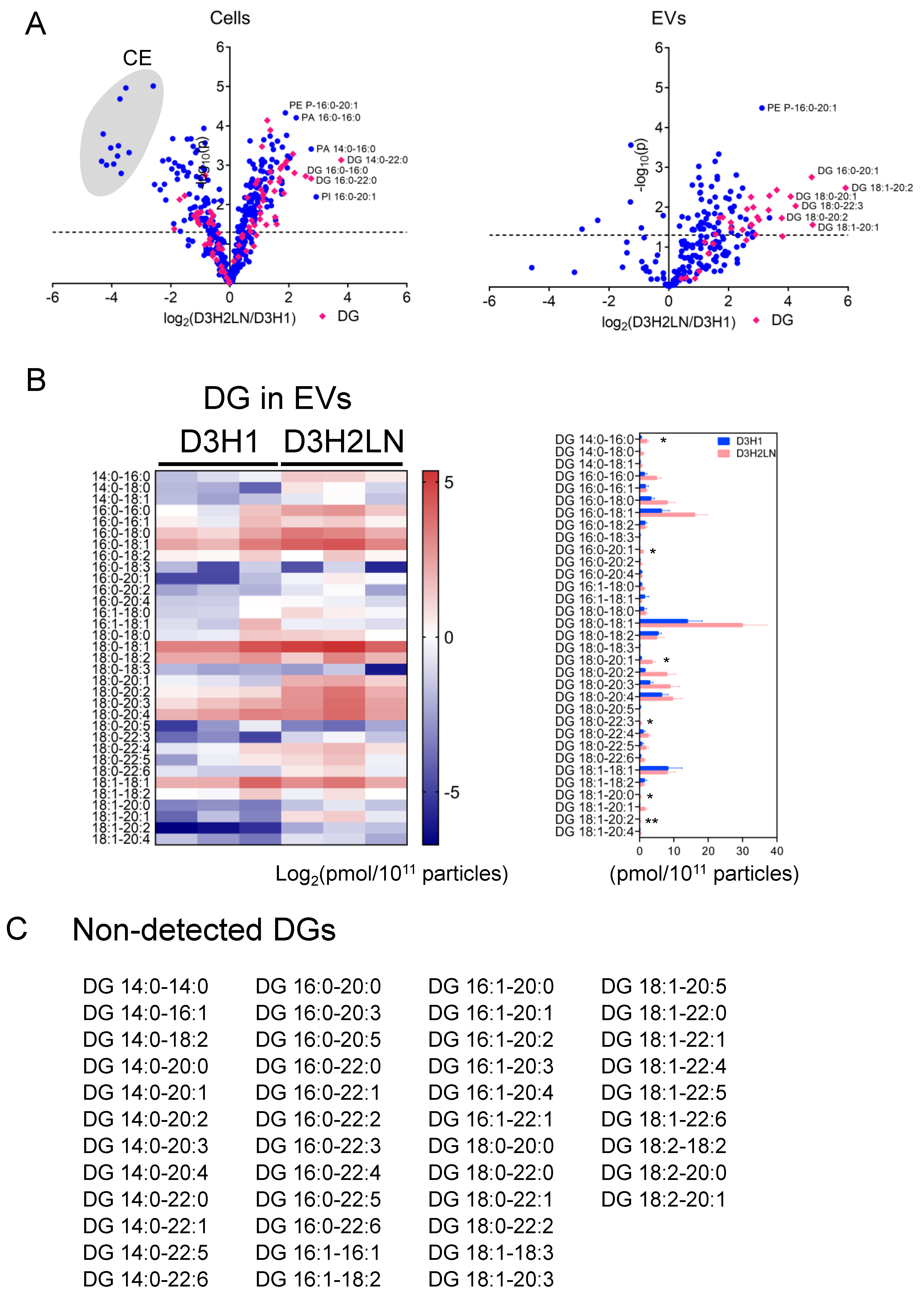
2.4. Comparison of Lipid Components of EVs from High- and Low-Metastatic TNBC
2.5. Comparison of Individual Lipid Species of EVs and Cells
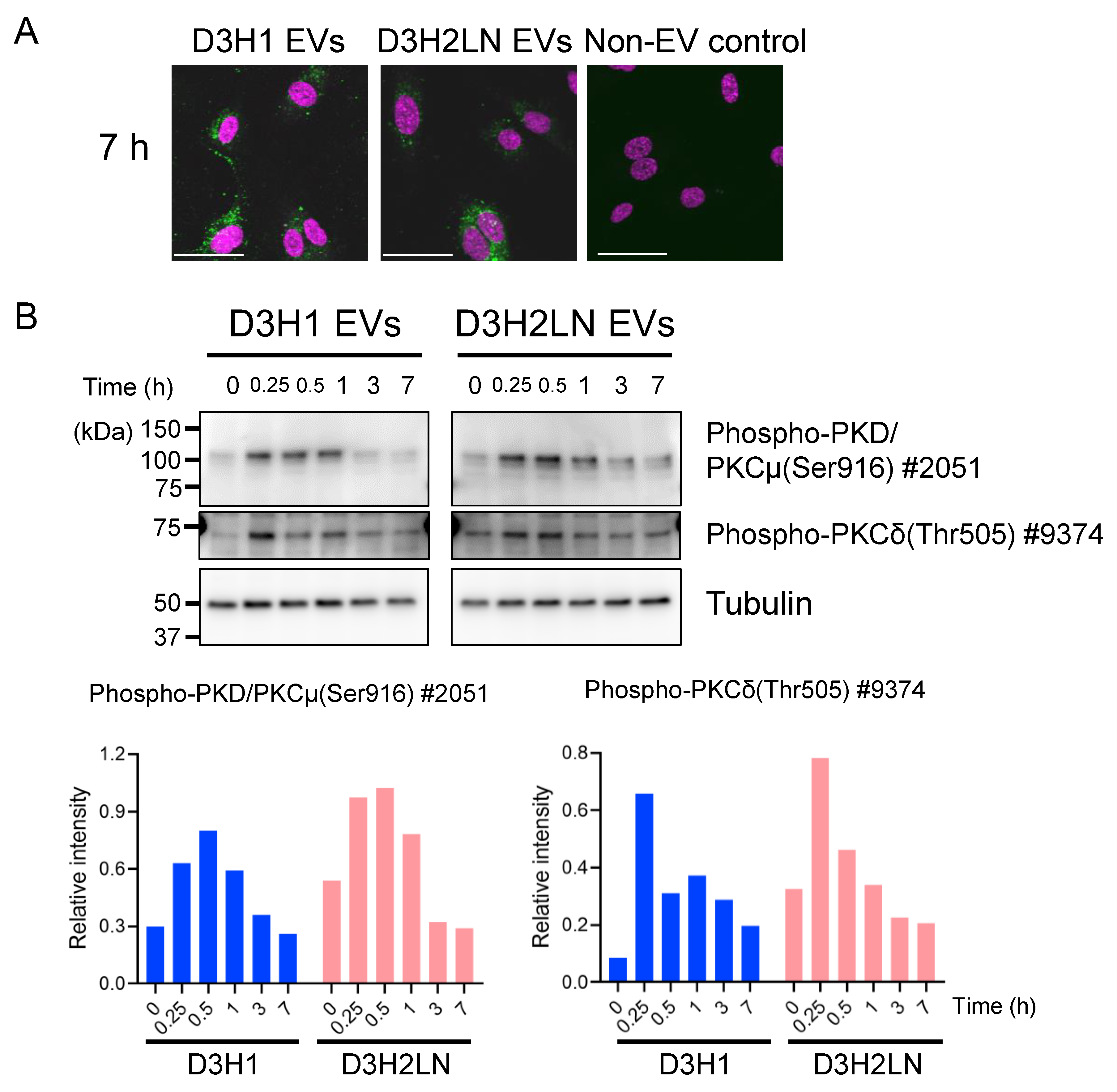
2.6. DGs in TNBC EVs Activated PKC Signaling Pathway of HUVECs
3. Discussion
4. Materials and Methods
4.1. Cell Lines And Cultures
4.2. Sample Preparation for Lipidomic Analysis
4.3. EV Isolation
4.4. Lipid Extraction
4.5. Lipidomic Analysis by SFC/QqQMS
4.6. Supplementation of EVs to HUVECs
4.7. Immunoblot
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yáñez-Mó, M.; Siljander, P.R.-M.; Andreu, Z.; Bedina Zavec, A.; Borràs, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 2015, 4, 27066. [Google Scholar] [CrossRef] [Green Version]
- Pathan, M.; Fonseka, P.; Chitti, S.V.; Kang, T.; Sanwlani, R.; Van Deun, J.; Hendrix, A.; Mathivanan, S. Vesiclepedia 2019: A compendium of RNA, proteins, lipids and metabolites in extracellular vesicles. Nucleic Acids Res. 2019, 47, D516–D519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Record, M.; Silvente-Poirot, S.; Poirot, M.; Wakelam, M.J.O. Extracellular vesicles: Lipids as key components of their biogenesis and functions. J. Lipid Res. 2018, 59, 1316–1324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skotland, T.; Sandvig, K.; Llorente, A. Lipids in exosomes: Current knowledge and the way forward. Prog. Lipid Res. 2017, 66, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Haraszti, R.A.; Didiot, M.C.; Sapp, E.; Leszyk, J.; Shaffer, S.A.; Rockwell, H.E.; Gao, F.; Narain, N.R.; DiFiglia, M.; Kiebish, M.A.; et al. High-resolution proteomic and lipidomic analysis of exosomes and microvesicles from different cell sources. J. Extracell. Vesicles 2016, 5, 32570. [Google Scholar] [CrossRef] [PubMed]
- Llorente, A.; Skotland, T.; Sylvänne, T.; Kauhanen, D.; Róg, T.; Orłowski, A.; Vattulainen, I.; Ekroos, K.; Sandvig, K. Molecular lipidomics of exosomes released by PC-3 prostate cancer cells. Biochim. Biophys. Acta 2013, 1831, 1302–1309. [Google Scholar] [CrossRef]
- Subra, C.; Laulagnier, K.; Perret, B.; Record, M. Exosome lipidomics unravels lipid sorting at the level of multivesicular bodies. Biochimie 2007, 89, 205–212. [Google Scholar] [CrossRef]
- Trajkovic, K.; Hsu, C.; Chiantia, S.; Rajendran, L.; Wenzel, D.; Wieland, F.; Schwille, P.; Brugger, B.; Simons, M. Ceramide Triggers Budding of Exosome Vesicles into Multivesicular Endosomes. Science 2008, 319, 1244–1247. [Google Scholar] [CrossRef]
