
Transcriptomics and Metabolomics Analyses Provide Novel Insights into Glucose-Induced Trophic Transition of the Marine Diatom Nitzschia laevis

 , ,
, ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
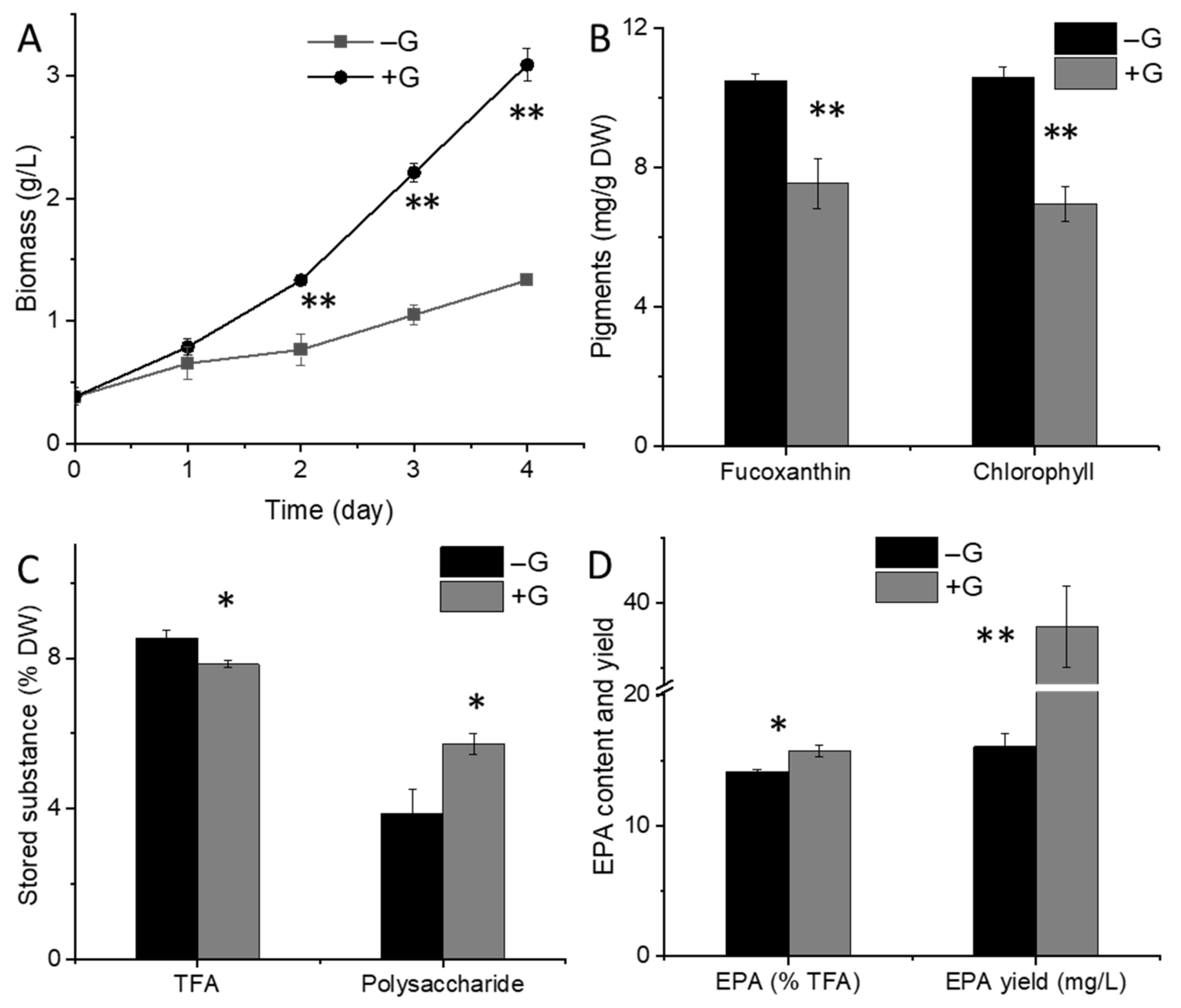
2.1. Physiological Changes in N. laevis under Glucose Addition
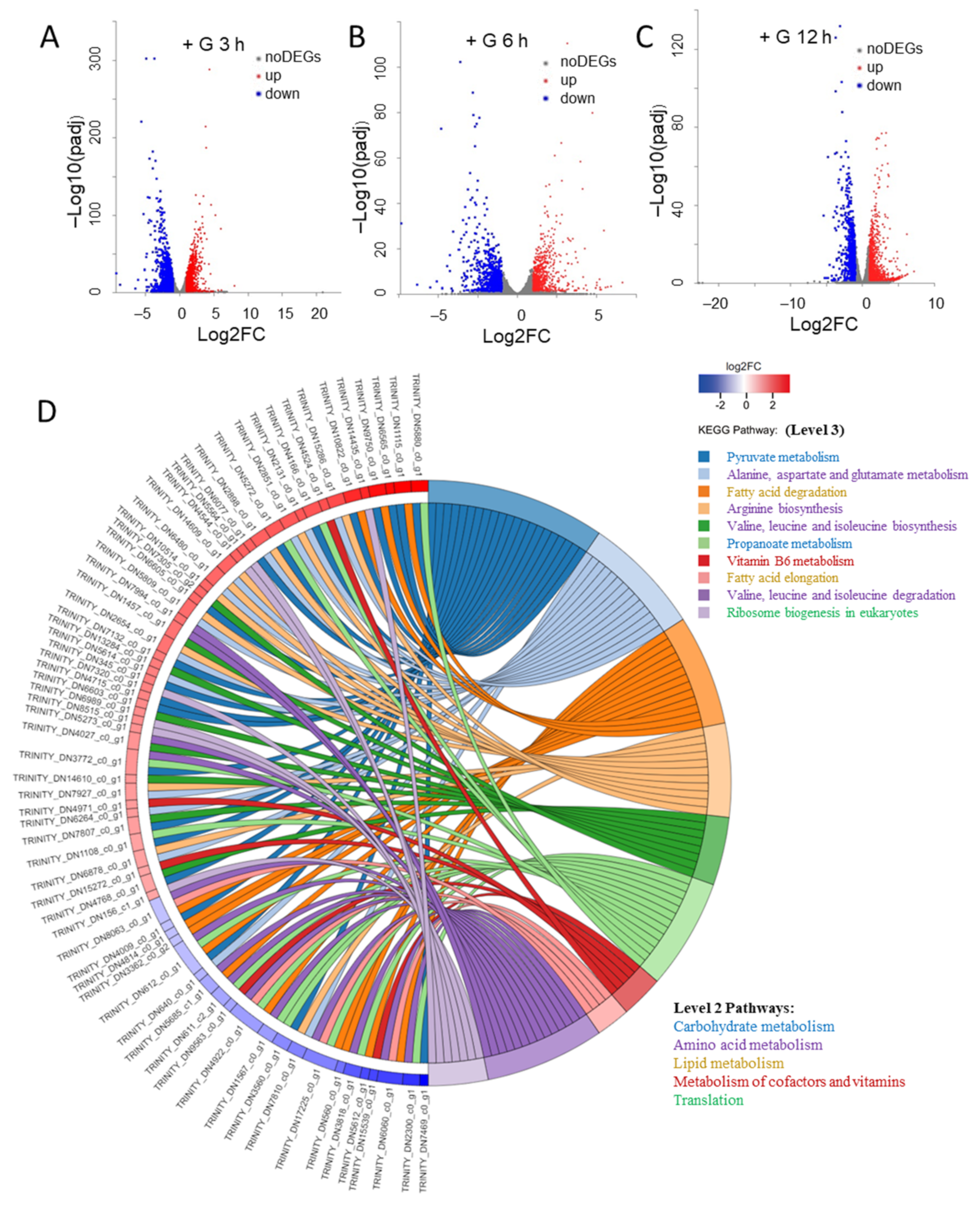
2.2. The Changes of Transcriptome during the Transition from Autotrophy to Mixotrophy
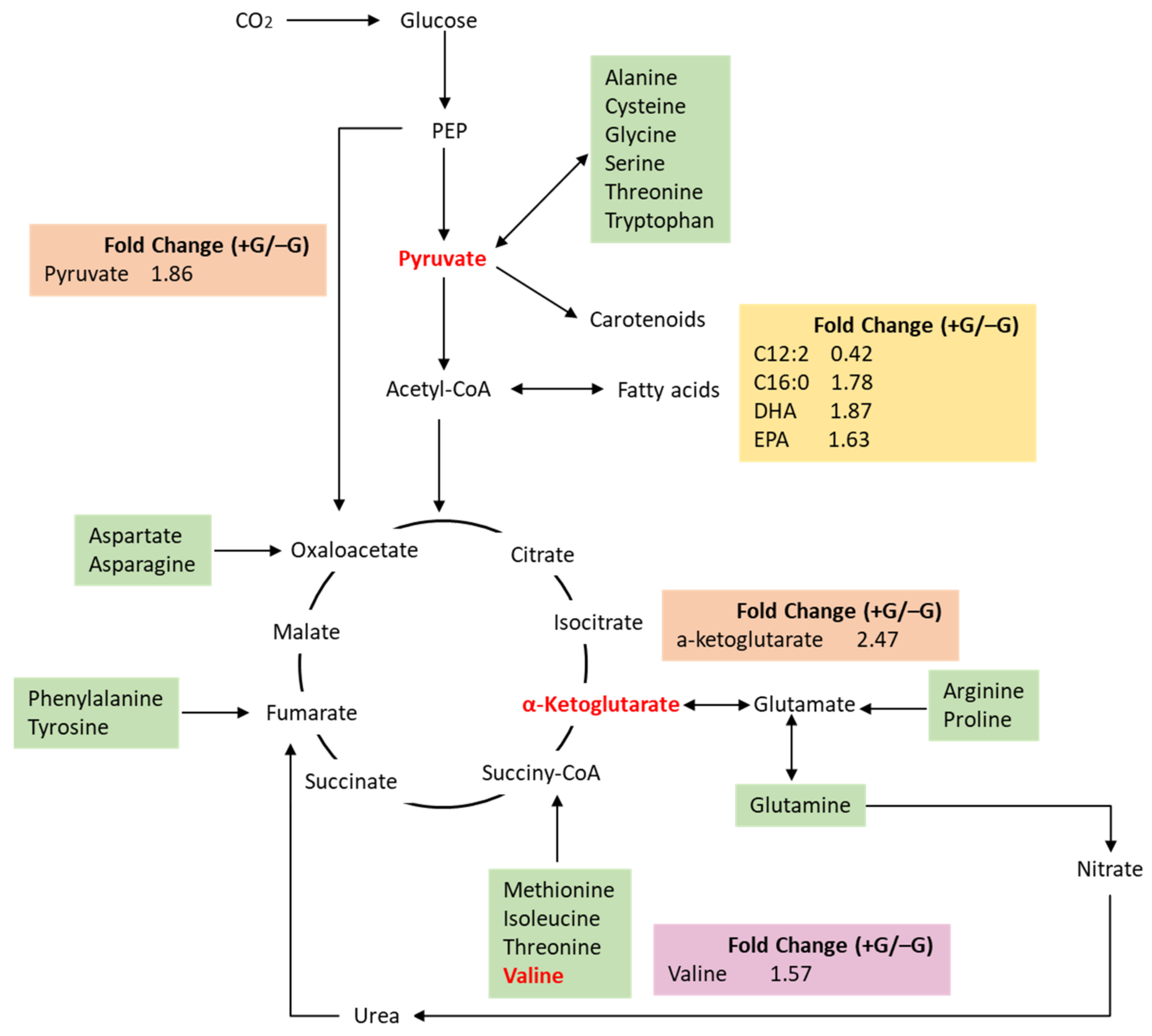
2.3. The Changes of Metabolites Profile during the Transition from Autotrophy to Mixotrophy
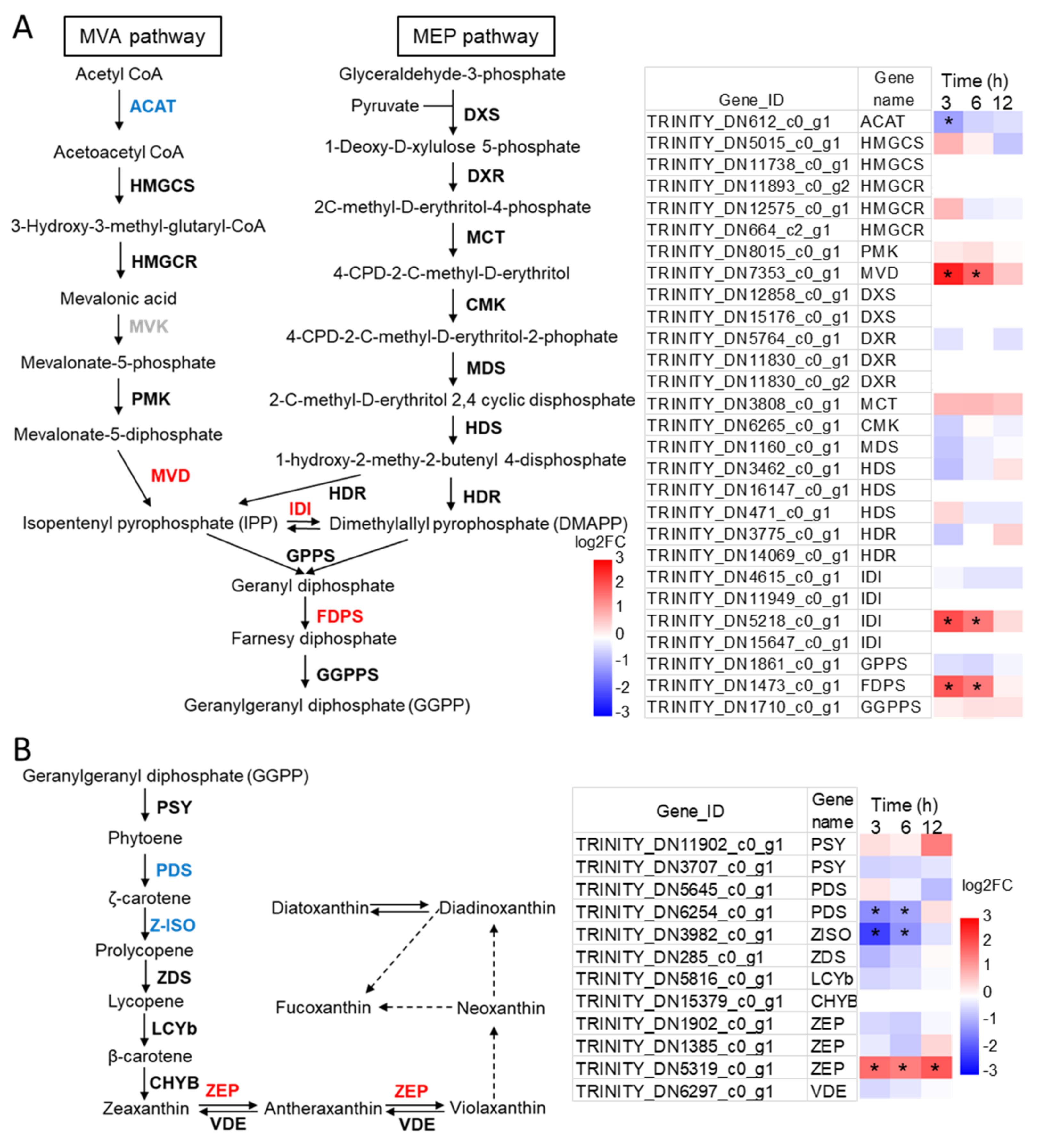
2.4. Identification of the Candidate Genes Involved in Carotenoids Biosynthesis and Their Expression Profiles
2.5. Alterations to FA Metabolism during the Transition from Autotrophy to Mixotrophy
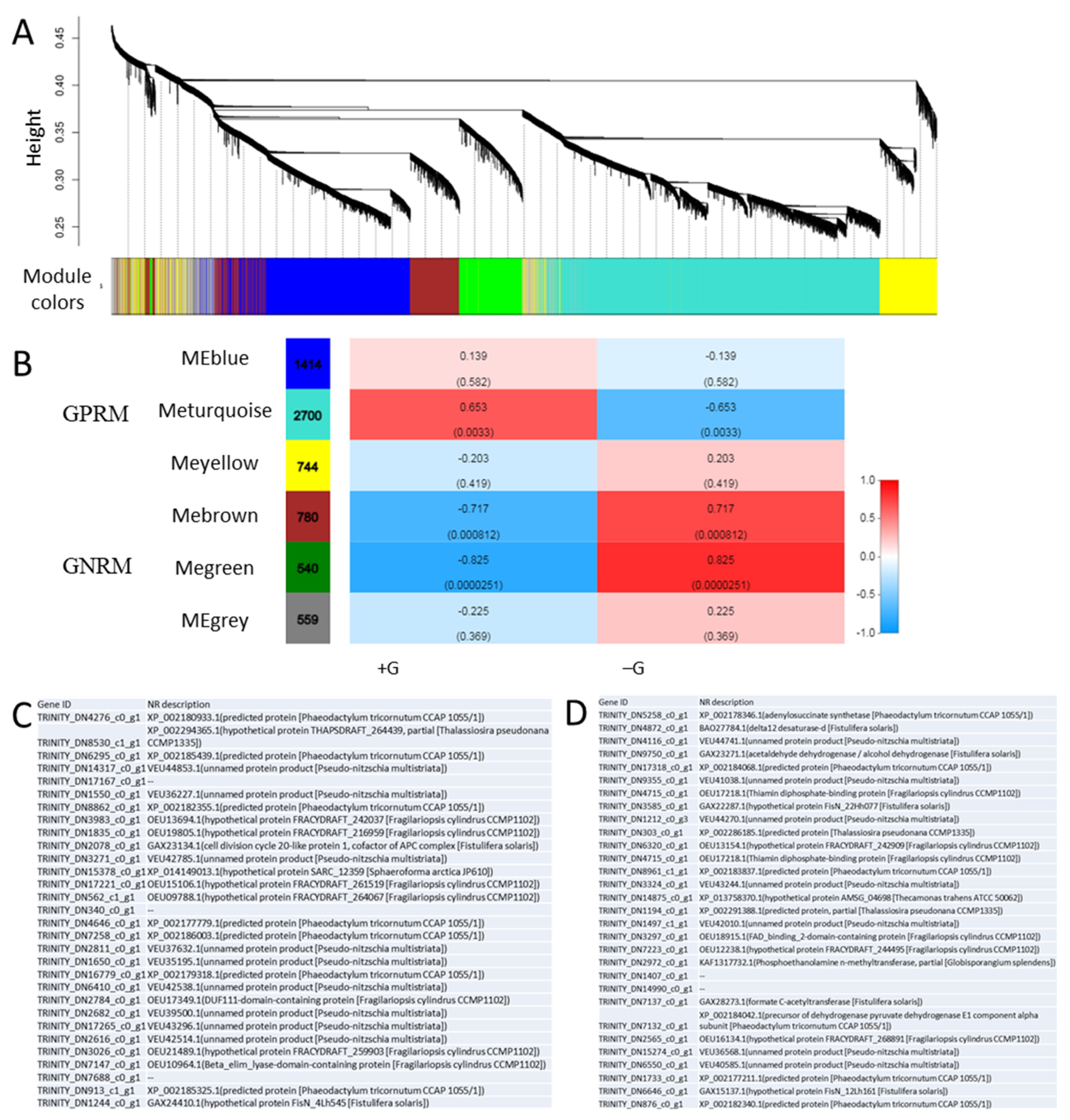
2.6. Weighted Gene Co-Expression Network Analysis (WGCNA)
3. Materials and Methods
3.1. Algal Strain and Cultivation Conditions
3.2. Lipids Analysis
3.3. Polysaccharides Content Analysis
3.4. Pigments in Photosynthesis Complexes
3.5. Transcriptomics Analysis
3.6. Weighted Gene Co-Expression Network (WGCNA)
3.7. Metabolomics Analysis
3.8. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Armbrust, E.V. The life of diatoms in the world’s oceans. Nature 2009, 459, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Falkowski, P.G.; Barber, R.T.; Smetacek, V.V. Biogeochemical Controls and Feedbacks on Ocean Primary Production. Science 1998, 281, 200–207. [Google Scholar] [CrossRef] [Green Version]
