Structural Stability, Electronic Structures, Mechanical Properties and Debye Temperature of Transition Metal Impurities in Tungsten: A First-Principles Study


Abstract
:1. Introduction
2. Model and Computational Details
3. Results and Discussion
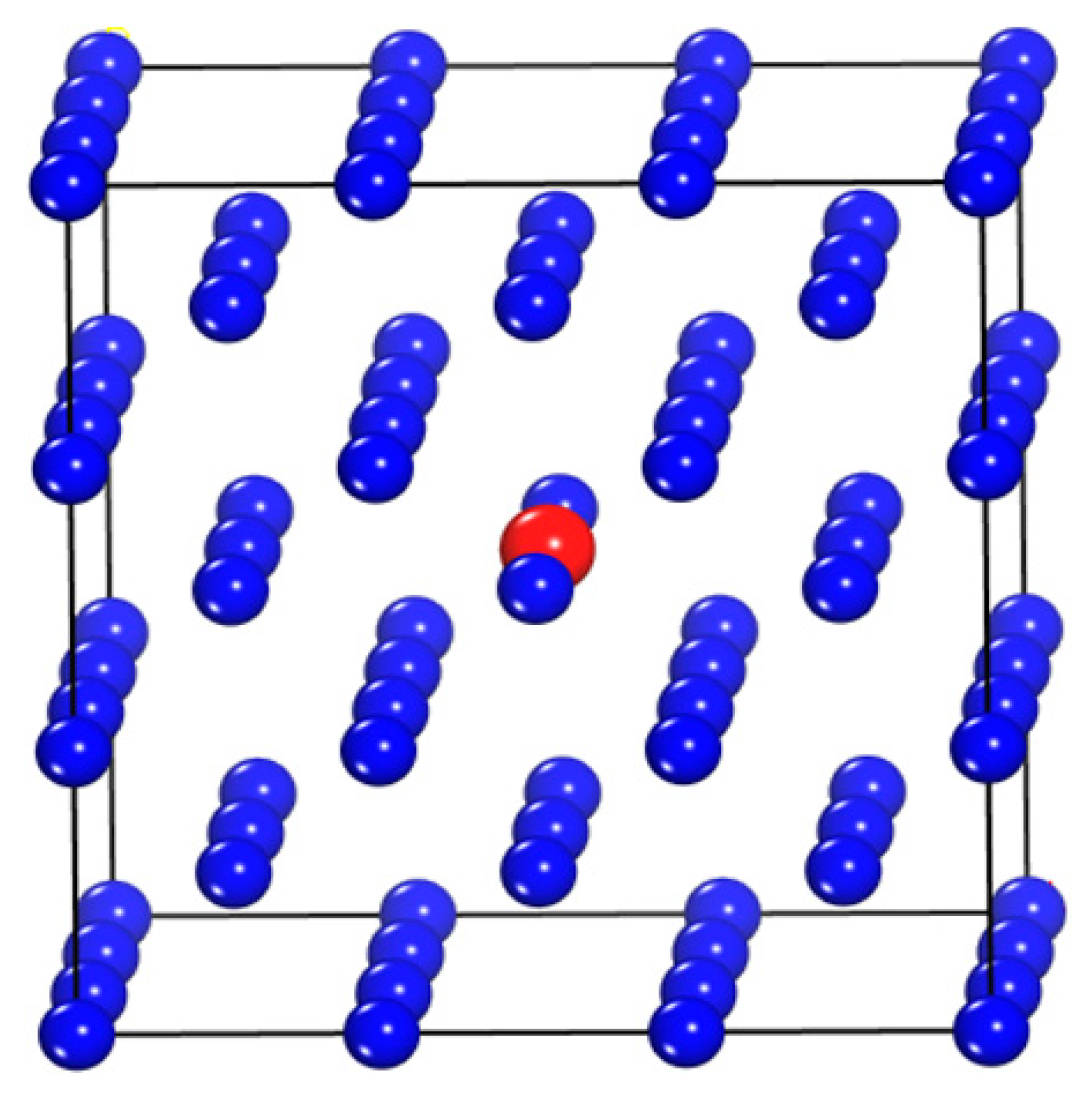
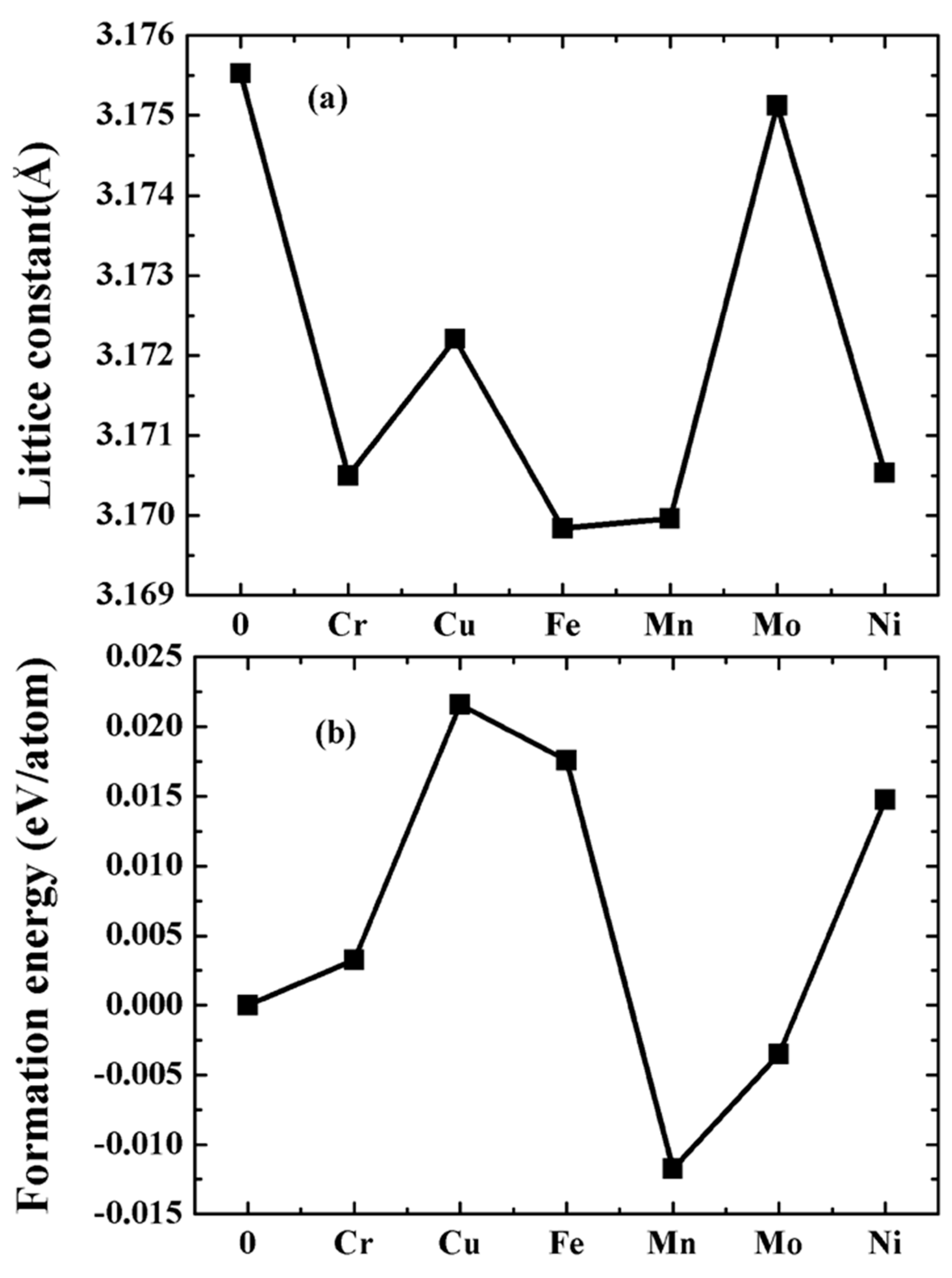
3.1. Crystal Configurations and Lattice Constants
3.2. Formation and Cohesive Energies
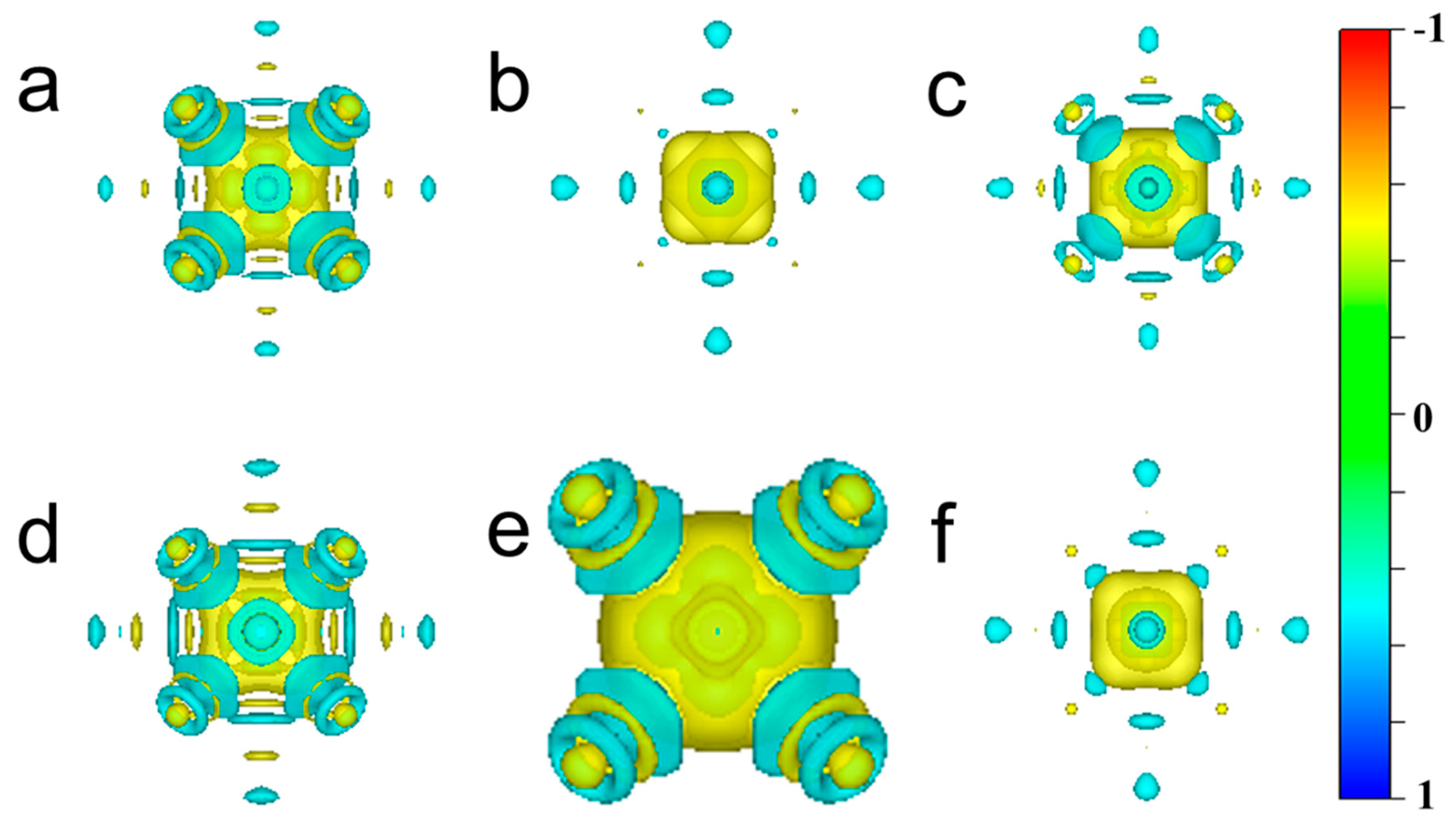
3.3. Charge Density
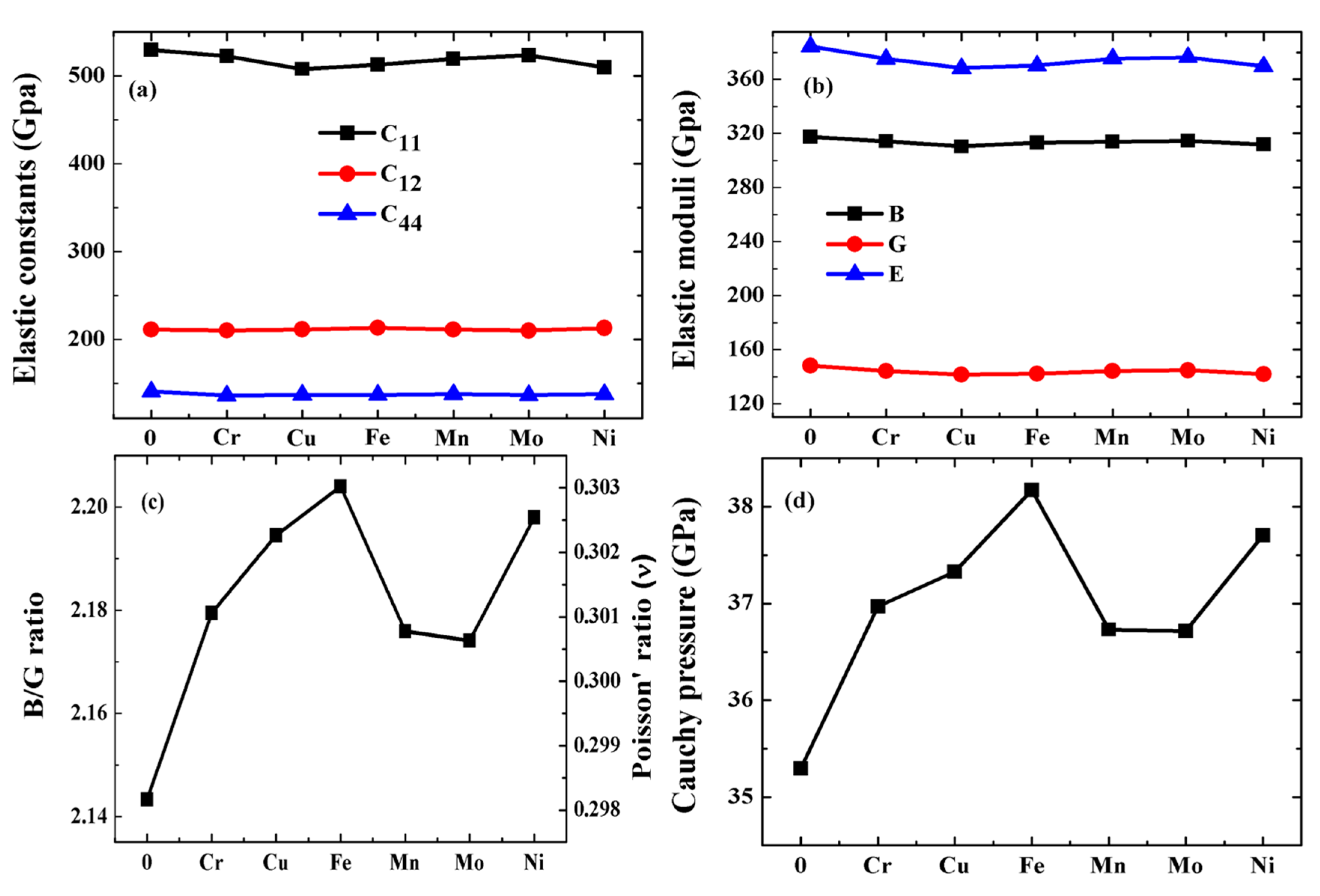