- Kosaka, N.; Iguchi, H.; Hagiwara, K.; Yoshioka, Y.; Takeshita, F.; Ochiya, T. Neutral sphingomyelinase 2 (nSMase2)-dependent exosomal transfer of angiogenic microRNAs regulate cancer cell metastasis. J. Biol. Chem. 2013, 288, 10849–10859. [Google Scholar] [CrossRef] [Green Version]
- Ghossoub, R.; Lembo, F.; Rubio, A.; Gaillard, C.B.; Bouchet, J.; Vitale, N.; Slavík, J.; Machala, M.; Zimmermann, P. Syntenin-ALIX exosome biogenesis and budding into multivesicular bodies are controlled by ARF6 and PLD2. Nat. Commun. 2014, 5, 3477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egea-Jimenez, A.L.; Zimmermann, P. Phospholipase D and phosphatidic acid in the biogenesis and cargo loading of extracellular vesicles. J. Lipid Res. 2018, 59, 1554–1560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blücher, C.; Stadler, S.C. Obesity and breast cancer: Current insights on the role of fatty acids and lipid metabolism in promoting breast cancer growth and progression. Front. Endocrinol. (Lausanne) 2017, 8, 293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gershuni, V.; Li, Y.R.; Williams, A.D.; So, A.; Steel, L.; Carrigan, E.; Tchou, J. Breast cancer subtype distribution is different in normal weight, overweight, and obese women. Breast Cancer Res. Treat. 2017, 163, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Dr., Abdelsalam, K.E.A.; Hassan, I.K.; Sadig, I.A. The role of developing breast cancer in alteration of serum lipid profile. J. Res. Med. Sci. 2012, 17, 562–565. [Google Scholar]
- Touvier, M.; Fassier, P.; His, M.; Norat, T.; Chan, D.S.M.; Blacher, J.; Hercberg, S.; Galan, P.; Druesne-Pecollo, N.; Latino-Martel, P. Cholesterol and breast cancer risk: A systematic review and meta-analysis of prospective studies. Br. J. Nutr. 2015, 114, 347–357. [Google Scholar] [CrossRef]
- Hilvo, M.; Denkert, C.; Lehtinen, L.; Müller, B.; Brockmöller, S.; Seppänen-Laakso, T.; Budczies, J.; Bucher, E.; Yetukuri, L.; Castillo, S.; et al. Novel theranostic opportunities offered by characterization of altered membrane lipid metabolism in breast cancer progression. Cancer Res. 2011, 71, 3236–3245. [Google Scholar] [CrossRef] [Green Version]
- Ozawa, P.M.M.; Alkhilaiwi, F.; Cavalli, I.J.; Malheiros, D.; de Souza Fonseca Ribeiro, E.M.; Cavalli, L.R. Extracellular vesicles from triple-negative breast cancer cells promote proliferation and drug resistance in non-tumorigenic breast cells. Breast Cancer Res. Treat. 2018, 172, 713–723. [Google Scholar] [CrossRef]