- Ragueneau, O.; Schultes, S.; Bidle, K.; Claquin, P.; Moriceau, B. Si and C interactions in the world ocean: Importance of ecological processes and implications for the role of diatoms in the biological pump. Glob. Biogeochem. Cycles 2006, 20. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharjya, R.; Kiran Marella, T.; Tiwari, A.; Saxena, A.; Kumar Singh, P.; Mishra, B. Bioprospecting of marine diatoms Thalassiosira, Skeletonema and Chaetoceros for lipids and other value-added products. Bioresour. Technol. 2020, 318, 124073. [Google Scholar] [CrossRef] [PubMed]
- Marella, T.K.; Tiwari, A. Marine diatom Thalassiosira weissflogii based biorefinery for co-production of eicosapentaenoic acid and fucoxanthin. Bioresour. Technol. 2020, 307, 123245. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Yuan, J.P.; Wu, C.F.; Wang, J.H. Fucoxanthin, a marine carotenoid present in brown seaweeds and diatoms: Metabolism and bioactivities relevant to human health. Mar. Drugs 2011, 9, 1806–1828. [Google Scholar] [CrossRef] [PubMed]
- Chuyen, H.V.; Eun, J.B. Marine carotenoids: Bioactivities and potential benefits to human health. Crit. Rev. Food Sci. 2017, 57, 2600–2610. [Google Scholar] [CrossRef]
- Endo, J.; Arita, M. Cardioprotective mechanism of omega-3 polyunsaturated fatty acids. J. Cardiol. 2016, 67, 22–27. [Google Scholar] [CrossRef] [Green Version]
- Kusaikin, M.I.; Ermakova, S.P.; Shevchenko, N.M.; Isakov, V.V.; Gorshkov, A.G.; Vereshchagin, A.L.; Grachev, M.A.; Zvyagintseva, T.N. Structural characteristics and antitumor activity of a new chrysolaminaran from the diatom alga Synedra acus. Chem. Nat. Compd. 2010, 46, 1–4. [Google Scholar] [CrossRef]
- D’Ippolito, G.; Sardo, A.; Paris, D.; Vella, F.M.; Adelfi, M.G.; Botte, P.; Gallo, C.; Fontana, A. Potential of lipid metabolism in marine diatoms for biofuel production. Biotechnol. Biofuels 2015, 8, 28. [Google Scholar] [CrossRef] [Green Version]
