
The Cyclic Imine Core Common to the Marine Macrocyclic Toxins Is Sufficient to Dictate Nicotinic Acetylcholine Receptor Antagonism

 , ,
, ,  , and
, and
Abstract
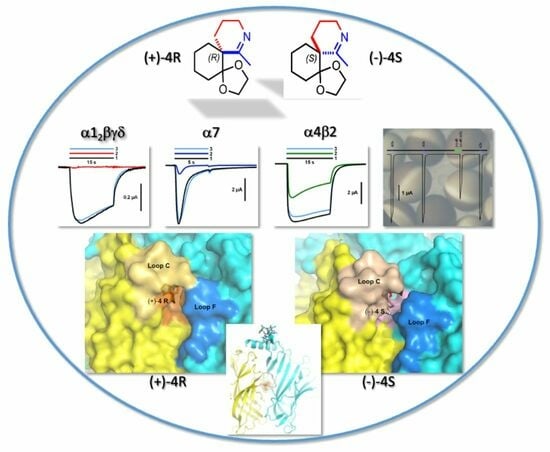
:
1. Introduction
2. Results and Discussion
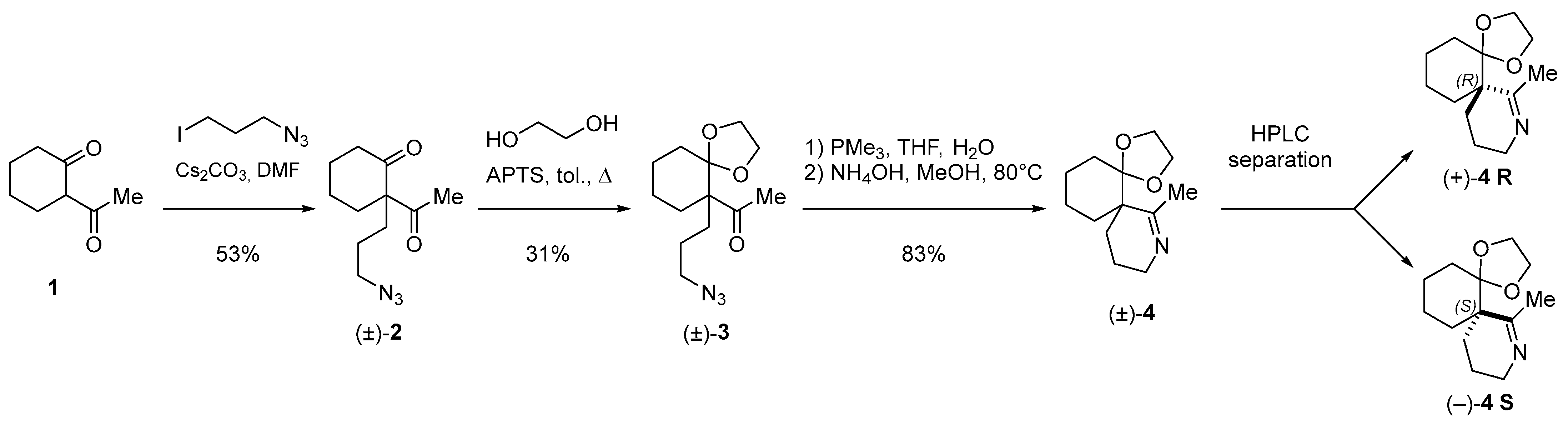
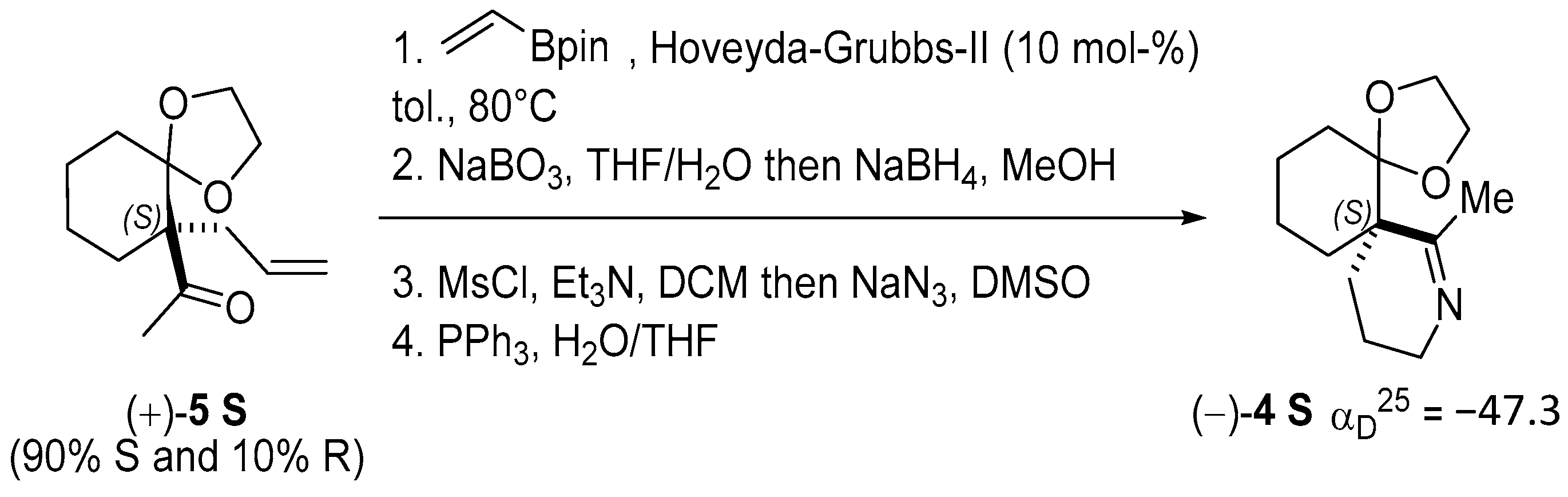
2.1. Chemical Synthesis and Characterization of the Spiroimine Enantiomers
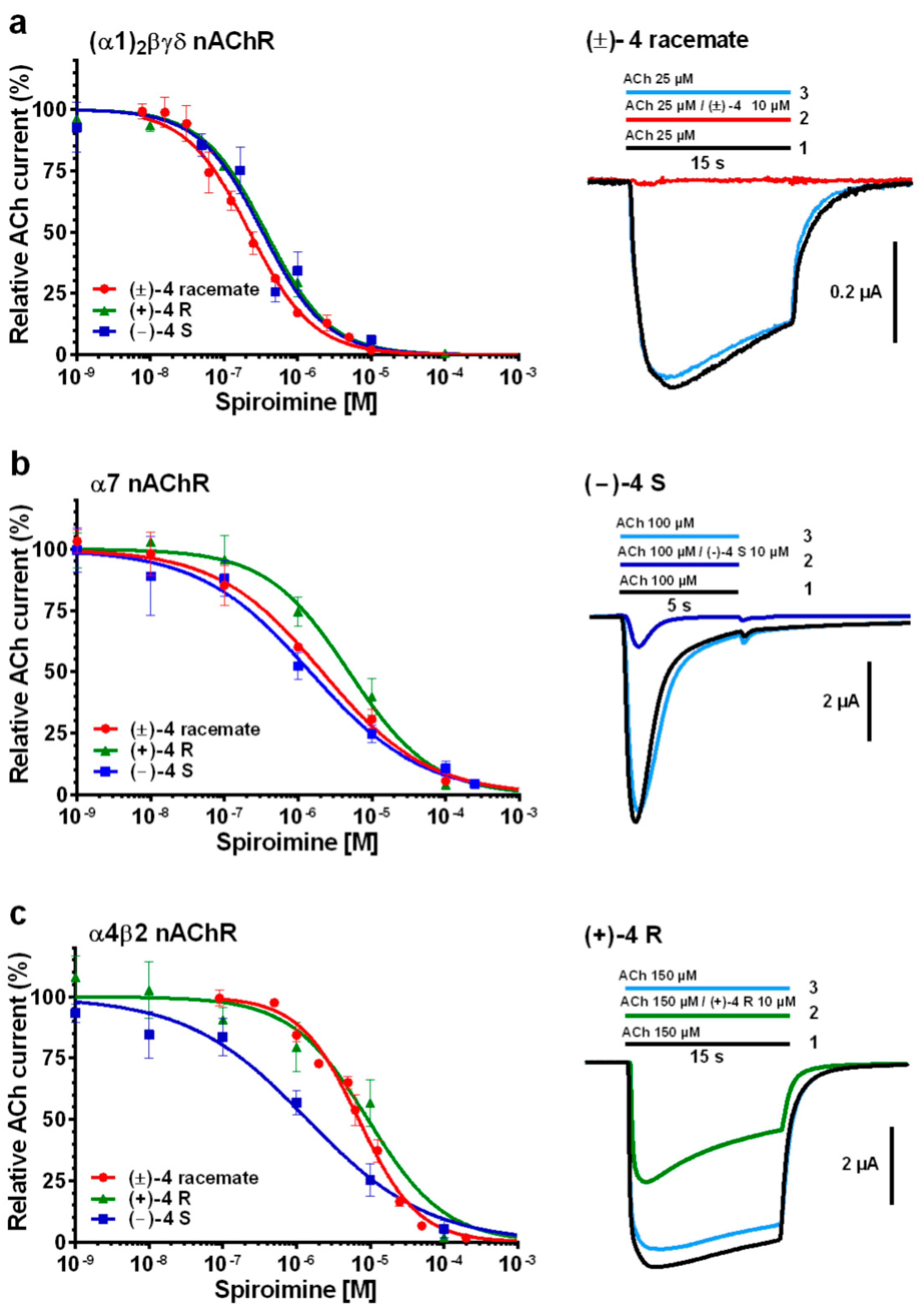
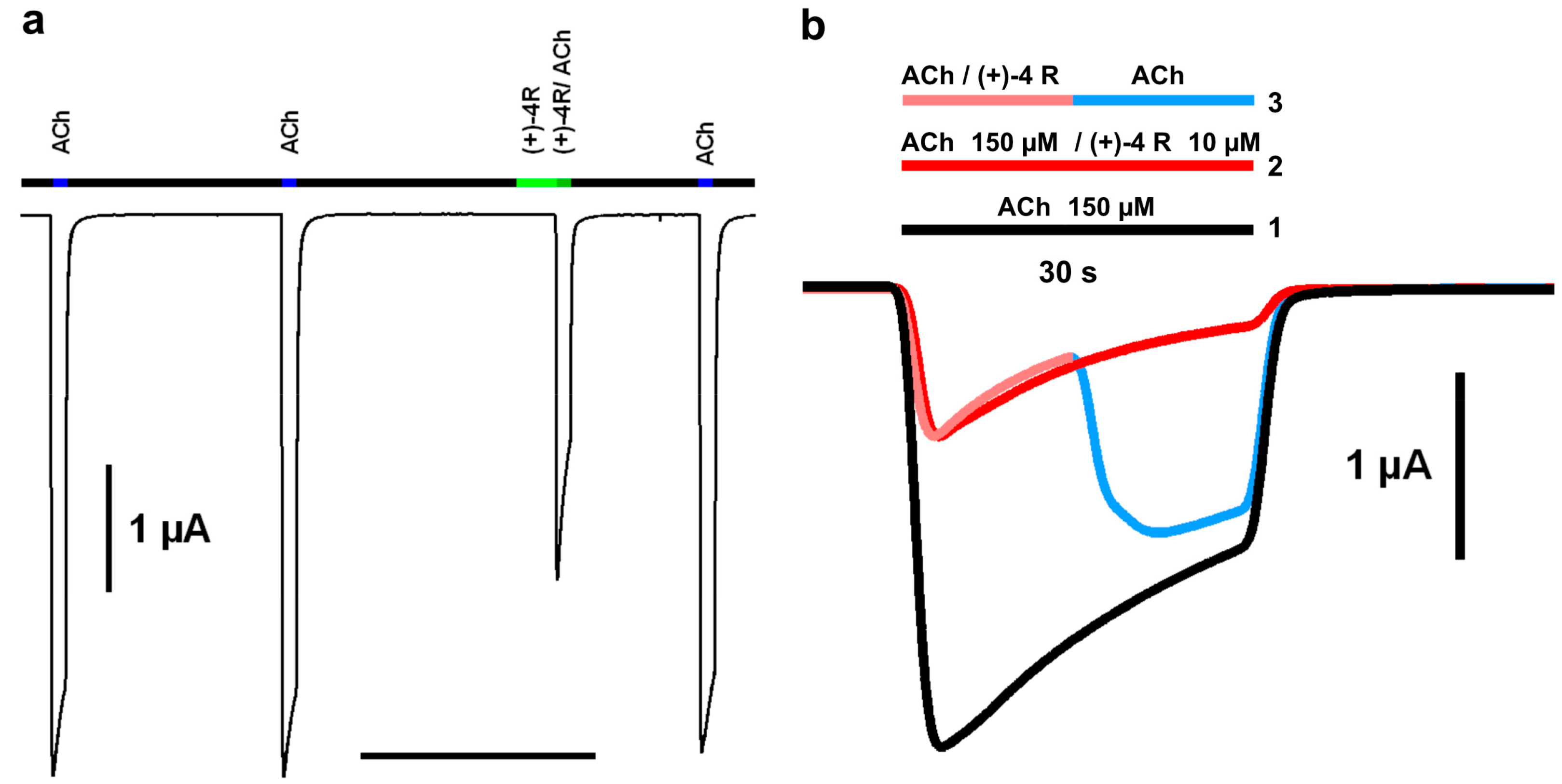
2.2. Functional Characteristics and nAChR Subtype Selectivity of the Spiroimine Racemate and Enantiomers
2.3. Binding Characteristics of the Spiroimine Enantiomers toward A- and L-AChBPs
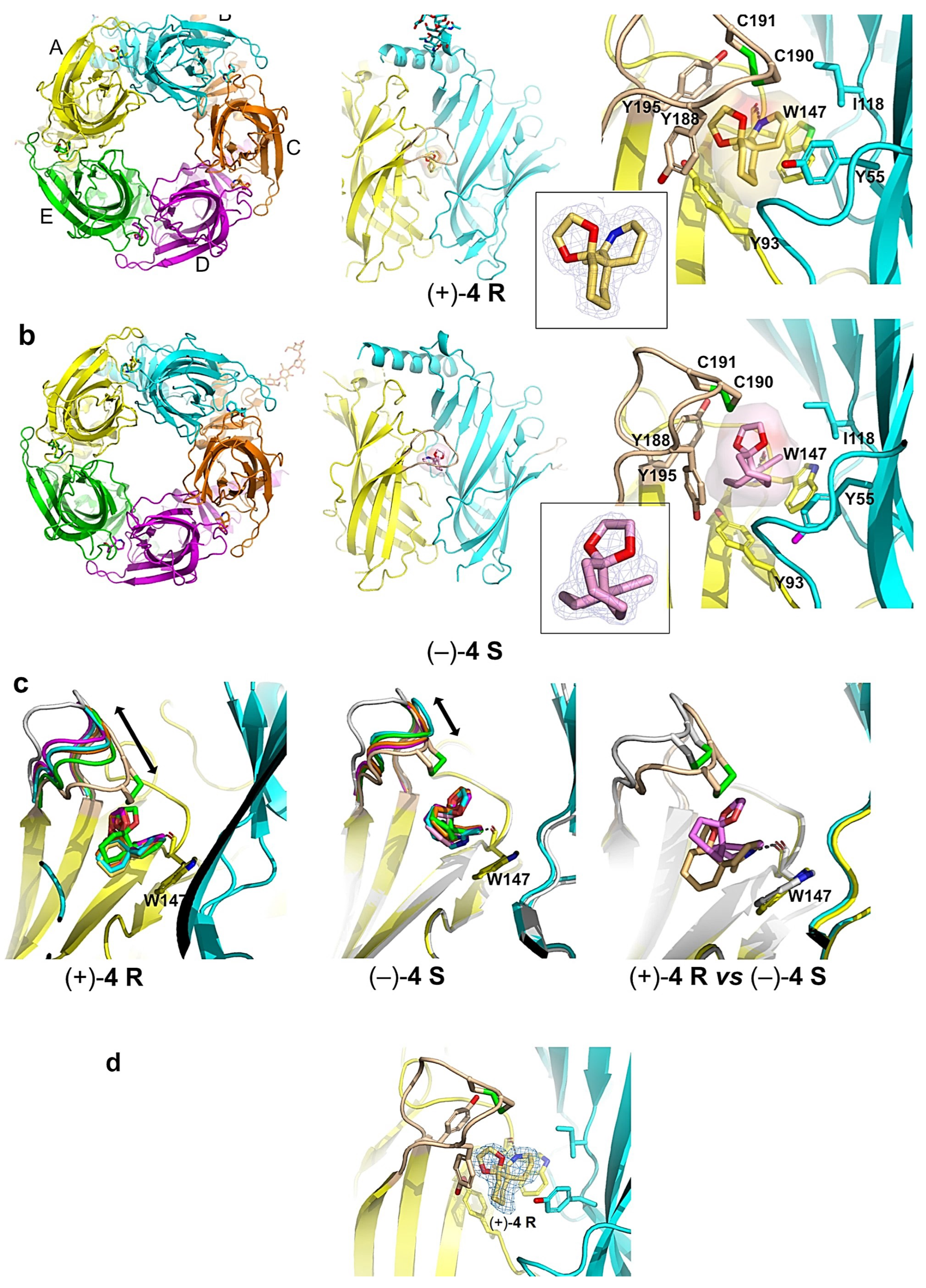
2.4. Overall View of the Crystalline Spiroimine-AChBP Complexes
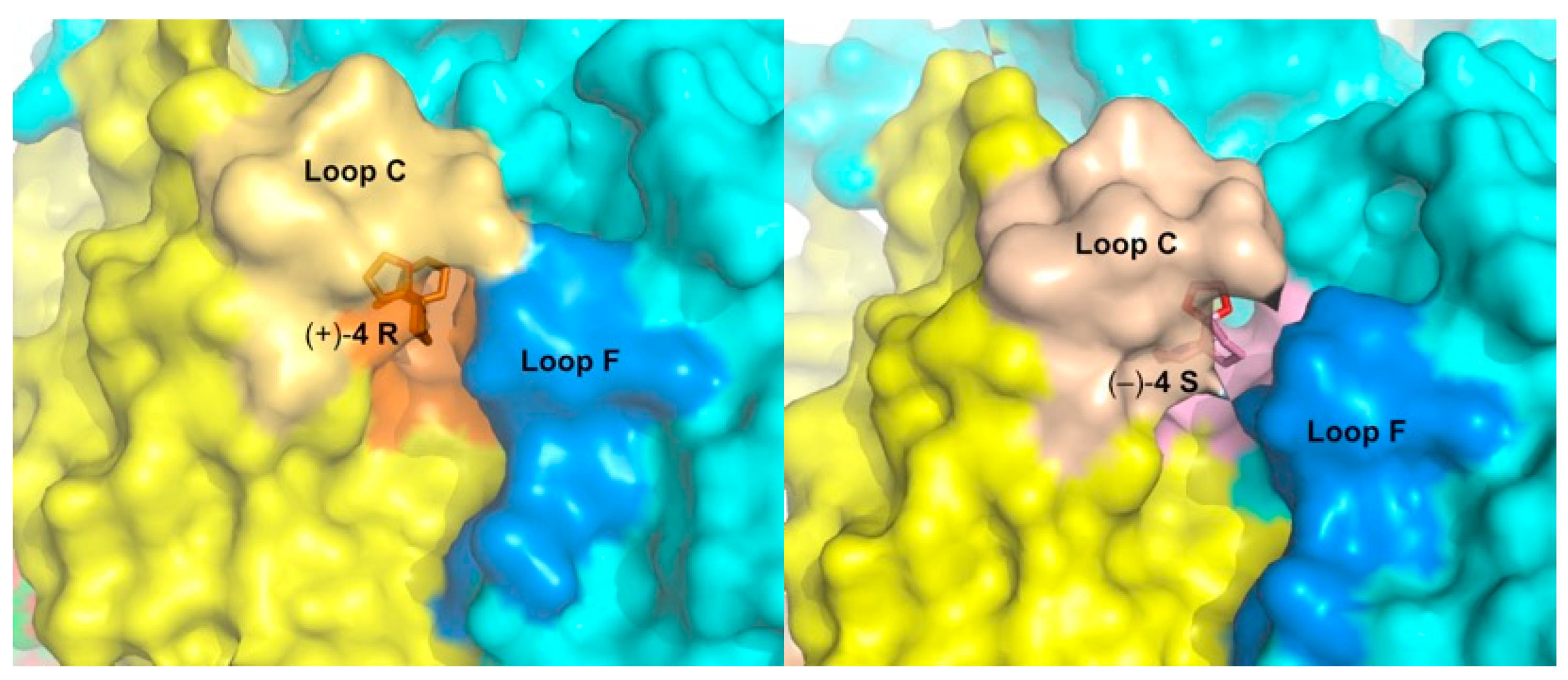
2.5. Detailed Description of the Crystalline Spiroimine-AChBP Complexes
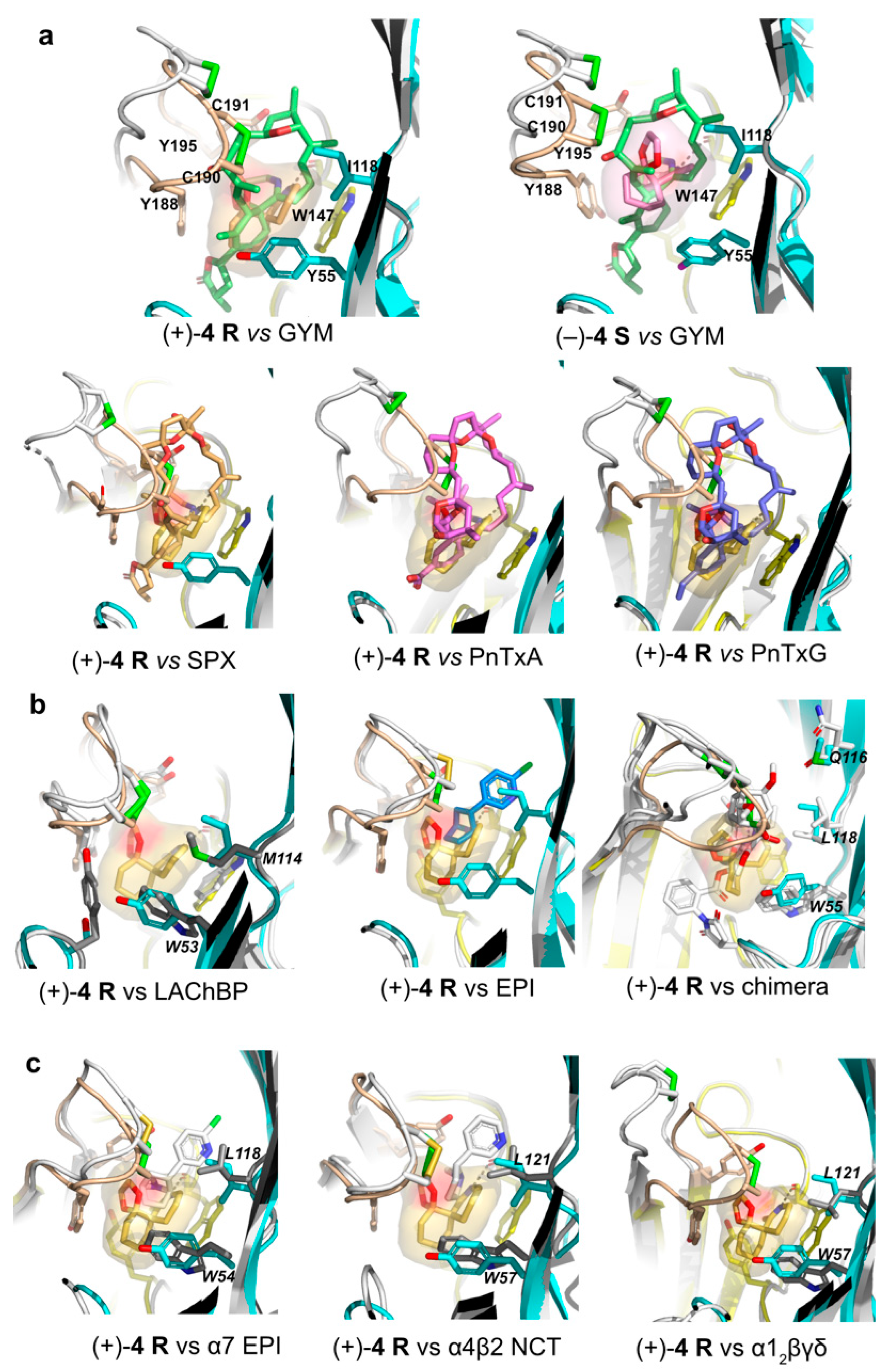
2.6. Structural Comparisons
3. Experimental Procedures
3.1. Chemical Synthesis, Separation, and Enantiomeric Characterization of the Spiroimines
3.2. Analysis of ACh-Evoked Currents
3.3. Ligand Binding to the AChBPs
3.4. Structure Determination and Refinement
3.5. Structural Analyses and Comparisons
3.6. Figures
4. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
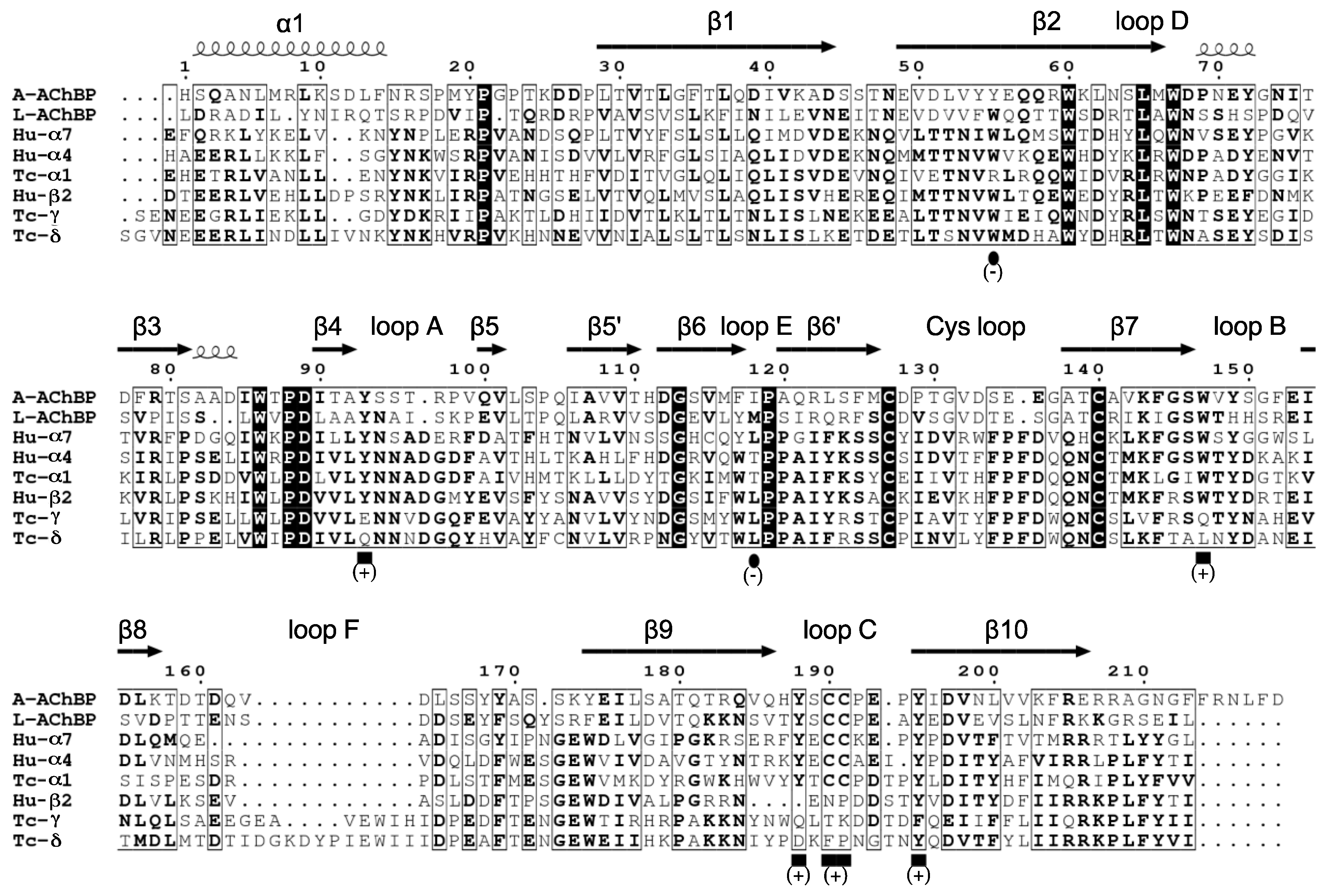
Receptors Used or Mentioned in this Study

Appendix B
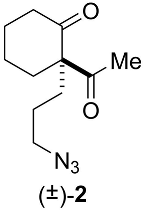
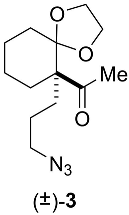
Appendix B.1. Chemical Synthesis of 2-acetyl-2-(3-azidopropyl)cyclohexanone (±)-2 (C11H17N3O2, 223.2760) (Figure A2)

Appendix B.2. Chemical Synthesis of 1-[6-(3-azidopropyl)-1,4-dioxaspiro[4.5]dec-6-yl]ethanone (±)-3 (C13H21N3O3, 267.3290) (Figure A3)

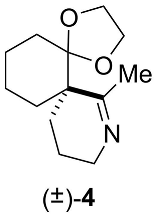
Appendix B.3. Chemical Synthesis of 7-methyl-1,4-dioxa-8-azadispiro[4.0.5.4]pentadec-7-ene (±)-4 (C13H21NO2, 223.3160) (Figure A4)

Appendix C
Appendix C.1. Live Animals and Biological Materials
Appendix C.2. Microtransplantation and Expression of the nAChRs in Xenopus Oocytes
Appendix C.3. Voltage-Clamp Recordings in Oocytes
Appendix D
Appendix D.1. Stable Expression and Purification of the AChBPs
Appendix D.2. Formation and Crystallization of the Spiroimine-AChBP Complexes and Data Collection
References
- Davidson, K.; Baker, C.; Higgins, C.; Higman, W.; Swan, S.; Veszelovszki, A.; Turner, A.D. Potential threats posed by new or emerging marine biotoxins in UK waters and examination of detection methodologies used for their control: Cyclic imines. Mar. Drugs 2015, 13, 7087–7112. [Google Scholar] [CrossRef] [PubMed]
- Stivala, C.E.; Benoit, E.; Aráoz, R.; Servent, D.; Novikov, A.; Molgó, J.; Zakarian, A. Synthesis and biology of cyclic imine toxins, an emerging class of potent, globally distributed marine toxins. Nat. Prod. Rep. 2015, 32, 411–435. [Google Scholar] [CrossRef] [PubMed]
- Molgó, J.; Marchot, P.; Aráoz, R.; Benoit, E.; Iorga, B.I.; Zakarian, A.; Taylor, P.; Bourne, Y.; Servent, D. Cyclic imine toxins from dinoflagellates: A growing family of potent antagonists of the nicotinic acetylcholine receptors. J. Neurochem. 2017, 142 (Suppl. S2), 41–51. [Google Scholar] [CrossRef] [PubMed]
- Farabegoli, F.; Blanco, L.; Rodríguez, L.P.; Vieites, J.M.; Cabado, A.G. Phycotoxins in marine shellfish: Origin, occurrence and effects on humans. Mar. Drugs 2018, 16, 188. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; An, H.J.; Kim, J.; Jeon, Y.J. Current situation of palytoxins and cyclic imines in Asia-pacific countries: Causative phytoplankton species and seafood poisoning. Int. J. Environ. Res. Public Health 2022, 19, 4921. [Google Scholar] [CrossRef] [PubMed]