- O’Brien, K.; Rani, S.; Corcoran, C.; Wallace, R.; Hughes, L.; Friel, A.M.; McDonnell, S.; Crown, J.; Radomski, M.W.; O’Driscoll, L. Exosomes from triple-negative breast cancer cells can transfer phenotypic traits representing their cells f origin to secondary cells. Eur. J. Cancer 2013, 49, 1845–1859. [Google Scholar] [CrossRef]
- Lowry, M.C.; Gallagher, W.M.; O’Driscoll, L. The Role of Exosomes in Breast Cancer. Clin. Chem. 2015, 61, 1457–1465. [Google Scholar] [CrossRef] [Green Version]
- Yu, D.-d.; Wu, Y.; Shen, H.Y.; Lv, M.M.; Chen, W.X.; Zhang, X.H.; Zhong, S.L.; Tang, J.H.; Zhao, J.H. Exosomes in development, metastasis and drug resistance of breast cancer. Cancer Sci. 2015, 106, 959–964. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Freitas, D.; Kim, H.S.; Fabijanic, K.; Li, Z.; Chen, H.; Mark, M.T.; Molina, H.; Martin, A.B.; Bojmar, L.; et al. Identification of distinct nanoparticles and subsets of extracellular vesicles by asymmetric flow field-flow fractionation. Nat. Cell Biol. 2018, 20, 332–343. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, D.E.; Hornig, Y.S.; Oei, Y.; Dusich, J.; Purchio, T. Bioluminescent human breast cancer cell lines that permit rapid and sensitive in vivodetection of mammary tumors and multiple metastases in immune deficient mice. Breast Cancer Res. 2005, 7, R444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishizuka, Y. The role of protein kinase C in cell surface signal transduction and tumour promotion. Nature 1984, 308, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Nishizuka, Y. Protein kinase C and lipid signaling for sustained cellular responses. FASEB J. 2018, 9, 484–496. [Google Scholar] [CrossRef]
- Brose, N.; Betz, A.; Wegmeyer, H. Divergent and convergent signaling by the diacylglycerol second messenger pathway in mammals. Curr. Opin. Neurobiol. 2004, 14, 328–340. [Google Scholar] [CrossRef] [PubMed]
- Griner, E.M.; Kazanietz, M.G. Protein kinase C and other diacylglycerol effectors in cancer. Nat. Rev. Cancer 2007, 7, 281–294. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Czerwinski, P.; Hortmann, M.; Sohn, H.Y.; Förstermann, U.; Li, H. Protein kinase C α promotes angiogenic activity of human endothelial cells via induction of vascular endothelial growth factor. Cardiovasc. Res. 2008, 78, 349–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toker, A. The biology and biochemistry of diacylglycerol signalling. EMBO Rep. 2005, 6, 310–314. [Google Scholar] [CrossRef]