- Smith, S.R.; Abbriano, R.M.; Hildebrand, M. Comparative analysis of diatom genomes reveals substantial differences in the organization of carbon partitioning pathways. Algal Res. 2012, 1, 2–16. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.C.; Daboussi, F. Genetic and metabolic engineering in diatoms. Philos. Trans. R. Soc. B 2017, 372, 20160411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falciatore, A.; Jaubert, M.; Bouly, J.P.; Bailleul, B.; Mock, T. Diatom Molecular Research Comes of Age: Model Species for Studying Phytoplankton Biology and Diversity. Plant Cell 2020, 32, 547–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowler, C.; Allen, A.E.; Badger, J.H.; Grimwood, J.; Jabbari, K.; Kuo, A.; Maheswari, U.; Martens, C.; Maumus, F.; Otillar, R.P.; et al. The Phaeodactylum genome reveals the evolutionary history of diatom genomes. Nature 2008, 456, 239–244. [Google Scholar] [CrossRef]
- Remmers, I.M.; D’Adamo, S.; Martens, D.E.; de Vos, R.C.H.; Mumm, R.; America, A.H.P.; Cordewener, J.H.G.; Bakker, L.V.; Peters, S.A.; Wijffels, R.H.; et al. Orchestration of transcriptome, proteome and metabolome in the diatom Phaeodactylum tricornutum during nitrogen limitation. Algal Res. 2018, 35, 33–49. [Google Scholar] [CrossRef]
- Matthijs, M.; Fabris, M.; Obata, T.; Foubert, I.; Franco-Zorrilla, J.M.; Solano, R.; Fernie, A.R.; Vyverman, W.; Goossens, A. The transcription factor bZIP14 regulates the TCA cycle in the diatom Phaeodactylum tricornutum. EMBO J. 2017, 36, 1559–1576. [Google Scholar] [CrossRef] [Green Version]