3.4. Mechanical Properties
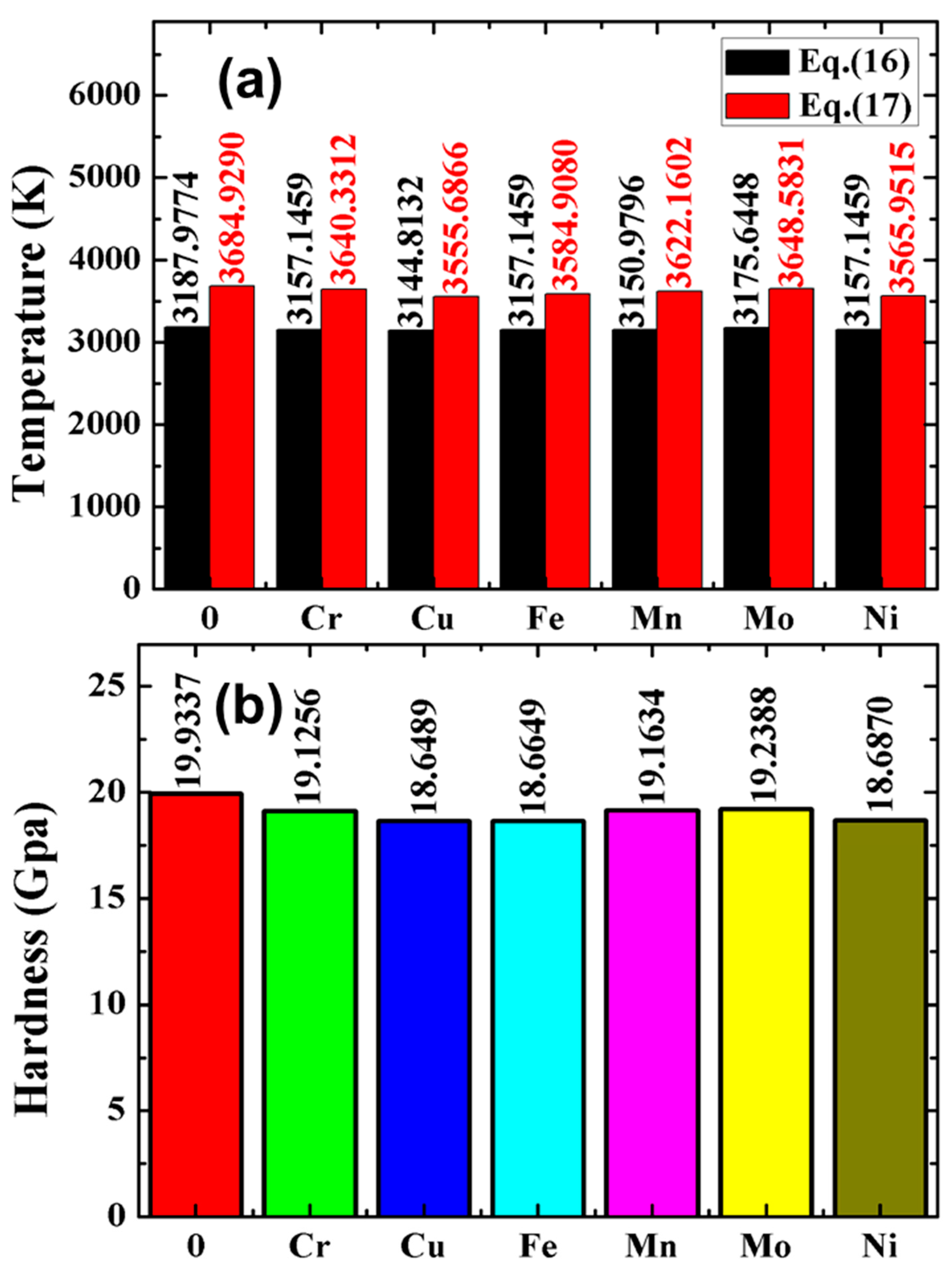
3.5. Melting Point and Hardness
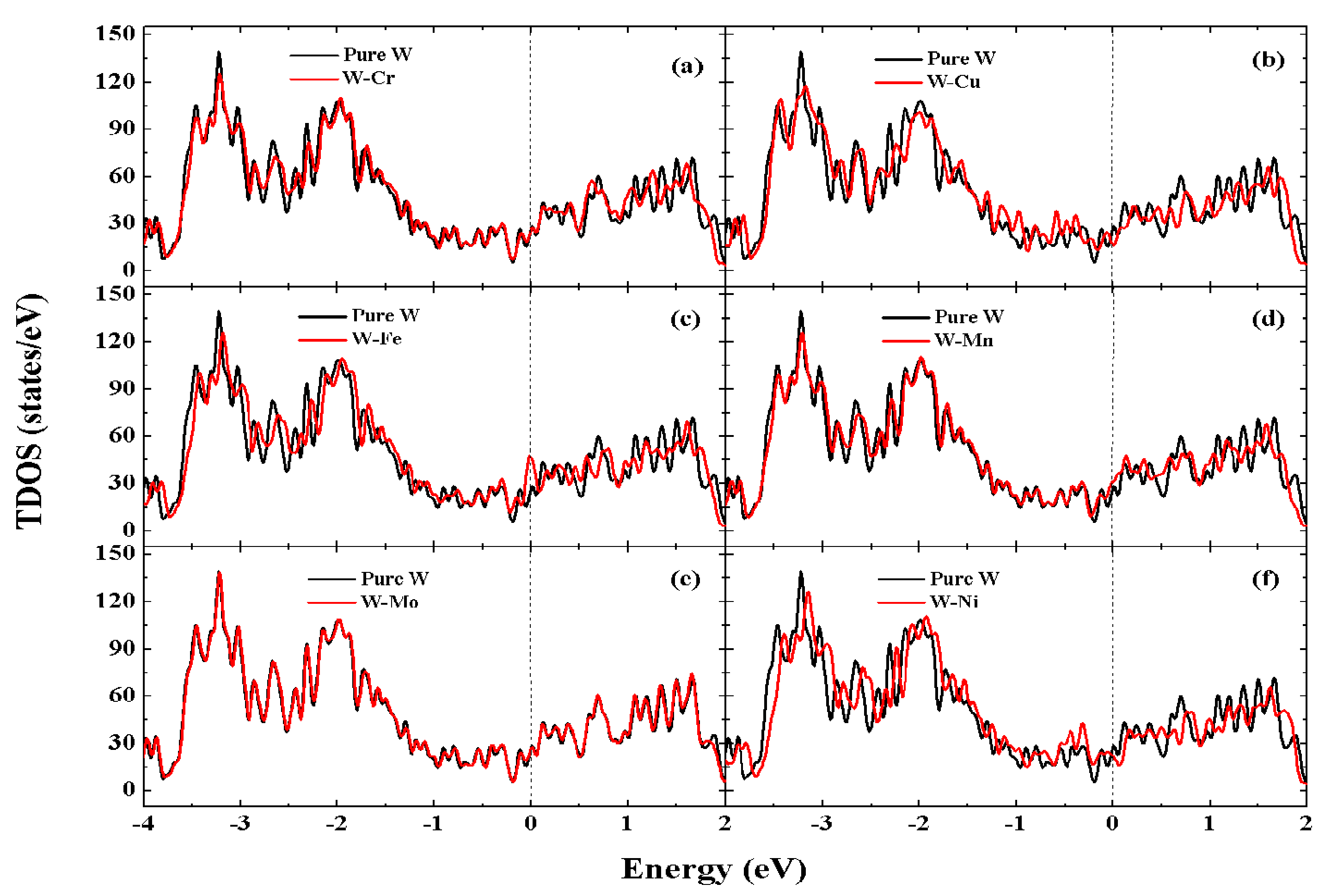
3.6. Electronic Structures
3.7. Debye Temperature
4. Summaries and Conclusions
- The W-TM alloys still maintain the bcc lattice, and have no structural phase transformation. Of note, however, the lattice constants of the W-Cu and W-Ni alloys are anomalous than that of other alloys. The lattice constants of the W-Cu and W-Ni alloys are higher than that of the W-Cr, W-Fe and W-Mn alloys.