- Johannes, F.J.; Prestle, J.; Eis, S.; Oberhagemann, P.; Pfizenmaier, K. PKCu is a novel, atypical member of the protein kinase C family. J. Biol. Chem. 1994, 269, 6140–6148. [Google Scholar]
- Rozengurt, E.; Rey, O.; Waldron, R.T. Protein kinase D signaling. J. Biol. Chem. 2005, 280, 13205–13208. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.J. PKD at the crossroads of DAG and PKC signaling. Trends Pharmacol. Sci. 2006, 27, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Ha, C.H.; Jin, Z.G. Protein kinase D1, a new molecular player in VEGF signaling and angiogenesis. Mol. Cells 2009, 28, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, I.M.; Zachary, I.C. Protein kinase D in vascular biology and angiogenesis. IUBMB Life 2011, 63, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.; Jin, Z.G. Protein kinase C-dependent protein kinase D activation modulates ERK signal pathway and endothelial cell proliferation by vascular endothelial growth factor. J. Biol. Chem. 2005, 280, 33262–33269. [Google Scholar] [CrossRef] [Green Version]
- Ha, C.H.; Wang, W.; Jhun, B.S.; Wong, C.; Hausser, A.; Pfizenmaier, K.; McKinsey, T.A.; Olson, E.N.; Jin, Z.-G. Protein Kinase D-dependent Phosphorylation and Nuclear Export of Histone Deacetylase 5 Mediates Vascular Endothelial Growth Factor-induced Gene Expression and Angiogenesis. J. Biol. Chem. 2008, 283, 14590–14599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, Q.; Wang, L.; Joe Zhao, Z.; Tang, H. Identification of protein kinase D2 as a pivotal regulator of endothelial cell proliferation, migration, and angiogenesis. J. Biol. Chem. 2009, 284, 799–806. [Google Scholar] [CrossRef] [Green Version]
- Qin, L.; Zeng, H.; Zhao, D. Requirement of protein kinase D tyrosine phosphorylation for VEGF-A 165-induced angiogenesis through its interaction and regulation of phospholipase Cγ phosphorylation. J. Biol. Chem. 2006, 281, 32550–32558. [Google Scholar] [CrossRef] [Green Version]
- Ren, B. Protein Kinase D1 Signaling in Angiogenic Gene Expression and VEGF-Mediated Angiogenesis. Front. Cell Dev. Biol. 2016, 4, 37. [Google Scholar] [CrossRef] [Green Version]