- Zaslavskaia, L.A.; Lippmeier, J.C.; Shih, C.; Ehrhardt, D.; Grossman, A.R.; Apt, K.E. Trophic obligate conversion of a photoautotrophic organism through metabolic engineering. Science 2001, 292, 2073–2075. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Sun, H.; Zhao, W.; Cheng, K.-W.; Chen, F.; Liu, B. A Hetero-Photoautotrophic Two-Stage Cultivation Process for Production of Fucoxanthin by the Marine Diatom Nitzschia laevis. Mar. Drugs 2018, 16, 219. [Google Scholar] [CrossRef] [Green Version]
- Wen, Z.Y.; Chen, F. Heterotrophic production of eicosapentaenoid acid by the diatom Nitzschia laevis: Effects of silicate and glucose. J. Ind. Microbiol. Biotechnol. 2000, 25, 218–224. [Google Scholar] [CrossRef]
- Lu, X.; Liu, B.; He, Y.; Guo, B.; Sun, H.; Chen, F. Novel insights into mixotrophic cultivation of Nitzschia laevis for co-production of fucoxanthin and eicosapentaenoic acid. Bioresour. Technol. 2019, 294, 122145. [Google Scholar] [CrossRef]
- Rolland, F.; Baena-Gonzalez, E.; Sheen, J. Sugar sensing and signaling in plants: Conserved and novel mechanisms. Annu. Rev. Plant Biol. 2006, 57, 675–709. [Google Scholar] [CrossRef] [Green Version]
- Roth, M.S.; Gallaher, S.D.; Westcott, D.J.; Iwai, M.; Louie, K.B.; Mueller, M.; Walter, A.; Foflonker, F.; Bowen, B.P.; Ataii, N.N.; et al. Regulation of Oxygenic Photosynthesis during Trophic Transitions in the Green Alga Chromochloris zofingiensis. Plant Cell 2019, 31, 579–601. [Google Scholar] [CrossRef] [Green Version]
- Matthias, J.; Claudia, B. Properties of photosystem I antenna protein complexes of the diatom Cyclotella meneghiniana. J. Exp. Bot. 2012, 63, 3673–3681. [Google Scholar]
- Wang, W.; Yu, L.J.; Xu, C.; Tomizaki, T.; Zhao, S.; Umena, Y.; Chen, X.; Qin, X.; Xin, Y.; Suga, M.; et al. Structural basis for blue-green light harvesting and energy dissipation in diatoms. Science 2019, 363, eaav0365. [Google Scholar] [CrossRef] [PubMed]