- Aasen, J.A.B.; Hardstaff, W.; Aune, T.; Quilliam, M.A. Discovery of fatty acid ester metabolites of spirolide toxins in mussels from Norway using liquid chromatography/tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2006, 20, 1531–1537. [Google Scholar] [CrossRef] [PubMed]
- McCarron, P.; Rourke, W.A.; Hardstaff, W.; Pooley, B.; Quilliam, M.A. Identification of pinnatoxins and discovery of their fatty acid ester metabolites in mussels (Mytilus edulis) from eastern Canada. J. Agric. Food Chem. 2012, 60, 1437–1746. [Google Scholar] [CrossRef] [PubMed]
- De la Iglesia, P.; McCarron, P.; Diogène, J.; Quilliam, M.A. Discovery of gymnodimine fatty acid ester metabolites in shellfish using liquid chromatography/mass spectrometry. Rapid Commun. Mass Spectrom. 2013, 27, 643–653. [Google Scholar] [CrossRef]
- Aráoz, R.; Barnes, P.; Séchet, V.; Delepierre, M.; Zinn-Justin, S.; Molgó, J.; Zakarian, A.; Hess, P.; Servent, D. Cyclic imine toxins survey in coastal european shellfish samples: Bioaccumulation and mode of action of 28-O-palmitoyl ester of pinnatoxin-G—First report of portimine-A bioaccumulation. Harmful Algae 2020, 98, 101887. [Google Scholar] [CrossRef]
- Ji, Y.; Che, Y.; Wright, E.J.; McCarron, P.; Hess, P.; Li, A. Fatty acid ester metabolites of gymnodimine in shellfish collected from China and in mussels (Mytilus galloprovincialis) exposed to Karenia selliformis. Harmful Algae 2020, 92, 101774. [Google Scholar] [CrossRef]
- Varriale, F.; Tartaglione, L.; Cinti, S.; Milandri, A.; Dall’Ara, S.; Calfapietra, A.; Dell’Aversano, C. Development of a data dependent acquisition-based approach for the identification of unknown fast-acting toxins and their ester metabolites. Talanta 2021, 224, 121842. [Google Scholar] [CrossRef] [PubMed]
- Kvrgic, K.; Lesic, T.; Aysal, A.I.; Dzafic, N.; Pleadin, J. Cyclic imines in shellfish and ascidians in the northern Adriatic Sea. Food Addit. Contam. Part B-Surveill. 2020, 14, 12–22. [Google Scholar] [CrossRef]
- Otero, P.; Silva, M. Emerging marine biotoxins in European waters: Potential risks and analytical challenges. Mar. Drugs 2022, 20, 199. [Google Scholar] [CrossRef] [PubMed]
- Finch, S.C.; Harwood, D.T.; Boundy, M.J.; Selwood, A.I. A review of cyclic imines in shellfish: Worldwide occurrence, toxicity and assessment of the risk to consumers. Mar. Drugs 2024, 22, 129. [Google Scholar] [CrossRef] [PubMed]
- Servent, D.; Malgorn, C.; Bernes, M.; Gil, S.; Simasotchi, C.; Hérard, A.S.; Delzescaux, T.; Thai, R.; Barbe, P.; Keck, M.; et al. First evidence that emerging pinnatoxin-G, a contaminant of shellfish, reaches the brain and crosses the placental barrier. Sci. Total Environ. 2021, 790, 148125. [Google Scholar] [CrossRef] [PubMed]
- Seki, T.; Satake, M.; MacKenzie, L.; Kaspar, H.F.; Yasumoto, T. Gymnodimine, a new marine toxin of unprecedented structure isolated from New Zealand oysters and the dinoflagellate, Gymnodinium sp. Tetrahedron Lett. 1995, 36, 7093–7096. [Google Scholar] [CrossRef]
- Miles, C.O.; Wilkins, A.L.; Stirling, D.J.; MacKenzie, A.L. New analogue of gymnodimine from a gymnodinium species. J. Agric. Food Chem. 2000, 48, 1373–1376. [Google Scholar] [CrossRef]
- Miles, C.O.; Wilkins, A.L.; Stirling, D.J.; MacKenzie, A.L. Gymnodimine C, an isomer of gymnodimine B, from Karenia selliformis. J. Agric. Food Chem. 2003, 51, 4838–4840. [Google Scholar] [CrossRef]
- Haywood, A.J.; Steidinger, K.A.; Truby, E.W.; Bergquist, P.R.; Bergquist, P.L.; Adamson, J.; MacKenzie, L. Comparative morphology and molecular phylogenetic analysis of three new species of the genus Karenia (Dinophyceae) from New Zealand. J. Phycol. 2004, 40, 165–179. [Google Scholar] [CrossRef]
- Tang, Z.; Qiu, J.; Wang, G.; Ji, Y.; Hess, P.; Li, A. Development of an efficient extraction method for harvesting gymnodimine-A from large-scale cultures of Karenia selliformis. Toxins 2021, 13, 793. [Google Scholar] [CrossRef]