- Stahelin, R.V.; Digman, M.A.; Medkova, M.; Ananthanarayanan, B.; Rafter, J.D.; Melowic, H.R.; Cho, W. Mechanism of diacylglycerol-induced membrane targeting and activation of protein kinase Cδ. J. Biol. Chem. 2004, 279, 29501–29512. [Google Scholar] [CrossRef] [Green Version]
- Tinsley, J.H.; Teasdale, N.R.; Yuan, S.Y. Involvement of PKCδ and PKD in pulmonary microvascular endothelial cell hyperpermeability. Am. J. Physiol. Physiol. 2004, 286, C105–C111. [Google Scholar] [CrossRef] [Green Version]
- Takeda, H.; Izumi, Y.; Takahashi, M.; Paxton, T.; Tamura, S.; Koike, T.; Yu, Y.; Kato, N.; Nagase, K.; Shiomi, M.; et al. Widely-targeted quantitative lipidomics method by supercritical fluid chromatography triple quadrupole mass spectrometry. J. Lipid Res. 2018, 59, 1283–1293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishida-Aoki, N.; Tominaga, N.; Takeshita, F.; Sonoda, H.; Yoshioka, Y.; Ochiya, T. Disruption of Circulating Extracellular Vesicles as a Novel Therapeutic Strategy against Cancer Metastasis. Mol. Ther. 2017, 25, 181–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, C.A.; Purushothaman, A.; Ramani, V.C.; Vlodavsky, I.; Sanderson, R.D. Heparanase regulates secretion, composition, and function of tumor cell-derived exosomes. J. Biol. Chem. 2013, 288, 10093–10099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maji, S.; Chaudhary, P.; Akopova, I.; Nguyen, P.M.; Hare, R.J.; Gryczynski, I.; Vishwanatha, J.K. Exosomal Annexin II Promotes Angiogenesis and Breast Cancer Metastasis. Mol. Cancer Res. 2017, 15, 93–105. [Google Scholar] [CrossRef] [Green Version]
- Gangoda, L.; Liem, M.; Ang, C.-S.; Keerthikumar, S.; Adda, C.G.; Parker, B.S.; Mathivanan, S. Proteomic Profiling of Exosomes Secreted by Breast Cancer Cells with Varying Metastatic Potential. Proteomics 2017, 17, 1600370. [Google Scholar] [CrossRef]
- Mori, T.; Takai, Y.; Yu, B.; Takahashi, J.; Nishizuka, Y.; Fujikura, T. Specificity of the fatty acyl moieties of diacylglycerol for the activation of calcium-activated, phospholipid-dependent protein kinase. J. Biochem. 1982, 91, 427–432. [Google Scholar] [CrossRef]
- Kaibuchi, K.; Takai, Y.; Sawamura, M.; Hoshijima, M.; Fujikura, T.; Nishizuka, Y. Synergistic functions of protein phosphorylation and calcium mobilization in platelet activation. J. Biol. Chem. 1983, 258, 6701–6704. [Google Scholar]
- Iglesias, T.; Waldron, R.T.; Rozengurt, E. Identification of in vivo phosphorylation sites required for protein kinase D activation. J. Biol. Chem. 1998, 273, 27662–27667. [Google Scholar] [CrossRef] [Green Version]