- Markou, G.; Angelidaki, I.; Georgakakis, D. Microalgal carbohydrates: An overview of the factors influencing carbohydrates production, and of main bioconversion technologies for production of biofuels. Appl. Microbiol. Biotechnol. 2012, 96, 631–645. [Google Scholar] [CrossRef]
- Kroth, P.G.; Chiovitti, A.; Gruber, A.; Martin-Jezequel, V.; Mock, T.; Parker, M.S.; Stanley, M.S.; Kaplan, A.; Caron, L.; Weber, T.; et al. A Model for Carbohydrate Metabolism in the Diatom Phaeodactylum tricornutum Deduced from Comparative Whole Genome Analysis. PLoS ONE 2008, 3, e1426. [Google Scholar] [CrossRef] [Green Version]
- Wen, Z.Y.; Chen, F. Production potential of eicosapentaenoic acid by the diatom Nitzschia laevis. Biotechnol. Lett. 2000, 22, 727–733. [Google Scholar] [CrossRef]
- Kathuria, S.V.; Chan, Y.H.; Nobrega, R.P.; Ozen, A.; Matthews, C.R. Clusters of isoleucine, leucine, and valine side chains define cores of stability in high-energy states of globular proteins: Sequence determinants of structure and stability. Protein Sci. 2016, 25, 662–675. [Google Scholar] [CrossRef] [Green Version]
- Lohr, M.; Im, C.S.; Grossman, A.R. Genome-based examination of chlorophyll and carotenoid biosynthesis in Chlamydomonas reinhardtii. Plant Physiol. 2005, 138, 490–515. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Liu, M.J.; Ding, W.; Liu, J. Novel Insights into Phosphorus Deprivation Boosted Lipid Synthesis in the Marine Alga Nannochloropsis oceanica without Compromising Biomass Production. J. Agric. Food Chem. 2020, 68, 11488–11502. [Google Scholar] [CrossRef]