- Van Wagoner, R.M.; Misner, I.; Tomas, C.R.; Wright, J.L.C. Occurrence of 12-methylgymnodimine in a spirolide-producing dinoflagellate Alexandrium peruvianum and the biogenetic implications. Tetrahedron Lett. 2011, 52, 4243–4246. [Google Scholar] [CrossRef]
- Van de Waal, D.B.; Tillmann, U.; Martens, H.; Krock, B.; van Scheppingen, Y.; John, U. Characterization of multiple isolates from an Alexandrium ostenfeldii bloom in The Netherlands. Harmful Algae 2015, 49, 94–104. [Google Scholar] [CrossRef]
- Anttila, M.; Strangman, W.; York, R.; Tomas, C.; Wright, J.L. Biosynthetic studies of 13-desmethylspirolide C produced by Alexandrium ostenfeldii (= A. peruvianum): Rationalization of the biosynthetic pathway following incorporation of (13)C-labeled methionine and application of the odd-even rule of methylation. J. Nat. Prod. 2016, 79, 484–489. [Google Scholar] [CrossRef] [PubMed]
- Harju, K.; Koskela, H.; Kremp, A.; Suikkanen, S.; de la Iglesia, P.; Miles, C.O.; Krock, B.; Vanninen, P. Identification of gymnodimine D and presence of gymnodimine variants in the dinoflagellate Alexandrium ostenfeldii from the Baltic Sea. Toxicon 2016, 112, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Martens, H.; Tillmann, U.; Harju, K.; Dell’Aversano, C.; Tartaglione, L.; Krock, B. Toxin variability estimations of 68 Alexandrium ostenfeldii (Dinophyceae) strains from The Netherlands reveal a novel abundant gymnodimine. Microorganisms 2017, 5, 29. [Google Scholar] [CrossRef]
- Zurhelle, C.; Nieva, J.; Tillmann, U.; Harder, T.; Krock, B.; Tebben, J. Identification of novel gymnodimines and spirolides from the marine dinoflagellate Alexandrium ostenfeldii. Mar. Drugs 2018, 16, 446. [Google Scholar] [CrossRef]
- Cembella, A.D.; Lewis, N.I.; Quilliam, M.A. The marine dinoflagellate Alexandrium ostenfeldii [Dinophyceae] as the causative organism of spirolide shellfish toxins. Phycologia 2000, 39, 67–74. [Google Scholar] [CrossRef]
- Hu, T.; Burton, I.W.; Cembella, A.D.; Curtis, J.M.; Quilliam, M.A.; Walter, J.A.; Wright, J.L. Characterization of spirolides a, c, and 13-desmethyl c, new marine toxins isolated from toxic plankton and contaminated shellfish. J. Nat. Prod. 2001, 64, 308–312. [Google Scholar] [CrossRef]
- Guéret, S.M.; Brimble, M.A. Spiroimine shellfish poisoning (SSP) and the spirolide family of shellfish toxins: Isolation, structure, biological activity and synthesis. Nat. Prod. Rep. 2010, 27, 1350–1366. [Google Scholar] [CrossRef]
- Molgó, J.; Benoit, E.; Aráoz, R.; Zakarian, A.; Iorga, B.I. Spirolides and cyclic imines: Toxicological profile. In Marine and Freshwater Toxins; Gopalakrishnakone, P., Haddad, V., Jr., Tubaro, A., Kim, E., Kem, W.R., Eds.; Springer: Dordrecht, The Netherlands, 2015; pp. 193–217. [Google Scholar] [CrossRef]
- Nieva, J.A.; Tebben, J.; Tillmann, U.; Wohlrab, S.; Krock, B. Mass spectrometry-based characterization of new spirolides from Alexandrium ostenfeldii (Dinophyceae). Mar. Drugs 2020, 18, 505. [Google Scholar] [CrossRef]
- Long, M.; Krock, B.; Castrec, J.; Tillmann, U. Unknown extracellular and bioactive metabolites of the genus Alexandrium: A review of overlooked toxins. Toxins 2021, 13, 905. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, L.; Smith, K.; Selwood, A.; McNabb, P.; van Ginkel, R.; Holland, P.; Munday, R. Production of pinnatoxins by a peridinoid dinoflagellate isolated from Northland, New Zealand. Harmful Algae 2010, 9, 384–389. [Google Scholar] [CrossRef]
- Rhodes, L.; Smith, K.; Selwood, A.; McNabb, P.; Munday, R.; Suda, S.; Molenaar, S.; Hallegraeff, G. Dinoflagellate Vulcanodinium rugosum identified as the causative organism of pinnatoxins in Australia, New Zealand and Japan. Phycologia 2011, 50, 624–628. [Google Scholar] [CrossRef] [PubMed]