- Matthews, S.A.; Rozengurt, E.; Cantrell, D. Characterization of serine 916 as an in vivo autophosphorylation site for protein kinase D/protein kinase Cμ. J. Biol. Chem. 1999, 274, 26543–26549. [Google Scholar] [CrossRef] [Green Version]
- Phuyal, S.; Skotland, T.; Hessvik, N.P.; Simolin, H.; Øverbye, A.; Brech, A.; Parton, R.G.; Ekroos, K.; Sandvig, K.; Llorente, A. The ether lipid precursor hexadecylglycerol stimulates the release and changes the composition of exosomes derived from PC-3 cells. J. Biol. Chem. 2015, 290, 4225–4237. [Google Scholar] [CrossRef] [Green Version]
- Fan, T.W.M.; Zhang, X.; Wang, C.; Yang, Y.; Kang, W.Y.; Arnold, S.; Higashi, R.M.; Liu, J.; Lane, A.N. Exosomal lipids for classifying early and late stage non-small cell lung cancer. Anal. Chim. Acta 2018, 1037, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Brzozowski, J.S.; Jankowski, H.; Bond, D.R.; McCague, S.B.; Munro, B.R.; Predebon, M.J.; Scarlett, C.J.; Skelding, K.A.; Weidenhofer, J. Lipidomic profiling of extracellular vesicles derived from prostate and prostate cancer cell lines. Lipids Health Dis. 2018, 17, 211. [Google Scholar] [CrossRef] [Green Version]
- Lydic, T.A.; Townsend, S.; Adda, C.G.; Collins, C.; Mathivanan, S.; Reid, G.E. Rapid and comprehensive “shotgun” lipidome profiling of colorectal cancer cell derived exosomes. Methods 2015, 87, 83–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skotland, T.; Hessvik, N.P.; Sandvig, K.; Llorente, A. Exosomal lipid composition and the role of ether lipids and phosphoinositides in exosome biology. J. Lipid Res. 2019, 60, 9–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Record, M.; Carayon, K.; Poirot, M.; Silvente-Poirot, S. Exosomes as new vesicular lipid transporters involved in cell-cell communication and various pathophysiologies. Biochim. Biophys. Acta 2014, 1841, 108–120. [Google Scholar] [CrossRef] [PubMed]
- Subra, C.; Grand, D.; Laulagnier, K.; Stella, A.; Lambeau, G.; Paillasse, M.; De Medina, P.; Monsarrat, B.; Perret, B.; Silvente-Poirot, S.; et al. Exosomes account for vesicle-mediated transcellular transport of activatable phospholipases and prostaglandins. J. Lipid Res. 2010, 51, 2105–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Podbielska, M.; Szulc, Z.M.; Kurowska, E.; Hogan, E.L.; Bielawski, J.; Bielawska, A.; Bhat, N.R. Cytokine-induced release of ceramide-enriched exosomes as a mediator of cell death signaling in an oligodendroglioma cell line. J. Lipid Res. 2016, 57, 2028–2039. [Google Scholar] [CrossRef] [Green Version]