- Lichtenthaler, H.K. The 1-deoxy-D-xylulose-5-phosphate pathway of isoprenoid biosynthesis in plants. Annu. Rev. Plant Phys. 1999, 50, 47–65. [Google Scholar] [CrossRef]
- Zhan, X.R.; Liao, X.Y.; Luo, X.J.; Zhu, Y.J.; Feng, S.G.; Yu, C.N.; Lu, J.J.; Shen, C.J.; Wang, H.Z. Comparative Metabolomic and Proteomic Analyses Reveal the Regulation Mechanism Underlying MeJA-Induced Bioactive Compound Accumulation in Cutleaf Groundcherry (Physalis angulata L.) Hairy Roots. J. Agric. Food Chem. 2018, 66, 6336–6347. [Google Scholar] [CrossRef] [PubMed]
- Ajikumar, P.K.; Tyo, K.; Carlsen, S.; Mucha, O.; Phon, T.H.; Stephanopoulos, G. Terpenoids: Opportunities for biosynthesis of natural product drugs using engineered microorganisms. Mol. Pharm. 2008, 5, 167–190. [Google Scholar] [CrossRef]
- Mao, X.; Lao, Y.; Sun, H.; Li, X.; Yu, J.; Chen, F. Time-resolved transcriptome analysis during transitions of sulfur nutritional status provides insight into triacylglycerol (TAG) and astaxanthin accumulation in the green alga Chromochloris zofingiensis. Biotechnol. Biofuels 2020, 13, 128. [Google Scholar] [CrossRef]
- Coesel, S.; Obornik, M.; Varela, J.; Falciatore, A.; Bowler, C. Evolutionary Origins and Functions of the Carotenoid Biosynthetic Pathway in Marine Diatoms. PLoS ONE 2008, 3, e2896. [Google Scholar] [CrossRef] [Green Version]
- Murchie, E.H.; Ruban, A.V. Dynamic non-photochemical quenching in plants: From molecular mechanism to productivity. Plant J. 2020, 101, 885–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertrand, M. Carotenoid biosynthesis in diatoms. Photosynth. Res. 2010, 106, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Athanasakoglou, A.; Kampranis, S.C. Diatom isoprenoids: Advances and biotechnological potential. Biotechnol. Adv. 2019, 37, 107417. [Google Scholar] [CrossRef]