- The W-Mo and W-Mn alloys have better alloying ability with strong interactions between W and Mo/Mn atoms. However, the alloying ability of the W-Cu, W-Fe, W-Cr and W-Ni is poor, and there is a weak chemical interaction between W and Cu/Cr/Fe/Ni atoms.
- The impurity elements Cr, Cu, Fe, Mn, Mo and Ni all affect the mechanical strength of pure tungsten metal, especially Cu and Ni. Fe element performs well in improving the ductility of pure W metal, followed by Ni, Cu, Cr and Mn, and the worst is Mo. The impurities Cu, Fe, Mn and Ni can improve the anisotropy of pure tungsten metal, while the impurities Cr and Mo result in more serious anisotropy of pure tungsten metal.
- The transition metal Cu impurity has the greatest influence on the melting point and hardness of pure W metal, while the transition metal Mo impurity has the least effect.
- The metallic bonding of the W-TM alloys is strengthened while covalent bonding is reduced. The metallicity of pure W metal can be enhanced by doping it with Fe or Mn, while doping with Cr, Cu, Mo and Ni reduces the metallicity of pure W metal, of which the W-Cu alloy has the worst metallicity.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tanabe, T.; Noda, N.; Nakamura, H. Review of high Z materials for PSI applications. J. Nucl. Mater. 1992, 196, 11–27. [Google Scholar] [CrossRef]
- Garcia-Rosales, C. Erosion processes in plasma-wall interactions. J. Nucl. Mater. 1994, 211, 202–214. [Google Scholar] [CrossRef]
- Chuyanov, V.A. ITER EDA project status. J. Nucl. Mater. 1996, 233, 4–8. [Google Scholar] [CrossRef]
- Ekman, M.; Persson, K.; Grimvall, G. Phase diagram and lattice instability in tungsten rhenium Alloys. J. Nucl. Mater. 2000, 278, 273–276. [Google Scholar] [CrossRef]
- Kim, Y.D.; Oh, N.L.; Oh, S.T.; Moon, I.H. Thermal conductivity of W-Cu composites at various Temperatures. Mater. Lett. 2001, 51, 420–424. [Google Scholar] [CrossRef]
- Dosovitskiy, G.A.; Samoilenkov, S.V. Thermal expansion of Ni-W, Ni-Cr, and Ni-Cr-W alloys between room temperature and 800 degrees c. Int. J. Thermophys. 2009, 30, 1931–1937. [Google Scholar] [CrossRef]
- Liu, B.X.; Huang, L.J.; Geng, L.; Wang, B.; Liu, C.; Zhang, W.C. Fabrication and superior ductility of laminated Ti-TiBW/Ti composites by diffusion welding. J. Alloys Compd. 2014, 602, 187–192. [Google Scholar] [CrossRef]
- Mutoh, Y.; Ichikawa, K.; Nagata, K.; Takeuchi, M. Effect of rhenium addition on fracture toughness of tungsten at elevated temperatures. J. Mater. Sci. 1995, 30, 770–775. [Google Scholar] [CrossRef]
- Jiang, D.Y.; Ouyang, C.Y.; Liu, S.Q. Mechanical properties of W-Ti alloys from first-principles Calculations. Fusion Eng. Des. 2016, 106, 34–39. [Google Scholar]
- Jiang, D.Y.; Wang, Q.L.; Hu, W.; Wei, Z.Q.; Tong, J.B.; Wan, H.Q. The effect of tantalum (Ta) doping on mechanical properties of tungsten (W): A first-principles study. J. Mater. Res. 2016, 31, 3401–3408. [Google Scholar] [CrossRef]
- Jiang, D.Y.; Zhou, Q.; Xue, L.; Wang, T.; Hu, J.F. First-principles study the phase stability and mechanical properties of binary W-Mo alloys. Fusion Eng. Des. 2018, 130, 56–61. [Google Scholar] [CrossRef]
- Jiang, D.Y.; Wang, T.; Huang, X.H.; Zou, X.Z.; Hu, J.F. Effect of Hf additions on phase stability and mechanical properties of binary W-Hf alloys: A first-principles study. Fusion Eng. Des. 2018, 137, 295–302. [Google Scholar] [CrossRef]
- Jiang, D.Y.; Zhou, Q.; Liu, W.H.; Wang, T.; Hu, J.F. First-principles study the structures and mechanical properties of binary W-V alloys. Phys. B Condens. Matter 2019, 552, 165–169. [Google Scholar] [CrossRef]
- Jiang, D.Y.; Xue, L.; Huang, X.M.; Wang, T.; Hu, J.F. Effect of Zr additions on crystal structures and mechanical properties of binary W-Zr alloys: A first-principles study. J. Mater. Res. 2019, 34, 290–300. [Google Scholar]
- Fukuzumi, S.; Yoshiie, T.; Satoh, Y.; Xu, Q.; Mori, H.; Kawai, M. Defect structural evolution in high purity tungsten irradiated with electrons using high voltage electron microscope. J. Nucl. Mater. 2005, 343, 308–312. [Google Scholar] [CrossRef]