- Nézan, E.; Chomérat, N. Vulcanodinium rugosum gen. nov., sp. nov. (dinophyceae): A new marine dinoflagellate from the French Mediterranean coast. Cryptogam. Algol. 2011, 32, 3–18. [Google Scholar] [CrossRef]
- Uemura, D.; Chou, T.; Haino, T.; Nagatsu, A.; Fukuzawa, S.; Zheng, S.Z.; Chen, H.S. Pinnatoxin-a—A toxic amphoteric macrocycle from the okinawan bivalve Pinna muricata. J. Am. Chem. Soc. 1995, 117, 1155–1156. [Google Scholar] [CrossRef]
- Selwood, A.I.; Miles, C.O.; Wilkins, A.L.; van Ginkel, R.; Munday, R.; Rise, F.; McNabb, P. Isolation, structural determination and acute toxicity of pinnatoxins E, F and G. J. Agric. Food Chem. 2010, 58, 6532–6542. [Google Scholar] [CrossRef] [PubMed]
- Selwood, A.I.; Wilkins, A.L.; Munday, R.; Gu, H.; Smith, K.F.; Rhodes, L.L.; Rise, F. Pinnatoxin H: A new pinnatoxin analogue from a South China Sea Vulcanodinium rugosum isolate. Tetrahedron Lett. 2014, 55, 5508–5510. [Google Scholar] [CrossRef]
- Fribley, A.M.; Xi, Y.; Makris, C.; Alves-de-Souza, C.; York, R.; Tomas, C.; Wright, J.L.C.; Strangman, W.K. Identification of portimine B, a new cell permeable spiroimine that induces apoptosis in oral squamous cell carcinoma. ACS Med. Chem. Lett. 2018, 10, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Selwood, A.I.; Wilkins, A.L.; Munday, R.; Shi, F.; Rhodes, L.L.; Holland, P.T. Portimine: A bioactive metabolite from the benthic dinoflagellate Vulcanodinium rugosum. Tetrahedron Lett. 2013, 4, 4705–4707. [Google Scholar] [CrossRef]
- Hermawan, I.; Higa, M.; Hutabarat, P.U.B.; Fujiwara, T.; Akiyama, K.; Kanamoto, A.; Haruyama, T.; Kobayashi, N.; Higashi, M.; Suda, S.; et al. Kabirimine, a new cyclic imine from an Okinawan dinoflagellate. Mar. Drugs 2019, 17, 353. [Google Scholar] [CrossRef]
- Takada, N.; Umemura, N.; Suenaga, K.; Uemura, D. Structural determination of pteriatoxins A, B and C, extremely potent toxins from the bivalve Pteria penguin. Tetrahedron Lett. 2001, 42, 3495–3497. [Google Scholar] [CrossRef]
- Hao, J.; Matsuura, F.; Kishi, Y.; Kita, M.; Uemura, D.; Asai, N.; Iwashita, T. Stereochemistry of pteriatoxins A, B, and C. J. Am. Chem. Soc. 2006, 128, 7742–7743. [Google Scholar] [CrossRef] [PubMed]
- Torigoe, K.; Murata, M.; Yasumoto, T.; Iwashita, T. Prorocentrolide, a toxic nitrogenous macrocycle from a marine dinoflagellate, Prorocentrum lima. J. Am. Chem. Soc. 1988, 110, 7876–7877. [Google Scholar] [CrossRef]
- Hu, T.; deFreitas, S.W.; Curtis, J.M.; Oshima, Y.; Walter, J.A.; Wright, J.L.C. Isolation and structure of prorocentrolide B, a fast-acting toxin from Prorocentrum maculosum. J. Nat. Prod. 1996, 59, 1010–1014. [Google Scholar] [CrossRef] [PubMed]
- Amar, M.; Aráoz, R.; Iorga, B.I.; Yasumoto, T.; Servent, D.; Molgó, J. Prorocentrolide-A from cultured Prorocentrum lima dinoflagellates collected in Japan blocks subtypes of nicotinic acetylcholine receptors. Toxins 2018, 10, 97. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Yang, A.R.; Yoo, Y.D.; Jeong, E.J.; Rho, J.R. Relative configurational assignment of 4-hydroxyprorocentrolide and prorocentrolide C isolated from a benthic dinoflagellate (Prorocentrum lima). J. Nat. Prod. 2019, 82, 1034–1039. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.-K.; Lee, G.-H.; Huang, R.; Chou, H.-N. Spiro-prorocentrimine, a novel macrocyclic lactone from a benthic Prorocentrum sp. of Taiwan. Tetrahedron Lett. 2001, 42, 1713–1716. [Google Scholar] [CrossRef]
- Molgó, J.; Aráoz, R.; Benoit, E.; Iorga, B. Cyclic imine toxins: Chemistry, origin, metabolism, pharmacology, toxicology and detection. In Seafood and Freshwater Toxins: Pharmacology, Physiology and Detection; Botana, L.M., Ed.; CRC Press: Boca Raton, FL, USA, 2014; pp. 951–989. [Google Scholar] [CrossRef]
- Otero, A.; Chapela, M.J.; Atanassova, M.; Vieites, J.M.; Cabado, A.G. Cyclic imines: Chemistry and mechanism of action: A review. Chem. Res. Toxicol. 2011, 24, 1817–1829. [Google Scholar] [CrossRef] [PubMed]