- Chen, H.H.; Jiang, J.G. Lipid Accumulation Mechanisms in Auto-and Heterotrophic Microalgae. J. Agric. Food Chem. 2017, 65, 8099–8110. [Google Scholar] [CrossRef]
- Garay, L.A.; Boundy-Mills, K.L.; German, J.B. Accumulation of High-Value Lipids in Single-Cell Microorganisms: A Mechanistic Approach and Future Perspectives. J. Agric. Food Chem. 2014, 62, 2709–2727. [Google Scholar] [CrossRef]
- Wu, T.; Fu, Y.L.; Shi, Y.; Li, Y.L.; Kou, Y.P.; Mao, X.M.; Liu, J. Functional Characterization of Long-Chain Acyl-CoA Synthetase Gene Family from the Oleaginous Alga Chromochloris zofingiensis. J. Agric. Food Chem. 2020, 68, 4473–4484. [Google Scholar] [CrossRef]
- Kevei, Z.; Baloban, M.; Da Ines, O.; Tiricz, H.; Kroll, A.; Regulski, K.; Mergaert, P.; Kondorosi, E. Conserved CDC20 cell cycle functions are carried out by two of the five isoforms in Arabidopsis thaliana. PLoS ONE 2011, 6, e20618. [Google Scholar] [CrossRef]
- Eguren, M.; Porlan, E.; Manchado, E.; Garcia-Higuera, I.; Canamero, M.; Farinas, I.; Malumbres, M. The APC/C cofactor Cdh1 prevents replicative stress and p53-dependent cell death in neural progenitors. Nat. Commun. 2013, 4, 2880. [Google Scholar] [CrossRef] [Green Version]
- Buttigieg, P.L.; Hankeln, W.; Kostadinov, I.; Kottmann, R.; Yilmaz, P.; Duhaime, M.B.; Glockner, F.O. Ecogenomic perspectives on domains of unknown function: Correlation-based exploration of marine metagenomes. PLoS ONE 2013, 8, e50869. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.H.; Taylor, M.C.; Barrow, R.A.; Croyal, M.; Masle, J. Loss of Phosphoethanolamine N-Methyltransferases Abolishes Phosphatidylcholine Synthesis and Is Lethal. Plant Physiol. 