- Kong, X.S.; Wu, X.B.; You, Y.W.; Liu, C.S.; Fang, Q.F.; Chen, J.L.; Luo, G.N.; Wang, Z.G. First-principles calculations of transition metal-solute interactions with point defects in tungsten. Acta Mater. 2014, 66, 172–183. [Google Scholar] [CrossRef]
- Qi, L.; Jin, Y.C.; Zhao, Y.H.; Yang, X.M.; Zhao, H.; Han, P.D. The structural, elastic, electronic properties and Debye temperature of Ni3Mo under pressure from first-principles. J. Alloys Compd. 2015, 621, 383–388. [Google Scholar] [CrossRef]
- Chen, Q.; Huang, Z.W.; Zhao, Z.D.; Hu, C.K. Thermal stabilities, elastic properties and electronic structures of B2-MgRE (RE = Sc, Y, La) by first-principles calculations. Comput. Mater. Sci. 2013, 67, 196–202. [Google Scholar] [CrossRef]
- Muzyk, M.; Nguyen-Manh, D.; Kurzydłowski, K.J.; Baluc, N.L.; Dudarev, S.L. Phase stability, point defects, and elastic properties of W-V and W-Ta alloys. Phys. Rev. B 2011, 84, 104115. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.J.; Shang, S.L.; Wang, Y.; Darling, K.A.; Butler, B.G.; Kecskes, L.J.; Liu, Z.K. Effects of alloying elements and temperature on the elastic properties of W-based alloys by first-principles calculations. J. Alloys Comp. 2016, 671, 267–275. [Google Scholar] [CrossRef] [Green Version]
- Giusepponi, S.; Celino, M. The ideal tensile strength of tungsten and tungsten alloys by first-principles calculations. J. Nucl. Mater. 2013, 435, 52–55. [Google Scholar] [CrossRef]
- Muzyk, M.; Nguyen-Manh, D.; Wróbel, J.; Kurzydłowski, K.J.; Baluc, N.L.; Dudarev, S.L. First-principles model for phase stability, radiation defects and elastic properties of W-Ta and W-V alloys. J. Nucl. Mater. 2013, 442, S680–S683. [Google Scholar] [CrossRef]
- Wei, N.; Jia, T.; Zhang, X.L.; Liu, T.; Zeng, Z.; Yang, X.Y. First-principles study of the phase stability and the mechanical properties of W-Ta and W-Re alloys. AIP Adv. 2014, 4, 057103. [Google Scholar] [CrossRef]
- Bertoldia, D.S.; Ramosa, S.B.; Guillermet, A.F. Interrelations between EOS parameters and cohesive energy of transition metals: Thermostatistical approach, ab initio calculations and analysis of “universality” features. J. Phys. Chem. Solids 2017, 107, 93–97. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for open-shell transition metals. Phys. Rev. B 1993, 48, 13115. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef] [PubMed]
- Blӧchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Perdew, J.P. Correlation hole of the spin-polarized electron gas, with exact small-wave-vector and high-density scaling. Phys. Rev. B 1991, 44, 13298. [Google Scholar] [CrossRef] [PubMed]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188. [Google Scholar] [CrossRef]
- Wallace, D.C. Thermoelastic Theory of Stressed Crystals and Higher-Order Elastic Constants. Solid State Phys. 1970, 25, 301–404. [Google Scholar]
- Zhao, J.J.; Winey, J.M.; Gupta, Y.M. First principles calculations of second- and third-order elastic constants for single crystals of arbitrary symmetry. Phys. Rev. B 2007, 75, 094105. [Google Scholar] [CrossRef]
- Voigt, W. über die Beziehung zwischen den beiden Elasticitӓts constanten isotroper Kӧrper. Ann. Physik 1889, 38, 573–587. [Google Scholar] [CrossRef]
- Reuss, A. Berechnung der Flieβgrenze von Mischkristallen auf Grupd der Plastizitäts bedingung für Einkristalle. Zamm Z. Angew. Math. Mech. 1929, 9, 49–58. [Google Scholar] [CrossRef]
- Hill, R. The elastic behaviour of a crystalline aggregate. Phys. Soc. Lond. Sect. A 1952, 65, 349–354. [Google Scholar] [CrossRef]
- Hill, R. Elastic properties of reinforced solids: Some theoretical principles. J. Mech. Phys. Solids 1963, 11, 357–372. [Google Scholar] [CrossRef]
- Anderson, O.L. A simplified method for calculating the Debye temperature from elastic constants. J. Phys. Chem. Solids 1963, 24, 909–917. [Google Scholar] [CrossRef]
- Wachter, P.; Filzmoser, M.; Rebizant, J. Electronic and elastic properties of the light actinide tellu-rides. Physica B 2001, 293, 199–223. [Google Scholar] [CrossRef]
- Jin, S.; Liu, Y.L.; Zhou, H.B.; Zhang, Y.; Lu, G.H. First-principles investigation on the effect of carbon on hydrogen trapping in tungsten. J. Nucl. Mater. 2011, 415, 709–712. [Google Scholar] [CrossRef]