- Kasheverov, I.; Kudryavtsev, D.; Shelukhina, I.; Nikolaev, G.; Utkin, Y.; Tsetlin, V. Marine origin ligands of nicotinic receptors: Low molecular compounds, peptides and proteins for fundamental research and practical applications. Biomolecules 2022, 12, 189. [Google Scholar] [CrossRef]
- Taylor, P. Agents acting at the neuromuscular junction and autonomic ganglia. In Goodman and Gilman’s The Pharmacological Basis of Therapeutics; Brunton, L.L., Lazo, J.S., Parker, K.L., Eds.; McGraw–Hill: New York, NY, USA, 2006; pp. 217–236. [Google Scholar]
- Changeux, J.P. The nicotinic acetylcholine receptor: The founding father of the pentameric ligand-gated ion channel superfamily. J. Biol. Chem. 2012, 287, 40207–40215. [Google Scholar] [CrossRef]
- Le Novère, N.; Corringer, P.J.; Changeux, J.P. The diversity of subunit composition in nAChRs: Evolutionary origins, physiologic and pharmacologic consequences. J. Neurobiol. 2002, 53, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Gharpure, A.; Noviello, C.M.; Hibbs, R.E. Progress in nicotinic receptor structural biology. Neuropharmacology 2020, 171, 108086. [Google Scholar] [CrossRef]
- Delgado-Vélez, M.; Quesada, O.; Villalobos-Santos, J.C.; Maldonado-Hernández, R.; Asmar-Rovira, G.; Stevens, R.C.; Lasalde-Dominicci, J.A. Pursuing high-resolution structures of nicotinic acetylcholine receptors: Lessons learned from five decades. Molecules 2021, 26, 5753. [Google Scholar] [CrossRef] [PubMed]
- Unwin, N. Nicotinic acetylcholine receptor and the structural basis of neuromuscular transmission: Insights from Torpedo postsynaptic membranes. Q. Rev. Biophys. 2013, 46, 283–322. [Google Scholar] [CrossRef] [PubMed]
- Changeux, J.P. The nicotinic acetylcholine receptor: A typical ‘allosteric machine’. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018, 373, 20170174. [Google Scholar] [CrossRef] [PubMed]
- Noviello, C.M.; Gharpure, A.; Mukhtasimova, N.; Cabuco, R.; Baxter, L.; Borek, D.; Sine, S.M.; Hibbs, R.E. Structure and gating mechanism of the α7 nicotinic acetylcholine receptor. Cell 2021, 184, 2121–2134.e13. [Google Scholar] [CrossRef] [PubMed]
- Morales-Perez, C.L.; Noviello, C.M.; Hibbs, R.E. X-ray structure of the human α4β2 nicotinic receptor. Nature 2016, 538, 411–415. [Google Scholar] [CrossRef]
- Rahman, M.M.; Teng, J.; Worrell, B.T.; Noviello, C.M.; Lee, M.; Karlin, A.; Stowell, M.H.B.; Hibbs, R.E. Structure of the native muscle-type nicotinic receptor and inhibition by snake venom toxins. Neuron 2020, 106, 952–962.e5. [Google Scholar] [CrossRef]
- Smit, A.B.; Syed, N.I.; Schaap, D.; van Minnen, J.; Klumperman, J.; Kits, K.S.; Lodder, H.; van der Schors, R.C.; van Elk, R.; Sorgedrager, B.; et al. A glia-derived acetylcholine-binding protein that modulates synaptic transmission. Nature 2001, 411, 261–268. [Google Scholar] [CrossRef]
- Brejc, K.; van Dijk, W.J.; Klaassen, R.V.; Schuurmans, M.; van Der Oost, J.; Smit, A.B.; Sixma, T.K. Crystal structure of an ACh-binding protein reveals the ligand-binding domain of nicotinic receptors. Nature 2001, 411, 269–276. [Google Scholar] [CrossRef]
- Hansen, S.B.; Talley, T.T.; Radić, Z.; Taylor, P. Structural and ligand recognition characteristics of an acetylcholine-binding protein from Aplysia californica. J. Biol. Chem. 2004, 279, 24197–24202. [Google Scholar] [CrossRef] [PubMed]
- Celie, P.H.; van Rossum-Fikkert, S.E.; van Dijk, W.J.; Brejc, K.; Smit, A.B.; Sixma, T.K. Nicotine and carbamylcholine binding to nicotinic acetylcholine receptors as studied in AChBP crystal structures. Neuron 2004, 41, 907–914. [Google Scholar] [CrossRef] [PubMed]