2019, 179, 124–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czumaj, A.; Sledzinski, T. Biological Role of Unsaturated Fatty Acid Desaturases in Health and Disease. Nutrients 2020, 12, 356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeffrey, S.W.; Humphrey, G.F. New Spectrophotometric Equations for Determining Chlorophylls a, B, C1 and C2 in Higher-Plants, Algae and Natural Phytoplankton. Biochem. Physiol. Pflanz. 1975, 167, 191–194. [Google Scholar] [CrossRef]
- Wagner, G.P.; Kin, K.; Lynch, V.J. Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples. Theory Biosci. 2012, 131, 281–285. [Google Scholar] [CrossRef]
- Wen, B.; Mei, Z.L.; Zeng, C.W.; Liu, S.Q. metaX: A flexible and comprehensive software for processing metabolomics data. BMC Bioinform. 2017, 18, 183. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mao, X.; Ge, M.; Wang, X.; Yu, J.; Li, X.; Liu, B.; Chen, F. Transcriptomics and Metabolomics Analyses Provide Novel Insights into Glucose-Induced Trophic Transition of the Marine Diatom Nitzschia laevis. Mar. Drugs 2021, 19, 426. https://doi.org/10.3390/md19080426
Mao X, Ge M, Wang X, Yu J, Li X, Liu B, Chen F. Transcriptomics and Metabolomics Analyses Provide Novel Insights into Glucose-Induced Trophic Transition of the Marine Diatom Nitzschia laevis. Marine Drugs. 2021; 19(8):426. https://doi.org/10.3390/md19080426
Chicago/Turabian StyleMao, Xuemei, Mengdie Ge, Xia Wang, Jianfeng Yu, Xiaojie Li, Bin Liu, and Feng Chen. 2021. "Transcriptomics and Metabolomics Analyses Provide Novel Insights into Glucose-Induced Trophic Transition of the Marine Diatom Nitzschia laevis" Marine Drugs 19, no. 8: 426. https://doi.org/10.3390/md19080426