- Kittel, B.C. Introduction to Solid State Physics, 7th ed.; Wiley: New York, NY, USA, 1996. [Google Scholar]
- Stathis, J.H.; Bolef, D.I. Elastic constants of tungsten between 4.2 and 77 K. J. Appl. Phys. 1980, 51, 4770. [Google Scholar] [CrossRef]
- Featherston, F.H.; Neighbors, J.R. Elastic Constants of tantalum, tungsten, and molybdenum. Phys. Rev. 1963, 130, 1324. [Google Scholar] [CrossRef]
- Sahu, B.R. Electronic structure and bonding of ultralight LiMg. Mater. Sci. Eng. B 1997, 49, 74–78. [Google Scholar] [CrossRef]
- Didukh, L. 3d-electrons contribution to cohesive energy of 3d-metals. Condens. Matter Phys. 2018, 21, 13701. [Google Scholar] [CrossRef]
- Zhang, Y.W.; Li, S.J.; Obbard, E.G.; Wang, H.; Wang, S.C.; Hao, Y.L.; Yang, R. Elastic properties of Ti-24Nb-4Zr-SSn single crystals with bcc crystal structure. Acta Mater. 2011, 59, 3081–3090. [Google Scholar] [CrossRef]
- Soderlind, P.; Eriksson, O.; Wills, J.M.; Boring, A.M. Theory of elastic constants of cubic transition metals and alloys. Phys. Rev. B 1993, 48, 5844–5851. [Google Scholar] [CrossRef] [PubMed]
- Einarsdotter, K.; Sadigh, B.; Grimvall, G.; Ozolins, V. Phonon Instabilities in fcc and bcc Tungsten. Phys. Rev. Lett. 1997, 79, 2073–2076. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.J.; Zhao, E.J.; Xiang, H.P.; Hao, X.F.; Liu, X.J.; Meng, J. Crystal structures and elastic properties of superhard IrN2 and IrN3 from first principles. Phys. Rev. B 2007, 76, 054115. [Google Scholar] [CrossRef]
- Liu, Q.J.; Liu, Z.T.; Feng, L.P.; Tian, H. First-principles study of structural, elastic, electronic and optical properties of orthorhombic NaAlF4. Comput. Mater. Sci. 2011, 50, 2822–2827. [Google Scholar] [CrossRef]
- Meradji, H.; Drablia, S.; Ghemid, S. First-principles elastic constants and electronic structure of BP, BAs, and BSb. Phys. Status Solidi B 2004, 241, 2881–2885. [Google Scholar] [CrossRef]
- Pugh, S.F. Relations between the elastic moduli and the plastic properties of polycrystalline pure Metals. Philos. Mag. A 1954, 45, 823–843. [Google Scholar] [CrossRef]
- Cao, Y.; Zhu, J.C.; Liu, Y.; Nong, Z.S.; Lai, Z.H. First-principles studies of the structural, elastic, electronic and thermal properties of Ni3Si. Comput. Mater. Sci. 2013, 69, 40–45. [Google Scholar] [CrossRef]
- Tang, B.Y.; Yu, W.Y.; Zeng, X.Q.; Ding, W.J.; Gray, M.F. First-principles study of the electronic structure and mechanical properties of CaMg2 Laves Phase. Mater. Sci. Eng. A 2008, 489, 444–450. [Google Scholar] [CrossRef]
- Li, C.H.; Hoe, J.L.; Wu, P. Empirical correlation between melting temperature and cohesive energy of binary Laves Phases. J. Phys. Chem. Solids 2003, 64, 201–212. [Google Scholar] [CrossRef]
- Xu, S.H.; Zhang, F.Q.; Peng, P.; Liu, J.S. First-principles calculations of structural stabilities and elastic properties of AB2 type intermetallics in ZA62 magnesium alloy. Acta Met. Sin. 2010, 46, 97–103. [Google Scholar]
- Coenen, J.W.; Philipps, V.; Brezinsek, S.; Pintsuk, G.; Tanabe, T.; Ueda, Y.; Samm, U.; TEXTOR Team. Analysis of structural changes and high-heat-flux tests on pre-damaged tungsten from tokamak melt experiments. Phys. Scr. 2011, T145, 014066. [Google Scholar] [CrossRef]
- Coenen, J.W.; Arnoux, G.; Bazylev, B.; Matthews, G.F.; Autricque, A.; Balboa, I.; Clever, M.; Dejarnac, R.; Coffey, I.; Corre, Y.; et al. ELM-induced transient tungsten melting in the JET divertor. Nucl. Fusion 2015, 55, 023010. [Google Scholar] [CrossRef]
- Richardson, R.C.D. The wear of metals by hard abrasives. Wear 1967, 10, 291–309. [Google Scholar] [CrossRef]
- Ji, Z.W.; Hu, C.H.; Wand, D.H. Mechanical ProPerties and chemical bonding of the Os-B system: A first-Principles study. Acta Mater. 2012, 60, 4208–4217. [Google Scholar] [CrossRef]
- Shi, Y.J.; Du, Y.L.; Chen, G.; Chen, G.L. First principle study on phase stability and electronic structure of YCu. Phys. Lett. A 2007, 368, 495–498. [Google Scholar] [CrossRef]
- Hu, Q.M.; Yang, R.; Xu, D.S.; Hao, Y.L.; Li, D.; Wu, W.T. Energetics and electronic structure of grain boundaries and surfaces of B- and H-doped Ni3Al. Phys. Rev. B 2003, 67, 224203. [Google Scholar] [CrossRef]