- Bourne, Y.; Talley, T.T.; Hansen, S.B.; Taylor, P.; Marchot, P. Crystal structure of a Cbtx-AChBP complex reveals essential interactions between snake alpha-neurotoxins and nicotinic receptors. EMBO J. 2005, 24, 1512–1522. [Google Scholar] [CrossRef] [PubMed]
- Hansen, S.B.; Sulzenbacher, G.; Huxford, T.; Marchot, P.; Taylor, P.; Bourne, Y. Structures of Aplysia AChBP complexes with nicotinic agonists and antagonists reveal distinctive binding interfaces and conformations. EMBO J. 2005, 24, 3635–3646. [Google Scholar] [CrossRef] [PubMed]
- Hibbs, R.E.; Sulzenbacher, G.; Shi, J.; Talley, T.T.; Conrod, S.; Kem, W.R.; Taylor, P.; Marchot, P.; Bourne, Y. Structural determinants for interaction of partial agonists with acetylcholine binding protein and neuronal alpha7 nicotinic acetylcholine receptor. EMBO J. 2009, 28, 3040–3051. [Google Scholar] [CrossRef] [PubMed]
- Shahsavar, A.; Gajhede, M.; Kastrup, J.S.; Balle, T. Structural studies of nicotinic acetylcholine receptors: Using acetylcholine-binding protein as a structural surrogate. Basic Clin. Pharmacol. Toxicol. 2016, 118, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Camacho-Hernandez, G.A.; Taylor, P. Lessons from nature: Structural studies and drug design driven by a homologous surrogate from invertebrates, AChBP. Neuropharmacology 2020, 179, 108108. [Google Scholar] [CrossRef] [PubMed]
- Bourne, Y.; Radić, Z.; Aráoz, R.; Talley, T.T.; Benoit, E.; Servent, D.; Taylor, P.; Molgó, J.; Marchot, P. Structural determinants in phycotoxins and AChBP conferring high affinity binding and nicotinic AChR antagonism. Proc. Natl. Acad. Sci. USA 2010, 107, 6076–6081. [Google Scholar] [CrossRef] [PubMed]
- Bourne, Y.; Sulzenbacher, G.; Radić, Z.; Aráoz, R.; Reynaud, M.; Benoit, E.; Zakarian, A.; Servent, D.; Molgó, J.; Taylor, P.; et al. Marine macrocyclic imines, pinnatoxins A and G: Structural determinants and functional properties to distinguish neuronal α7 from muscle α1(2)βγδ nAChRs. Structure 2015, 23, 1106–1115. [Google Scholar] [CrossRef]
- Toumieux, S.; Beniazza, R.; Desvergnes, V.; Aráoz, R.; Molgó, J.; Landais, Y. Synthesis of the gymnodimine tetrahydrofuran core through a Ueno-Stork radical cyclization. Org. Biomol. Chem. 2011, 9, 3726–3732. [Google Scholar] [CrossRef]
- Duroure, L.; Jousseaume, T.; Aráoz, R.; Barré, E.; Retailleau, P.; Chabaud, L.; Molgó, J.; Guillou, C. 6,6-Spiroimine analogs of (−)-gymnodimine A: Synthesis and biological evaluation on nicotinic acetylcholine receptors. Org. Biomol. Chem. 2011, 9, 8112–8118. [Google Scholar] [CrossRef] [PubMed]
- Alonso, E.; Otero, P.; Vale, C.; Alfonso, A.; Antelo, A.; Giménez-Llort, L.; Chabaud, L.; Guillou, C.; Botana, L.M. Benefit of 13-desmethyl spirolide C treatment in triple transgenic mouse model of Alzheimer disease: Beta-amyloid and neuronal markers improvement. Curr. Alzheimer Res. 2013, 10, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Kanehira, K.; Hagihara, T.; Kumada, M.J. Asymmetric synthesis catalyzed by chiral ferrocenylphosphine-transition metal complexes. 5. Palladium-catalyzed asymmetric allylation of active methine compounds. J. Org. Chem. 1998, 53, 113–120. [Google Scholar] [CrossRef]
- Hauser, T.A.; Hepler, C.D.; Kombo, D.C.; Grinevich, V.P.; Kiser, M.N.; Hooker, D.N.; Zhang, J.; Mountfort, D.; Selwood, A.; Akireddy, S.R.; et al. Comparison of acetylcholine receptor interactions of the marine toxins, 13-desmethylspirolide C and gymnodimine. Neuropharmacology 2012, 62, 2239–2250. [Google Scholar] [CrossRef]
- Tan, K.P.; Singh, K.; Hazra, A.; Madhusudhan, M.S. Peptide bond planarity constrains hydrogen bond geometry and influences secondary structure conformations. Curr. Res. Struct. Biol. 2020, 3, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Bourne, Y.; Radić, Z.; Taylor, P.; Marchot, P. Conformational remodeling of femtomolar inhibitor-acetylcholinesterase complexes in the crystalline state. J. Am. Chem. Soc. 2010, 132, 18292–18300. [Google Scholar] [CrossRef] [PubMed]