- Kakazu, E.; Mauer, A.S.; Yin, M.; Malhi, H. Hepatocytes release ceramide-enriched pro-inflammatory extracellular vesicles in an IRE1α-dependent manner. J. Lipid Res. 2016, 57, 233–245. [Google Scholar] [CrossRef] [Green Version]
- Almena, M.; Mérida, I. Shaping up the membrane: Diacylglycerol coordinates spatial orientation of signaling. Trends Biochem. Sci. 2011, 36, 593–603. [Google Scholar] [CrossRef]
- Spitaler, M.; Cantrell, D.A. Protein kinase C and beyond. Nat. Immunol. 2004, 5, 785–790. [Google Scholar] [CrossRef]
- Park, J.-E.; Kim, Y.-I.; Yi, A.-K. Protein Kinase D1: A New Component in TLR9 Signaling. J. Immunol. 2008, 181, 2044–2055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Gera, N.; Li, H.; Yun, M.; Zhang, L.; Wang, Y.; Wang, Q.J.; Jin, T. GPCR-mediated PLCβγ/PKCβ/PKD signaling pathway regulates the cofilin phosphatase slingshot 2 in neutrophil chemotaxis. Mol. Biol. Cell 2015, 26, 874–886. [Google Scholar] [CrossRef] [PubMed]
- Davidson-Moncada, J.K.; Lopez-Lluch, G.; Segal, A.W.; Dekker, L.V. Involvement of protein kinase D in Fcγ-receptor activation of the NADPH oxidase in neutrophils. Biochem. J. 2002, 363, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Bligh, E.G.; Dyer, W.J. A Rapid Method of Total Lipid Extraction and Purification. Can. J. Biochem. Physiol. 2010, 37, 911–917. [Google Scholar] [CrossRef] [Green Version]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nishida-Aoki, N.; Izumi, Y.; Takeda, H.; Takahashi, M.; Ochiya, T.; Bamba, T. Lipidomic Analysis of Cells and Extracellular Vesicles from High- and Low-Metastatic Triple-Negative Breast Cancer. Metabolites 2020, 10, 67. https://doi.org/10.3390/metabo10020067
Nishida-Aoki N, Izumi Y, Takeda H, Takahashi M, Ochiya T, Bamba T. Lipidomic Analysis of Cells and Extracellular Vesicles from High- and Low-Metastatic Triple-Negative Breast Cancer. Metabolites. 2020; 10(2):67. https://doi.org/10.3390/metabo10020067
Chicago/Turabian StyleNishida-Aoki, Nao, Yoshihiro Izumi, Hiroaki Takeda, Masatomo Takahashi, Takahiro Ochiya, and Takeshi Bamba. 2020. "Lipidomic Analysis of Cells and Extracellular Vesicles from High- and Low-Metastatic Triple-Negative Breast Cancer" Metabolites 10, no. 2: 67. https://doi.org/10.3390/metabo10020067