- Yang, X.M.; Hou, H.; Zhao, Y.H.; Yang, L.; Han, P.D. Preparation of lamellar carbon matrix for sulfur as cathode material of lithium-sulfur batteries. Comput. Mater. Sci. 2014, 84, 374–382. [Google Scholar] [CrossRef]
- Yan, M.F.; Chen, H.T. Structural, elastic and electronic properties of Cr2N: A first-principles study. Comput. Mater. Sci. 2014, 88, 81–85. [Google Scholar] [CrossRef]
- Li, Y.F.; Gao, Y.M.; Xiao, B. Theoretical study on the stability, elasticity, hardness and electronic structures of W-C binary compounds. J. Alloys Comp. 2010, 502, 28–37. [Google Scholar] [CrossRef]
- Huang, Z.W.; Zhao, Y.H.; Hou, H.; Han, P.D. Electronic structural, elastic properties and thermodynamics of Mg17Al12, Mg2Si and A12Y phases from first-principles calculations. Physica B 2012, 407, 1075–1081. [Google Scholar] [CrossRef]
- Yu, C.F.; Jiang, X.F.; Cheng, P.F.; Zhu, C.J. Correlations among linear expansion coefficient, Debye temperature and Young’s modulus of metals. Phys. Exp. 2012, 32, 37–40. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Change of Total Energy |
|---|---|
| Composition | C11 (GPa) | C12 (GPa) | C44 (GPa) | V (Å3) | LC (Å) | Ef (eV/atom) | |
|---|---|---|---|---|---|---|---|
| Pure W | 529.937 | 211.189 | 140.594 | 32.022 | 3.1755 | 0.00000 | 8.58829 |
| Experiment | 533.9 [40] | 205.1 [40] | 163.3 [40] | 3.165 [40] | |||
| Experiment | 532.55 [41] | 204.95 [41] | 163.13 [41] | 3.165 [41] | |||
| Experiment | 533 [45] | 205 [45] | 163 [45] | ||||
| Theory | 553 [45,46] | 207 [45,46] | 163 [45,46] | 8.45 [24] | |||
| W53Cr1 | 522.391 | 210.153 | 136.216 | 31.870 | 3.1705 | 0.00324 | 8.50110 |
| W53Cu1 | 508.069 | 211.512 | 136.858 | 31.921 | 3.1722 | 0.02157 | 8.46980 |
| W53Fe1 | 513.013 | 213.208 | 136.862 | 31.850 | 3.1698 | 0.01760 | 8.50635 |
| W53Mn1 | 519.316 | 211.177 | 137.715 | 31.853 | 3.1700 | -0.01173 | 8.49174 |
| W53Mo1 | 523.787 | 210.048 | 136.617 | 32.010 | 3.1751 | -0.00352 | 8.55048 |
| W53Ni1 | 509.806 | 212.978 | 137.574 | 31.871 | 3.1705 | 0.01477 | 8.50091 |
| Composition | B (GPa) | G (GPa) | E (GPa) | B/G | ν | C’ (GPa) | A |
| Pure W | 317.438 | 148.106 | 384.518 | 2.1433 | 0.2981 | 35.2972 | 0.882 |
| W53Cr1 | 314.232 | 144.177 | 375.155 | 2.1795 | 0.3010 | 36.9685 | 0.873 |
| W53Cu1 | 310.364 | 141.426 | 368.332 | 2.1945 | 0.3022 | 37.3271 | 0.923 |
| W53Fe1 | 313.143 | 142.078 | 370.240 | 2.2040 | 0.3029 | 38.1730 | 0.913 |
| W53Mn1 | 313.890 | 144.257 | 375.280 | 2.1759 | 0.3007 | 36.7310 | 0.894 |
| W53Mo1 | 314.627 | 144.718 | 376.439 | 2.1741 | 0.3006 | 36.7151 | 0.871 |
| W53Ni1 | 311.920 | 141.910 | 369.669 | 2.1980 | 0.3025 | 37.7018 | 0.927 |
| Composition | ρ (g·cm−3) | M (g/mol) | Acoustic Velocity (m·s−1) | ΘD (K) | |||
|---|---|---|---|---|---|---|---|
| vs | vl | vm | This Work | Ref. [65] | |||
| Pure W | 19.0738 | 183.8400 | 2786.56 | 5195.75 | 3111.78 | 367.579 | 333.4 |
| W53Cr1 | 18.9097 | 181.3984 | 2761.25 | 5175.28 | 3084.65 | 364.949 | — |
| W53Cu1 | 18.9015 | 181.6122 | 2735.38 | 5137.75 | 3056.21 | 361.389 | — |
| W53Fe1 | 18.9288 | 181.4696 | 2739.70 | 5152.78 | 3061.32 | 362.263 | — |
| W53Mn1 | 18.9251 | 181.4529 | 2760.89 | 5171.97 | 3084.14 | 364.951 | — |
| W53Mo1 | 18.9117 | 182.2122 | 2766.28 | 5180.71 | 3090.11 | 365.062 | — |
| W53Ni1 | 18.9220 | 181.5224 | 2738.57 | 5146.28 | 3059.88 | 362.014 | — |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, D.; Wu, M.; Liu, D.; Li, F.; Chai, M.; Liu, S. Structural Stability, Electronic Structures, Mechanical Properties and Debye Temperature of Transition Metal Impurities in Tungsten: A First-Principles Study. Metals 2019, 9, 967. https://doi.org/10.3390/met9090967
Jiang D, Wu M, Liu D, Li F, Chai M, Liu S. Structural Stability, Electronic Structures, Mechanical Properties and Debye Temperature of Transition Metal Impurities in Tungsten: A First-Principles Study. Metals. 2019; 9(9):967. https://doi.org/10.3390/met9090967
Chicago/Turabian StyleJiang, Diyou, Musheng Wu, Desheng Liu, Fangfang Li, Minggang Chai, and Sanqiu Liu. 2019. "Structural Stability, Electronic Structures, Mechanical Properties and Debye Temperature of Transition Metal Impurities in Tungsten: A First-Principles Study" Metals 9, no. 9: 967. https://doi.org/10.3390/met9090967