- Nemecz, Á.; Taylor, P. Creating an α7 nicotinic acetylcholine recognition domain from the acetylcholine-binding protein: Crystallographic and ligand selectivity analyses. J. Biol. Chem. 2011, 286, 42555–42565. [Google Scholar] [CrossRef] [PubMed]
- Kaczanowska, K.; Camacho Hernandez, G.A.; Bendiks, L.; Kohs, L.; Cornejo-Bravo, J.M.; Harel, M.; Finn, M.G.; Taylor, P. Substituted 2-aminopyrimidines selective for α7-nicotinic acetylcholine receptor activation and association with acetylcholine binding proteins. J. Am. Chem. Soc. 2017, 139, 3676–3684. [Google Scholar] [CrossRef] [PubMed]
- Neudert, G.; Klebe, G. DSX: A knowledge-based scoring function for the assessment of protein-ligand complexes. J. Chem. Inf. Model. 2011, 51, 2731–2745. [Google Scholar] [CrossRef]
- Rahman, M.M.; Basta, T.; Teng, J.; Lee, M.; Worrell, B.T.; Stowell, M.H.B.; Hibbs, R.E. Structural mechanism of muscle nicotinic receptor desensitization and block by curare. Nat. Struct. Mol. Biol. 2022, 29, 386–394. [Google Scholar] [CrossRef]
- McCoy, A.J. Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr. D Biol. Crystallogr. 2007, 63, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Murshudov, G.N.; Vagin, A.A.; Dodson, E.J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 1997, 53, 240–255. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [PubMed]
- CCP4. The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. D. Biol. Crystallogr. 1994, 50, 760–763. [Google Scholar] [CrossRef] [PubMed]
- Krissinel, E.; Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef] [PubMed]
- Davis, I.W.; Leaver-Fay, A.; Chen, V.B.; Block, J.N.; Kapral, G.J.; Wang, X.; Murray, L.W.; Arendall, W.B.; Snoeyink, J.; Richardson, J.S.; et al. MolProbity: All-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007, 35, W375–W383. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger, LLC. The PyMOL Molecular Graphics System, Version 1.5.0.4; Schrödinger, LLC: New York, NY, USA, 2010. [Google Scholar]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.; Shyong, Y.J.; Samskey, N.; Ho, K.Y.; Radić, Z.; Fenical, W.; Sharpless, K.B.; Kovarik, Z.; Camacho-Hernandez, G.A. Ligand design for human acetylcholinesterase and nicotinic acetylcholine receptors, extending beyond the conventional and canonical. J. Neurochem. 2021, 158, 1217–1222. [Google Scholar] [CrossRef] [PubMed]
- Kem, W.R.; Andrud, K.; Bruno, G.; Xing, H.; Soti, F.; Talley, T.T.; Taylor, P. Interactions of nereistoxin and its analogs with vertebrate nicotinic acetylcholine receptors and molluscan ACh binding proteins. Mar. Drugs 2022, 20, 49. [Google Scholar] [CrossRef]
- Sine, S.M.; Taylor, P. Relationship between reversible antagonist occupancy and the functional capacity of the acetylcholine receptor. J. Biol. Chem. 1981, 56, 6692–6699. [Google Scholar] [CrossRef]
- Bouzat, C. New insights into the structural bases of activation of Cys-loop receptors. J. Physiol. Paris 2012, 106, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Dumont, J.N. Oogenesis in Xenopus laevis (Daudin). I. Stages of oocyte development in laboratory maintained animals. J. Morphol. 1972, 136, 153–179. [Google Scholar] [CrossRef] [PubMed]
- Villeneuve, P.; Esnault, G.; Benoit, E.; Molgó, J.; Aráoz, R. A monitoring study of repetitive surgical oocyte harvest in Xenopus laevis. In Toxins and Ion Transfers; Collection Meetings in Toxinology; Barbier, J., Benoit, E., Gilles, N., Ladant, D., Martin-Eauclaire, M.F., Mattei, C., Molgó, J., Popoff, M.R., Servent, D., Eds.; SFET Publication: Gif-sur-Yvette, France, 2011; pp. 173–178. ISSN 1760-6004. Available online: http://sfet.asso.fr/international/e-book-rt/e-book-rt.html (accessed on 27 November 2011).
- Kharrat, R.; Servent, D.; Girard, E.; Ouanounou, G.; Amar, M.; Marrouchi, R.; Benoit, E.; Molgó, J. The marine phycotoxin gymnodimine targets muscular and neuronal nicotinic acetylcholine receptor subtypes with high affinity. J. Neurochem. 2008, 107, 952–963. [Google Scholar] [CrossRef] [PubMed]
- Krieger, F.; Mourot, A.; Aráoz, R.; Kotzyba-Hibert, F.; Molgó, J.; Bamberg, E.; Goeldner, M. Fluorescent agonists for the Torpedo nicotinic acetylcholine receptor. ChemBioChem 2008, 9, 1146–1153. [Google Scholar] [CrossRef] [PubMed]
- Aráoz, R.; Ouanounou, G.; Iorga, B.I.; Goudet, A.; Alili, D.; Amar, M.; Benoit, E.; Molgó, J.; Servent, D. The neurotoxic effect of 13,19-didesmethyl and 13-desmethyl spirolide C phycotoxins is mainly mediated by nicotinic rather than muscarinic acetylcholine receptors. Toxicol. Sci. 2015, 147, 156–167. [Google Scholar] [CrossRef]
- Miledi, R.; Palma, E.; Eusebi, F. Microtransplantation of neurotransmitter receptors from cells to Xenopus oocyte membranes: New procedure for ion channel studies. Methods Mol. Biol. 2006, 322, 347–355. [Google Scholar] [CrossRef]
- Aráoz, R.; Servent, D.; Molgó, J.; Iorga, B.I.; Fruchart-Gaillard, C.; Benoit, E.; Gu, Z.; Stivala, C.; Zakarian, A. Total synthesis of pinnatoxins A and G, and revision of the mode of action of pinnatoxin A. J. Am. Chem. Soc. 2011, 133, 10499–10511. [Google Scholar] [CrossRef]
- Sands, S.B.; Costa, A.C.; Patrick, J.W. Barium permeability of neuronal nicotinic receptor alpha 7 expressed in Xenopus oocytes. Biophys. J. 1993, 65, 2614–2621. [Google Scholar] [CrossRef] [PubMed]
- Cieslikiewicz-Bouet, M.; Naldi, M.; Bartolini, M.; Pérez, B.; Servent, D.; Jean, L.; Aráoz, R.; Renard, P.-Y. Functional characterization of multifunctional ligands targeting acetylcholinesterase and alpha 7 nicotinic acetylcholine receptor. Biochem. Pharmacol. 2020, 177, 114010. [Google Scholar] [CrossRef]
- Kabsch, W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Evans, P. Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 2006, 62, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Evans, P.R.; Murshudov, G.N. How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr. 2013, 69, 1204–1214. [Google Scholar] [CrossRef] [PubMed]
- French, S.; Wilson, K. On the treatment of negative intensity observations. Acta Crystallogr. 1978, 34, 517–525. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| nAChR Subtype | IC50 (µM) a and Hill Slope b | ||
|---|---|---|---|
| Racemic (±)-4 | Spiromine (+)-4 R | Spiroimine (−)-4 S | |
| Torpedo α12βγδ | 0.24 (0.19–0.26) a | 0.37 (0.29–0.47) a | 0.33 (0.18–0.61) a |
| ~1 b | ~1 b | ~1 b | |
| Human α7 | 2.11 (1.39–3.21) a | 4.88 (3.16–7.51) a | 1.45 (0.89–2.36) a |
| 0.61 (0.47–0.75) b | 0.78 (0.54–1.01) b | 0.58 (0.44–0.72) b | |
| Human α4β2 | 6.96 (5.77–8.39) a | 9.31 (3.26–26.59) a | 1.41 (0.63–3.15) a |
| 1.08 (0.86–1.29) b | 0.83 (0.15–1.52) b | 0.53 (0.32–0.74) b | |
| AChBP Subtype | Kinetic/Equilibrium Parameters | Racemic (±)-4 | Spiroimine (+)-4 R | Spiroimine (−)-4 S |
|---|---|---|---|---|
| L-AChBP | kon (109 M−1min−1) a | 3.6 ± 0.6 | n.d. | 7.8 ± 0.1 |
| koff (103 min−1) a | 6.2 ± 1.6 | n.d. | 3.2 ± 1.1 | |
| koff/GAL (103 min−1) a | 6.7 ± 0.6 | n.d. | 6.2 ± 0.1 | |
| Kd/(koff/kon) (μM) b | 1.7 | n.a. | 0.41 | |
| Kd/SFeq (μM) c | 0.32/15 d | 12 | 0.32 | |
| Kd/SPAeq (μM) a | 2.6 ± 1.9 | 12 ± 3 | (3.4 ± 0.3) e | |
| A-AChBP | kon (109 M−1 min−1) a | 6.3 ± 1.2 | 7.4 ± 0.4 | 6.1 ± 0 |
| koff (103 min−1) a | 18 ± 2 | 16 ± 1 | 26 ± 1 | |
| koff/GAL (103 min−1) a | 15 ± 0 | 14 ± 1 | 24 ± 1 | |
| Kd/(koff/kon) (μM) b | 2.9 | 2.2 | 4.3 | |
| Kd/SFeq (μM) c | 4.2 | 3.1 | 5.4 | |
| Kd/SPAeq (μM) a | 2.4 ± 1.5 | 2.3 ± 0.4 | 2.4 ± 0.5 |
| Interface | DSX Score (kJ/mol) | ||
|---|---|---|---|
| (+)-4 R Complex | (−)-4 S Complex | (±)-4 Complex | |
| (A*-B) | −94.628 a | −77.480 b | −76.901 b |
| (B*-C) | −77.500 b | −69.469 b | −70.878 b |
| (C*-D) | −82.139 b | −67.328 b | −74.733 b |
| (D*-E) | −83.353 b | −64.331 b | −76.814 b |
| (E*-A) | −86.909 b | −68.729 b | −78.242 b |
| Mean score ± SD (n = 5) | −84.906 ± 6.392 | −69.467 ± 4.892 | −75.514 ± 2.879 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bourne, Y.; Sulzenbacher, G.; Chabaud, L.; Aráoz, R.; Radić, Z.; Conrod, S.; Taylor, P.; Guillou, C.; Molgó, J.; Marchot, P. The Cyclic Imine Core Common to the Marine Macrocyclic Toxins Is Sufficient to Dictate Nicotinic Acetylcholine Receptor Antagonism. Mar. Drugs 2024, 22, 149. https://doi.org/10.3390/md22040149
Bourne Y, Sulzenbacher G, Chabaud L, Aráoz R, Radić Z, Conrod S, Taylor P, Guillou C, Molgó J, Marchot P. The Cyclic Imine Core Common to the Marine Macrocyclic Toxins Is Sufficient to Dictate Nicotinic Acetylcholine Receptor Antagonism. Marine Drugs. 2024; 22(4):149. https://doi.org/10.3390/md22040149
Chicago/Turabian StyleBourne, Yves, Gerlind Sulzenbacher, Laurent Chabaud, Rómulo Aráoz, Zoran Radić, Sandrine Conrod, Palmer Taylor, Catherine Guillou, Jordi Molgó, and Pascale Marchot. 2024. "The Cyclic Imine Core Common to the Marine Macrocyclic Toxins Is Sufficient to Dictate Nicotinic Acetylcholine Receptor Antagonism" Marine Drugs 22, no. 4: 149. https://doi.org/10.3390/md22040149